Optimization of a Pre-MEKC Separation SPE Procedure for Steroid Molecules in Human Urine Samples

Abstract
:1. Introduction
2. Results and Discussion
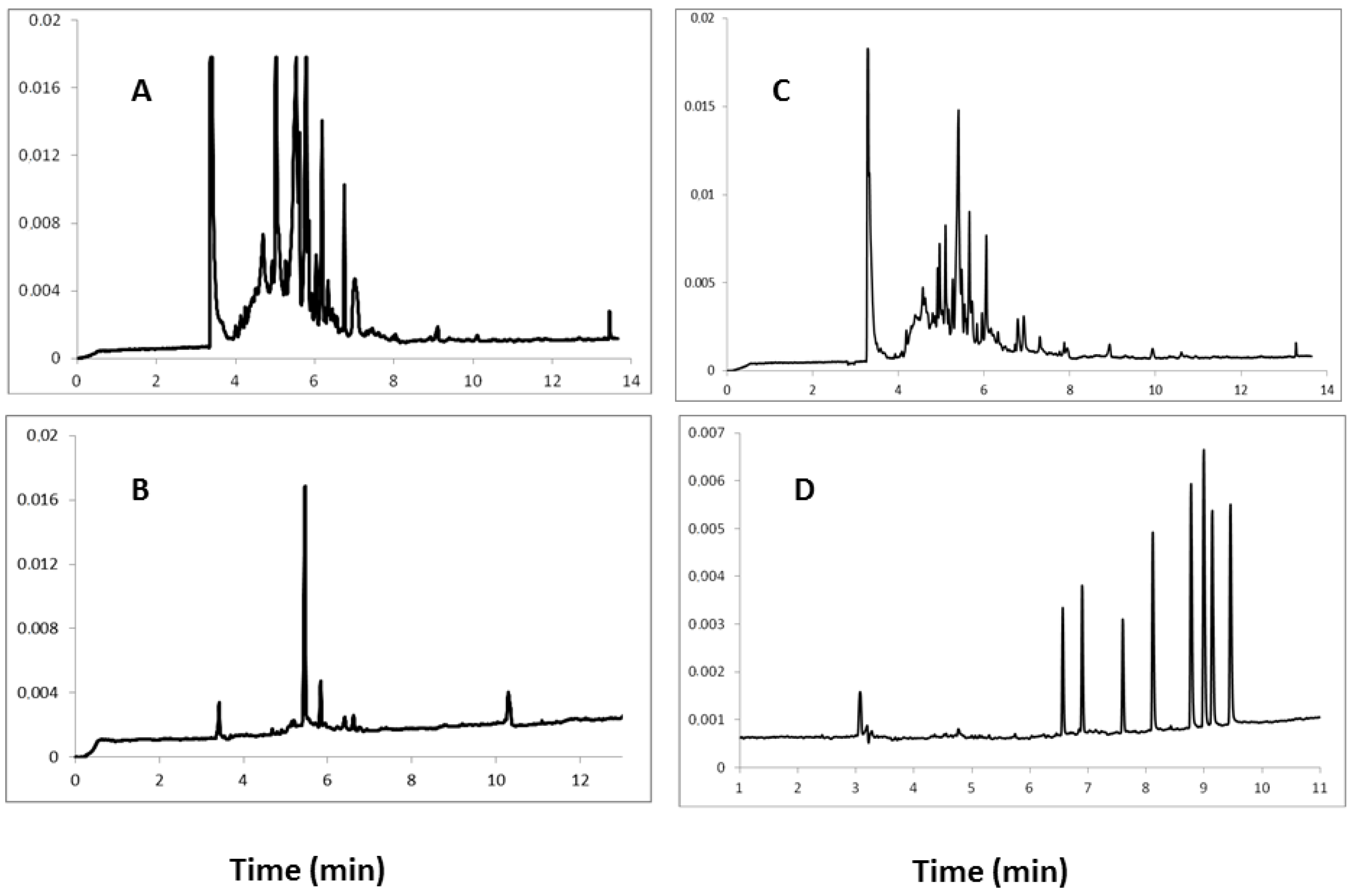
2.1. Comparison and Optimization of the Extraction Procedure

2.2. Optimization of Electrophoretic Conditions
2.2.1. Separation Buffer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer composition | Observations |
|---|---|
| 10 mM Na2B4O7, 50 mM SDS + 20% MeOH | Satisfactory separation was achieved with applied voltage 23 kV, generated current 45–50 μA. Relatively long analysis time; high and sharp peaks of analytes; the phenomenon of “tailing” peaks caused by too high content of methanol. |
| 20 mM Na2B4O7, 50 mM SDS + 20% MeOH | Separation was deemed unsatisfactory, peaks with a broad base, interfering with each other, just as in the previous test buffer phenomenon of “tailing”, the current slightly higher than in the buffer before test. The analysis time was shortened slightly. |
| 10 mM Na2B4O7, 50 mM SDS | The peaks were very narrow, but overlapping. During the analysis of eight analyte standards five overlapping peaks were obtained within a short analysis time. |
| 10 mM Na2B4O7, 50 mM SDS + 15% MeOH | The peaks were very sharp, well-separated from each other, but a relatively long analysis time (16 min) was necessary. |
