Novel Conformationally Constrained Analogues of Agomelatine as New Melatoninergic Ligands

Abstract
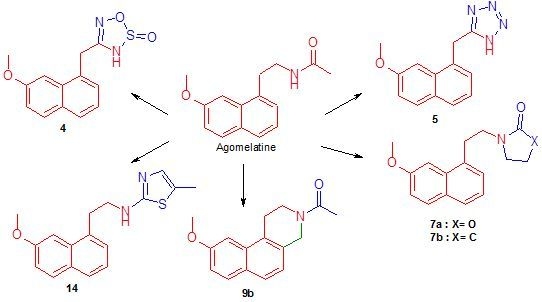
:
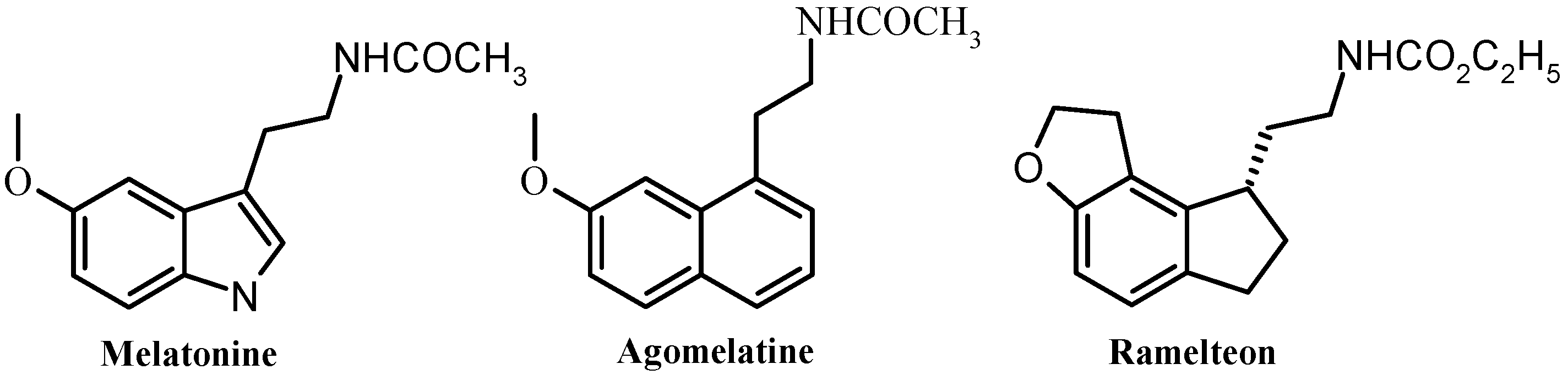
1. Introduction

2. Results and Discussion
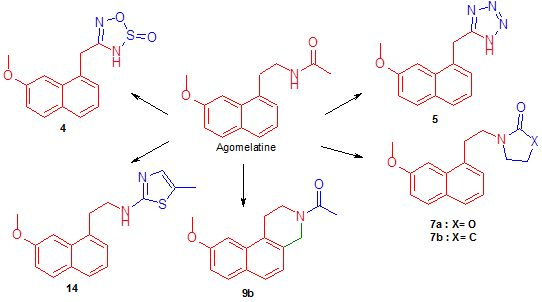
2.1. Chemistry
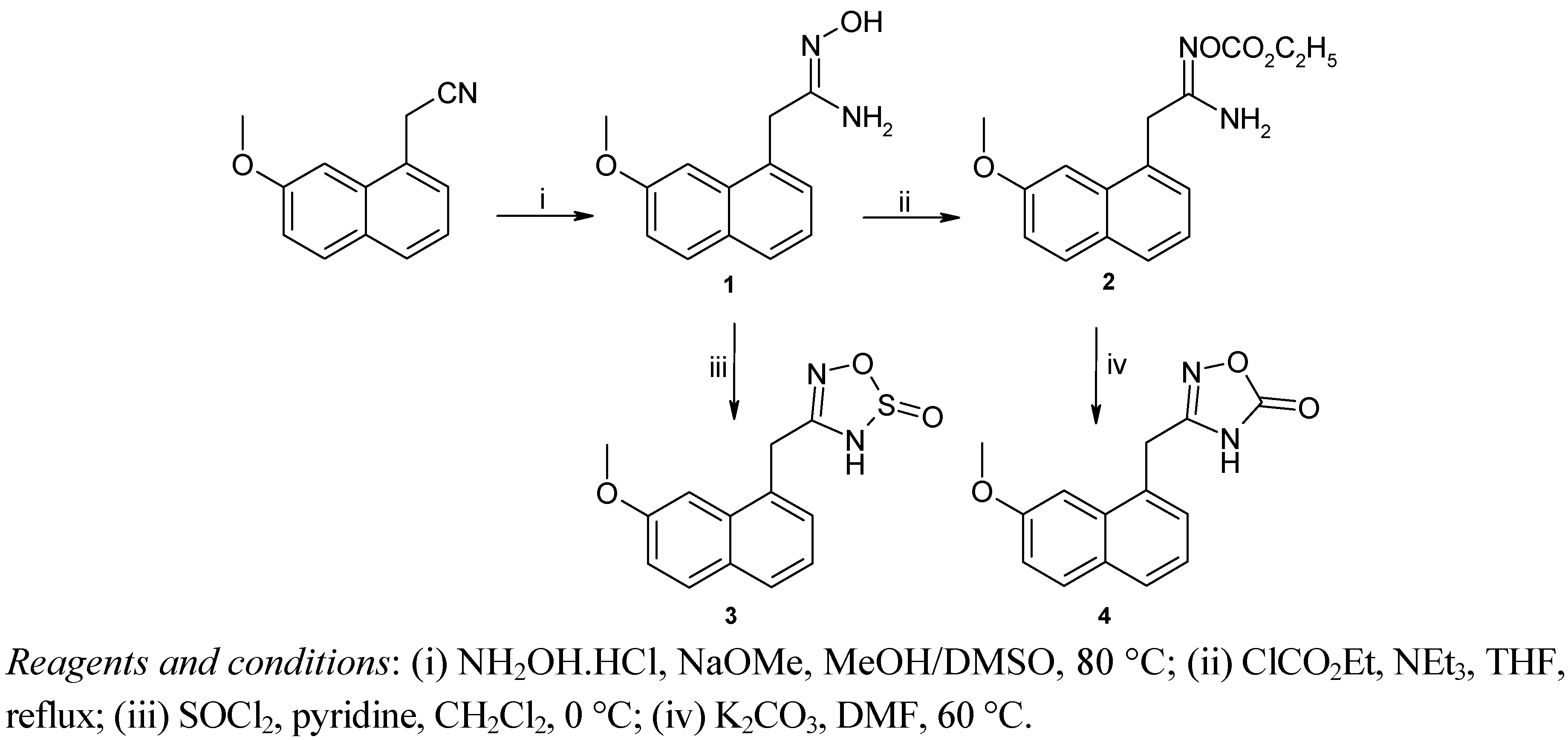


2.2. Pharmacology
2.2.1. Reagents and Chemicals
2.2.2. Assays for MT1 and MT2 Receptor Subtypes
2.3. Discussion

{kind=link}
{kind=link}
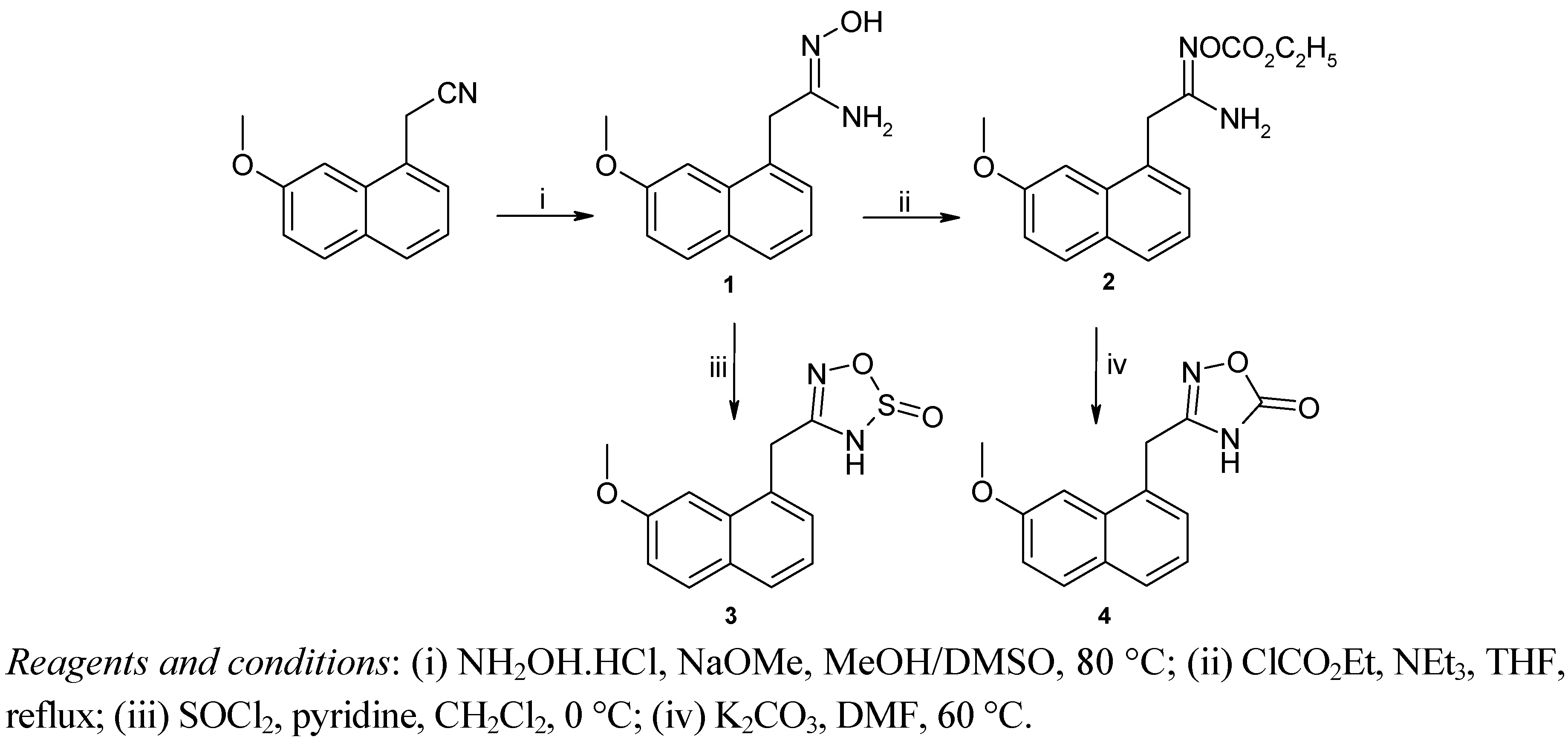{kind=link}
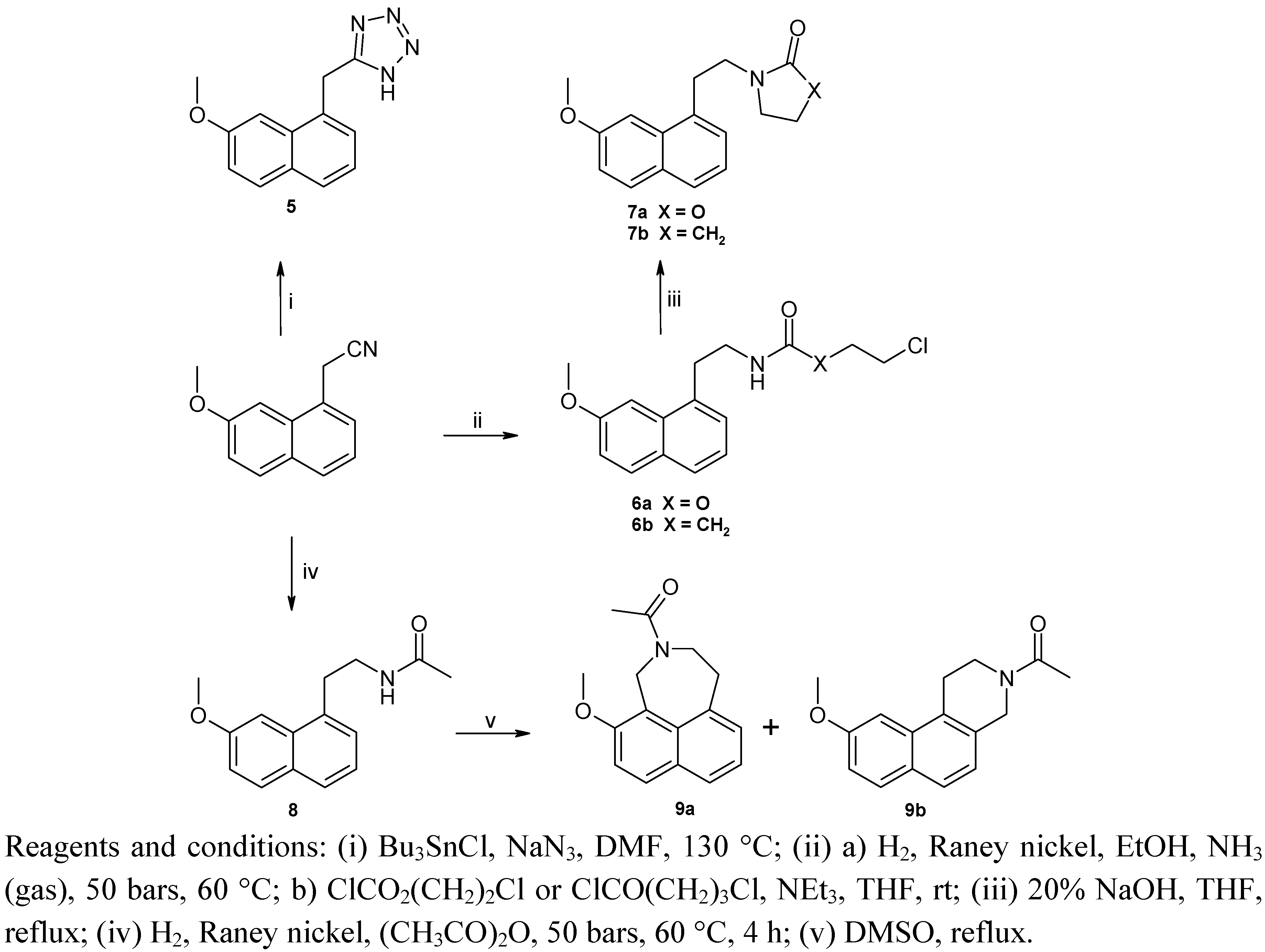{kind=link}
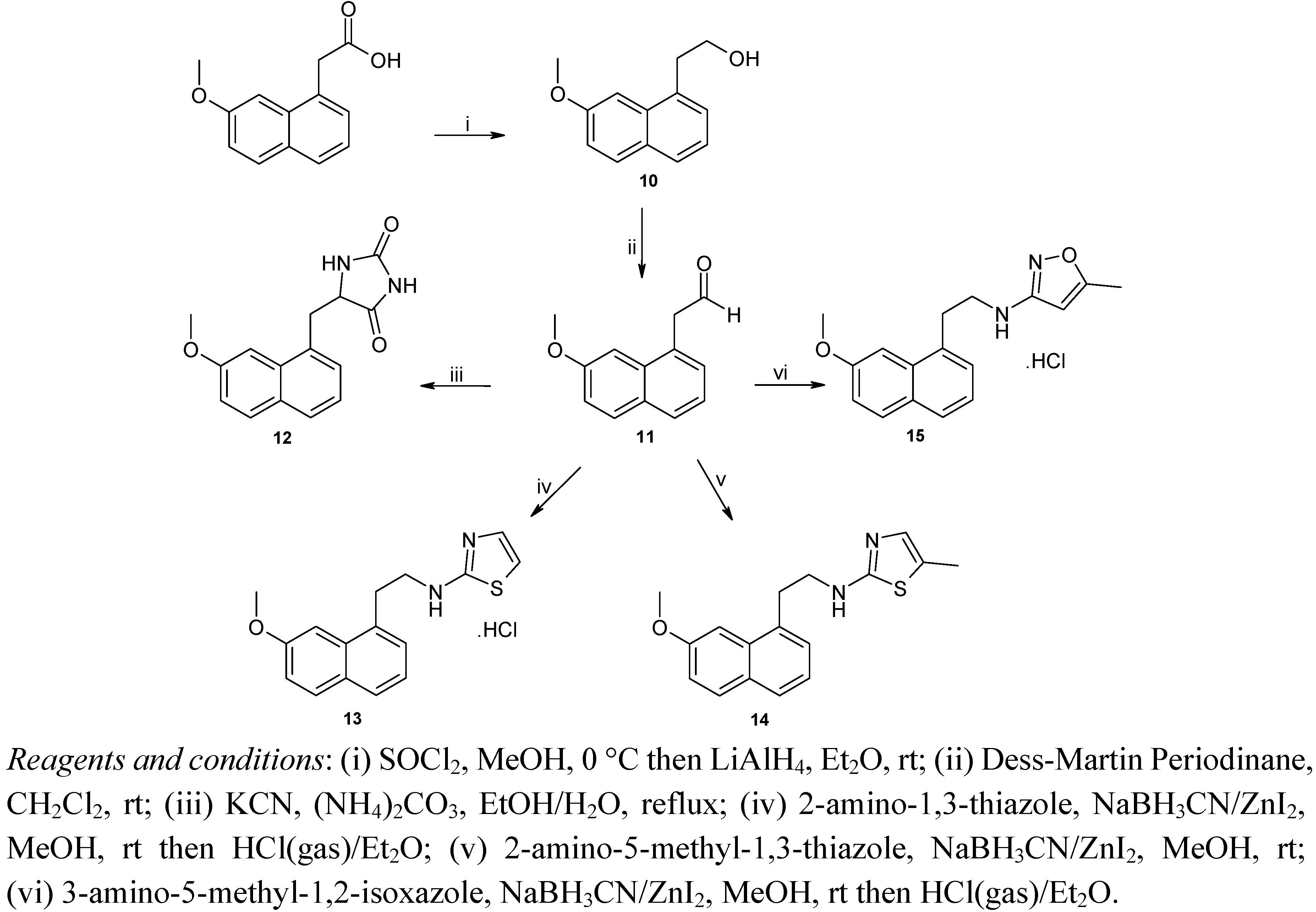{kind=link}
| Compound | Ki (nM) MT1 | Ki (nM) MT2 | S (MT1/MT2) |
|---|---|---|---|
| Melatonin | 0.2 ± 0.02 | 0.3 ± 0.03 | 0.17 |
| Agomelatine | 0.1 ± 0.01 | 0.12 ± 0.02 | 0.83 |
| 3 | >1,000 | 1300 | >0.77 |
| 4 | 80.0 ± 16.0 | 25.2 ± 10.7 | 3 |
| 5 | 380 ± 114 | 190 ± 84 | 2 |
| 7a | 113 ± 18 | 6.0 ± 0.2 | 19 |
| 7b | 68.5 ± 18.2 | 2.1 ± 0.01 | 33 |
| 9a | 2500 ± 57 | 469 ± 44 | 5.4 |
| 9b | 3000 ± 27 | 800 ± 59 | 3.75 |
| 12 | 2170 ± 63 | 141 ± 31 | 15.4 |
| 13 | 33.9 ± 8.1 | 12.5 ± 0.4 | 3 |
| 14 | 389 ± 121 | 354 ± 32 | 1 |
| 15 | 48.4 ± 11.2 | 20.3 ± 3.9 | 2 |
3. Experimental
3.1. General
3.2. General Protocol for the Preparation of Compounds 9a and 9b
3.3. General Protocol for the Preparation of Compounds 13–15
4. Conclusions
Acknowledgments
References
- Lerner, A.B.; Case, J.D.; Heinzelman, R.V. Structure of melatonin. J. Am. Chem. Soc. 1959, 81, 6084–6085. [Google Scholar]