| 10 mM Na2B4O7, 50 mM SDS + 10% MeOH | The peaks were very sharp, narrow, symmetrical, non-overlapping, and excellently separated. This buffer proved to be the most optimal separation electrolyte. |
| 10 mM NaH2PO4, 25 mM SDS (pH 3.0) | Separation was not achieved after 30 min. |
2.2.2. Capillary Parameters and Conditioning
2.2.3. Injection
2.2.4. Analytical Wavelengths

2.2.5. Temperature of Analysis
2.2.6. Voltage
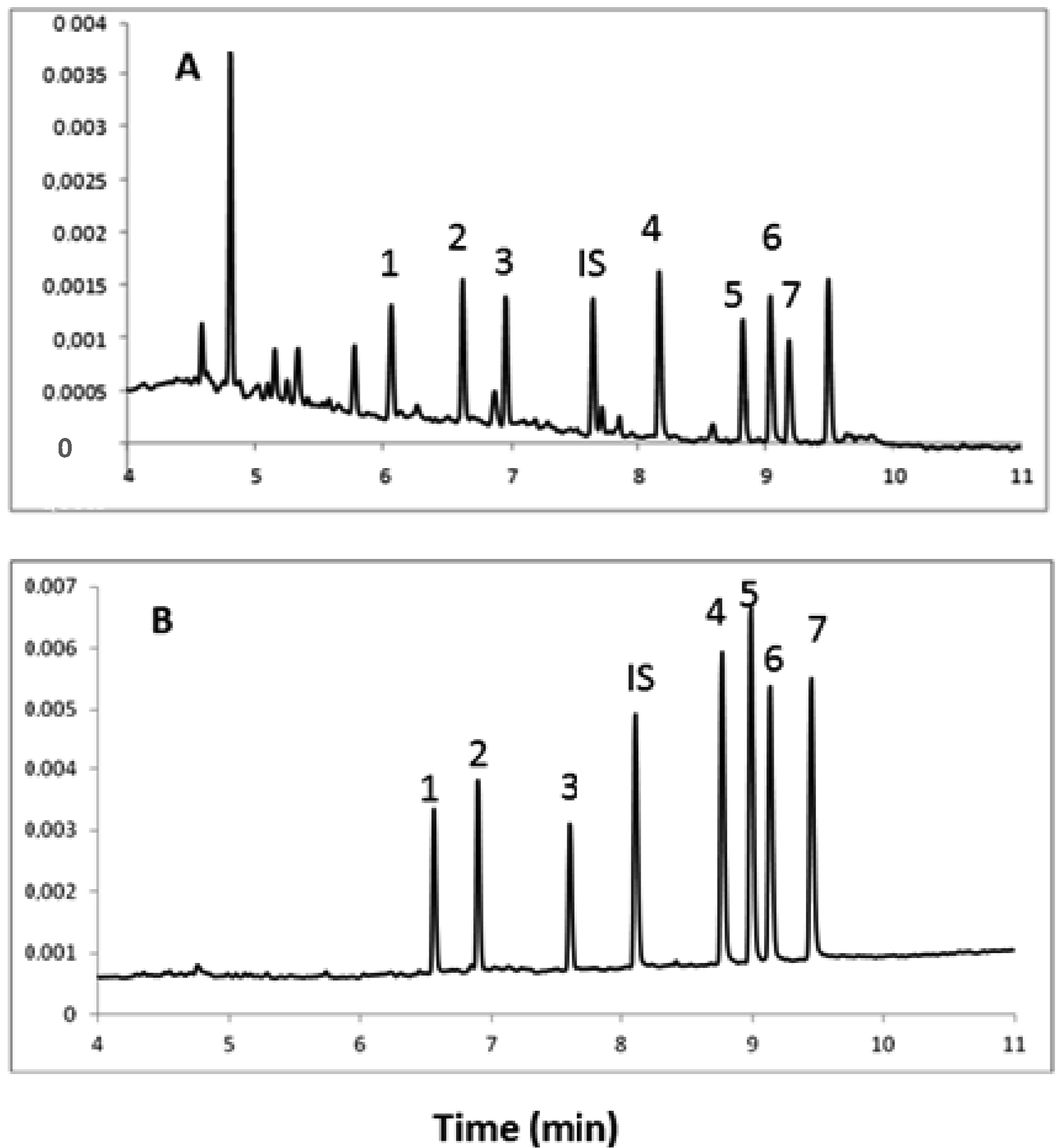
2.3. Validation of the Method
2.3.1. Specificity
2.3.2. Linearity

| Compound | Intercept (±SD) | Slope (±SD) | r2 | LOD [ng mL−1] | LOQ [ng mL−1] |
|---|---|---|---|---|---|
| Cortisone | 0.0025 (7.1 × 10−6) | 0.0417 (0.0029) | 0.9999 | 1.5 | 5 |
| Cortisol | 0.0035 (2.2 × 10−5) | 0.0163 (0.0091) | 0.9999 | 1.5 | 5 |
| Corticosterone | 0.0024 (2.0 × 10−5) | 0.0961 (0.008) | 0.9996 | 1.5 | 5 |
| Testosterone | 0.0029 (1.9 × 10−5) | 0.079 (0.0078) | 0.9997 | 1.5 | 5 |
| 17α-Methyltestosterone | 0.0029 (1.6 × 10−5) | 0.0617 (0.0066) | 0.9998 | 1.5 | 5 |
| Epitestosterone | 0.004 (3.1 × 10−5) | −0.0008 (0.0125) | 0.9996 | 1.5 | 5 |
| Progesterone | 0.0021 (2.4 × 10−5) | 0.0056(0.0113) | 0.9995 | 7 | 25 |
2.3.3. Limits of Detection and Limit of Quantitation
2.3.4. Precision
| Steroid Nominal concentration [ng mL−1] | Intra-day precision (n = 6) | Inter-day precision (n = 6) | Recovery (%) | ||
|---|---|---|---|---|---|
| Concentration found [ng mL−1] (±SD) | RSD (%) | Concentration found [ng mL−1] (±SD) | RSD (%) | ||