- Guerrero, J.M.; Reiter, R.J. Melatonin-Immune System Relationships. Curr. Top. Med. Chem. 2002, 2, 167–169. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Mansana, M.I.; Benloucif, S. Molecular pharmacology and function of melatonin receptor subtype. Adv. Exp. Med. Biol. 1999, 460, 181–190. [Google Scholar]
- Malpaux, B.; Migaud, M.; Tricoire, H.; Chemineau, P. Biology of Mammalian Photoperiodism and the Critical Role of the Pineal Gland and Melatonin. J. Biol. Rhythms. 2001, 16, 336–347. [Google Scholar] [CrossRef]
- Reiter, R.J. The melatonin rhythm: Both a clock and a calendar. Experientia 1993, 49, 654–664. [Google Scholar] [CrossRef]
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. MT1 melatonin receptors mediate somatic, behavioral, and reproductive neuroendocrine responses to photoperiod and melatonin in Siberian hamsters. J. Biol. Chem. 2000, 275, 31311–31317. [Google Scholar]
- Vella, F.; Ferry, G.; Delagrange, P.; Boutin, J.A. NRH: Quinone reductase 2: An enzyme of surprises and mysteries. Biochem. Pharmacol. 2005, 71, 1–12. [Google Scholar] [CrossRef]
- Lotufo, C.M.C.; Lopes, C.; Dubocovich, M.L.; Farsky, S.H.P.; Markus, R.P. Melatonin and N-acetylserotonin inhibit leukocyte rolling and adhesion to rat microcirculation. Eur. J. Pharmacol. 2001, 430, 351–357. [Google Scholar] [CrossRef]
- Pintor, J.; Martin, L.; Pelaez, T.; Hoyle, C.H.V.; Peral, A. Involvement of melatonin MT3 receptors in the regulation of intraocular pressure in rabbits. Eur. J. Pharmacol. 2001, 416, 251–254. [Google Scholar] [CrossRef]
- Zlotos, D.P. Recent Advances in Melatonin Receptor Ligands. Arch. Pharm. Chem. Life Sci. 2005, 338, 229–247. [Google Scholar] [CrossRef]
- Garratt, P.J.; Tsotinis, A. Synthesis of compounds as melatonin agonists and antagonists. Mini-Rev. Med. Chem. 2007, 7, 1075–1088. [Google Scholar] [CrossRef]
- Carocci, A.; Catalano, A.; Lovece, A.; Lentini, G.; Duranti, A.; Lucini, V.; Pannacci, M.; Scaglione, F.; Franchini, C. Design, synthesis, and pharmacological effects of structurally simple ligands for MT1 and MT2 melatonin receptors. Bioorg. Med. Chem. 2010, 18, 6496–6511. [Google Scholar] [CrossRef]
- Tsotinis, A.; Afroudakis, P.A.; Davidson, K.; Prashar, A.; Sugden, D. Design, Synthesis, and Melatoninergic Activity of New Azido- and Isothiocyanato-Substituted Indoles. J. Med. Chem. 2007, 50, 6436–6440. [Google Scholar] [CrossRef]
- Nonno, R.; Pannacci, M.; Lucini, V.; Angeloni, D.; Fraschini, F.; Stankov, B.M. Ligand efficacy and potency at recombinant human MT2 melatonin receptors: Evidence for agonist activity of some MT1-antagonists. Br. J. Pharmacol. 1999, 127, 1288–1294. [Google Scholar] [CrossRef]
- Fisher, S.P.; Sugden, D. Sleep-promoting action of IIK7, a selective MT2 melatonin receptor agonist in the rat. Neurosci. Lett. 2009, 457, 93–96. [Google Scholar] [CrossRef]
- Koike, T.; Hoashi, Y.; Takai, T.; Nakayama, M.; Yukuhiro, N.; Ishikawa, T.; Hirai, K.; Uchikawa, O. 1,6-Dihydro-2H-indeno[5,4-b]furan derivatives: Design, synthesis and pharmacological characterization of a novel class of highly potent MT2-selective agonists. J. Med. Chem. 2011, 54, 3436–3444. [Google Scholar] [CrossRef]
- Ettaoussi, M.; Sabaouni, A.; Rami, M.; Boutin, J.A.; Delagrange, P.; Renard, P.; Spedding, M.; Caignard, D.H.; Berthelot, P.; Yous, S. Design, synthesis and pharmacological evaluation of new series of naphthalenicanalogues as melatoninergic (MT1/MT2) and serotoninergic 5-HT2C dual ligands (I). Eur. J. Med. Chem. 2012, 49, 310–323. [Google Scholar]
- Markl, C.; Attia, M.I.; Julius, J.; Sethi, S.; Witt-Enderby, P.A.; Zlotos, D.P. Synthesis and pharmacological evaluation of 1,2,3,4-tetrahydropyrazino [1,2-a]indole and 2-[(phenylmethyl-amino)methyl]-1H-indole analogues as novel melatoninergic ligands. Bioorg. Med. Chem. 2009, 17, 4583–4594. [Google Scholar] [CrossRef]
- Attia, M.I.; Witt-Enderby, P.A.; Julius, J. Synthesis and pharmacological evaluation of pentacyclic 6a,7-dihydrodiindole and 2,3-dihydrodiindole derivatives as novel melatoninergic ligands. Bioorg. Med. Chem. 2008, 16, 7654–7661. [Google Scholar] [CrossRef]
- Simpson, D.; Curran, M.P. Ramelteon: A review of its use in Insomnia. Drugs 2008, 68, 1901–1919. [Google Scholar] [CrossRef]
- Montgomery, S.A. Major depressive disorders: Clinical efficacy and tolerability of agomelatine, a new melatonergicagonist. Eur. Neuropsychopharmacol. 2006, 16, S633–S638. [Google Scholar] [CrossRef]
- Dagyte, G.; Trentani, A.; Postema, F.; Luiten, P.G.; Den Boer, J.A.; Gabriel, C.; Mocaër, E.; Meerlo, P.; Van der Zee, E. The novel antidepressant agomelatine normalizes hippocampal neuronal activity and promotes neurogenesis in chronically stressed rats. CNS Neurosci. Ther. 2010, 16, 195–207. [Google Scholar] [CrossRef]
- Dagyte, G.; Luiten, P.G.; De Jager, T.; Gabriel, C.; Mocaër, E.; Den Boer, J.A.; Van der Zee, E. Chronic stress and antidepressant agomelatine induce region-specific changes in synapsin I expression in the rat brain. J. Neurosci. Res. 2011, 89, 1646–1657. [Google Scholar] [CrossRef]
- Mathé-Allainmat, M.; Andrieux, J.; Langlois, M. Recent developments in melatonin receptor ligands. Expert Opin. Ther. Pat. 1997, 7, 1447–1458. [Google Scholar] [CrossRef]
- Marot, C.; Chavatte, P.; Morin-Allory, L.; Guillaumet, G.; Viaud-Massuard, M.C.; Renard, P.; Lesieur, D.; Michel, A. Pharmacophoric Search and 3D-QSAR Comparative Molecular Field Analysis Studies on Agonists of Melatonin Sheep Receptors. J. Med. Chem. 1998, 41, 4453–4465. [Google Scholar] [CrossRef]
- Mésangeau, C.; Pérès, B.; Descamps-François, C.; Chavatte, P.; Audinot, V.; Coumailleau, S.; Boutin, J.A.; Delagrange, P. Design, synthesis and pharmacologicalevaluation of novelnaphthalenicderivatives as selective MT1 melatoninergic ligands. Bioorg. Med. Chem. 2010, 18, 3426–3436. [Google Scholar] [CrossRef]
- Descamps-François, C.; Yous, S.; Chavatte, P.; Audinot, V.; Bonnaud, A.; Boutin, J.A.; Delagrange, P.; Bennejean, C.; Renard, P.; Lesieur, D. Design and synthesis of naphthalenic dimers as selective MT1 melatoninergic ligands. J. Med. Chem. 2003, 46, 1127–1129. [Google Scholar] [CrossRef]
- Mésangeau, C.; Fraise, M.; Delagrange, P.; Caignard, D.H.; Boutin, J.A.; Berthelot, P.; Yous, S. Preparation and pharmacological evaluation of a novel series of 2-(phenylthio) benzo[b]thiophenes as selective MT2 receptor ligands. Eur. J. Med. Chem. 2011, 46, 1835–1840. [Google Scholar] [CrossRef]
- Durieux, S.; Chanu, A.; Bochu, C.; Audinot, V.; Coumailleau, S.; Boutin, J.A.; Delagrange, P.; Caignard, D.H.; Bennejean, C.; Renard, P.; et al. Design and synthesis of 3-phenyltetrahydronaphthalenic derivatives as new selective MT2 melatoninergic ligands. Part II. Bioorg. Med. Chem. 2009, 17, 2963–2974. [Google Scholar] [CrossRef]
- Poissonnier-Durieux, S.; Ettaoussi, M.; Pérès, B.; Boutin, J.A.; Audinot, V.; Bennejean, C.; Delagrange, P.; Caignard, D.H.; Renard, P.; Berthelot, P.; et al. Synthesis of 3-phenylnaphthalenic derivatives as new selective MT2 melatoninergic ligands. Bioorg. Med. Chem. 2008, 16, 8339–8348. [Google Scholar]
- Yous, S.; Durieux-Poissonnier, S.; Lipka-Belloli, E.; Guelzim, H.; Bochu, C.; Audinot, V.; Boutin, J.A.; Delagrange, P.; Bennejean, C.; Renard, P.; et al. Design and synthesis of 3-phenyl tetrahydronaphthalenic derivatives as new selective MT2 melatoninergic ligands. Bioorg. Med. Chem. 2003, 11, 753–759. [Google Scholar] [CrossRef]
- Leclerc, V.; Ettaoussi, M.; Rami, M.; Farce, A.; Boutin, J.A.; Delagrange, P.; Caignard, D.H.; Renard, P.; Berthelot, P.; Yous, S. Design and synthesis of naphthalenic derivatives as new ligands at the melatoninbinding site MT3. Eur. J. Med. Chem. 2011, 46, 1622–1629. [Google Scholar] [CrossRef]
- Ettaoussi, M.; Péres, B.; Klupsch, F.; Delagrange, P.; Boutin, J.A.; Renard, P.; Caignard, D.H.; Chavatte, P.; Berthelot, P.; Lesieur, D.; et al. Design and synthesis of benzofuranic derivatives as new ligandsat the melatonin-binding site MT3. Bioorg. Med. Chem. 2008, 16, 4954–4562. [Google Scholar] [CrossRef]
- Leclerc, V.; Yous, S.; Delagrange, P.; Boutin, J.A.; Renard, P.; Lesieur, D. Synthesis of Nitroindole Derivatives with High Affinity and Selectivity for Melatoninergic Binding Sites MT3. J. Med. Chem. 2002, 45, 1853–1859. [Google Scholar] [CrossRef]
- Yous, S.; Andrieux, J.; Howell, H.E.; Morgan, P.J.; Renard, P.; Pfeiffer, B.; Lessieur, D.; Guardiola-Lemaitre, B. Novel naphthalenic ligands with high affinity for the melatonin receptor. J. Med. Chem. 1992, 35, 1484–1486. [Google Scholar] [CrossRef]
- Assogba, L.; Ahamada-Himidi, A.; Meddad-Bel Habich, N.; Aoun, D.; Boukli, L.; Massicot, F.; Mounier, C.M.; Huet, J.; Lamouri, A.; Ombetta, J.E.; et al. Inhibition ofsecretoryphospholipase A2. Design, synthesis and structure–activity relationship studies starting from 4-tetradecyloxy-benzamidine to obtain specific inhibitors of group II sPLA2s. Eur. J. Med. Chem. 2005, 40, 850–861. [Google Scholar] [CrossRef]
- Boukli, L.; Touaibia, M.; Meddad-Belhabich, N.; Djimdé, A.; Park, C.H.; Kim, J.J.; Yoon, J.H.; Lamouri, A.; Heymans, F. Design of new potent and selective secretory phospholipase A2 inhibitors. Part 5: Synthesis and biological activity of 1-alkyl-4-[4,5-dihydro-1,2,4-[4H]-oxadiazol-5-one-3-ylmethylbenz-4′-yl(oyl)] piperazines. Bioorg. Med. Chem. 2008, 16, 1242–1253. [Google Scholar] [CrossRef]
- Lenda, F.; Guenoun, F.; Tazi, B.; Ben Larbi, N.; Allouchi, H.; Martinez, J.; Lamaty, F. Synthesis of New Tetrazole-Substituted Pyroaminoadipic and Pipecolic Acid Derivatives. Eur. J. Org. Chem. 2005, 326–333. [Google Scholar]
- McKay, A.F.; Braun, R.O. Cyclizationsof β-Chloroethyl Substituted Amminocarbonic Acids. J. Org. Chem. 1951, 16, 1829–1832. [Google Scholar] [CrossRef]
- Depreux, P.; Lesieur, D.; Ait Mansour, H.; Morgan, P.; Howell, H.E.; Renard, P.; Caignard, D.H.; Pfeiffer, B.; Delagrange, P.; Guardiola, B.; et al. Synthesis and Structure-Activity Relationships of Novel Naphthalenic and Bioisosteric Related Amidic Derivatives as Melatonin Receptor Ligands. J. Med. Chem. 1994, 37, 3231–3239. [Google Scholar] [CrossRef]
- Mésangeau, C.; Yous, S.; Pérèz, B.; Lesieur, D.; Besson, T. Pictet–Spengler heterocyclizations via microwave-assisted degradation of DMSO. Tetrahedron Lett. 2005, 46, 2465–2468. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar] [CrossRef]
- Audinot, V.; Mailliet, F.; Lahaye-Brasseur, C.; Bonnaud, A.; Le Gall, A.; Amossé, C.; Dromaint, S.; Rodriguez, M.; Nagel, N.; Galizzi, J.P.; et al. New selective ligands of human cloned melatonin MT1 and MT2 receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 2003, 36, 7553–7561. [Google Scholar]
- Sample Availability: Samples of the compounds 5, 13, 14 and 15 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rami, M.; Landagaray, E.; Ettaoussi, M.; Boukhalfa, K.; Caignard, D.-H.; Delagrange, P.; Berthelot, P.; Yous, S. Novel Conformationally Constrained Analogues of Agomelatine as New Melatoninergic Ligands. Molecules 2013, 18, 154-166. https://doi.org/10.3390/molecules18010154
Rami M, Landagaray E, Ettaoussi M, Boukhalfa K, Caignard D-H, Delagrange P, Berthelot P, Yous S. Novel Conformationally Constrained Analogues of Agomelatine as New Melatoninergic Ligands. Molecules. 2013; 18(1):154-166. https://doi.org/10.3390/molecules18010154
Chicago/Turabian StyleRami, Marouan, Elodie Landagaray, Mohamed Ettaoussi, Koussayla Boukhalfa, Daniel-Henri Caignard, Philippe Delagrange, Pascal Berthelot, and Saïd Yous. 2013. "Novel Conformationally Constrained Analogues of Agomelatine as New Melatoninergic Ligands" Molecules 18, no. 1: 154-166. https://doi.org/10.3390/molecules18010154