| Cortisone | |||||
| 10 | 10.1 (±0.7) | 7.0 | 10.7 (±1.1) | 8.5 | 100.5 |
| 100 | 103.0(±3.4) | 3.3 | 105.1 (±4.1) | 3.9 | 103.0 |
| 500 | 493.6 (±6.0) | 1.2 | 509.0 (±4.9) | 1.0 | 98.7 |
| Cortisol | |||||
| 10 | 11.4 (±0.6) | 5.1 | 10.9 (±0.9) | 8.4 | 114.0 |
| 100 | 101.4 (±2.9) | 2.9 | 106.3 (±4.5) | 4.2 | 101.4 |
| 500 | 497.5 (±10.5) | 2.1 | 505.1 (±5.9) | 1.2 | 99.5 |
| Corticosterone | |||||
| 10 | 11.0 (±0.9) | 8.3 | 11.3 (±1.0) | 8.8 | 110.3 |
| 100 | 108.4 (±8.4) | 7.8 | 91.1 (±7.1) | 7.9 | 108.4 |
| 500 | 502.0 (±1.3) | 0.3 | 507.1 (±4.5) | 0.9 | 100.4 |
| Testosterone | |||||
| 10 | 9.9 (±0.9) | 9.1 | 10.4 (±1.0) | 9.5 | 99.1 |
| 100 | 97.9 (±4.7) | 4.8 | 93.7 (±6.2) | 6.6 | 97.9 |
| 500 | 498.9 (±6.3) | 1.3 | 504.4 (±7.3) | 1.4 | 99.8 |
| 17α-Methylotestosterone | |||||
| 10 | 10.9 (±1.0) | 9.2 | 11.1 (±1.1) | 9.7 | 108.6 |
| 100 | 102.2 (±5.7) | 5.6 | 106.1 (±7.1) | 6.7 | 102.2 |
| 500 | 497.7 (±10.7) | 2.2 | 507.3 (±18.5) | 3.7 | 99.5 |
| Epitestosterone | |||||
| 10 | 10.7 (±1.0) | 9.2 | 10.8 (±1.0) | 9.2 | 106.8 |
| 100 | 108.8 (±5.3) | 4.9 | 109.3 (±6.1) | 5.6 | 108.8 |
| 500 | 502.9 (±17.6) | 3.5 | 509.7 (±19.5) | 3.8 | 100.6 |
| Progesterone | |||||
| 25 | 28.6 (±2.5) | 8.7 | 29.8 (±2.9) | 9.7 | 114.2 |
| 100 | 101.8 (±5.4) | 5.3 | 106.4 (±6.2) | 5.8 | 101.8 |
| 500 | 486.0 (±18.4) | 3.8 | 510.6 (±19.5) | 3.8 | 97.2 |
2.3.5. Recovery
2.3.6. Stability
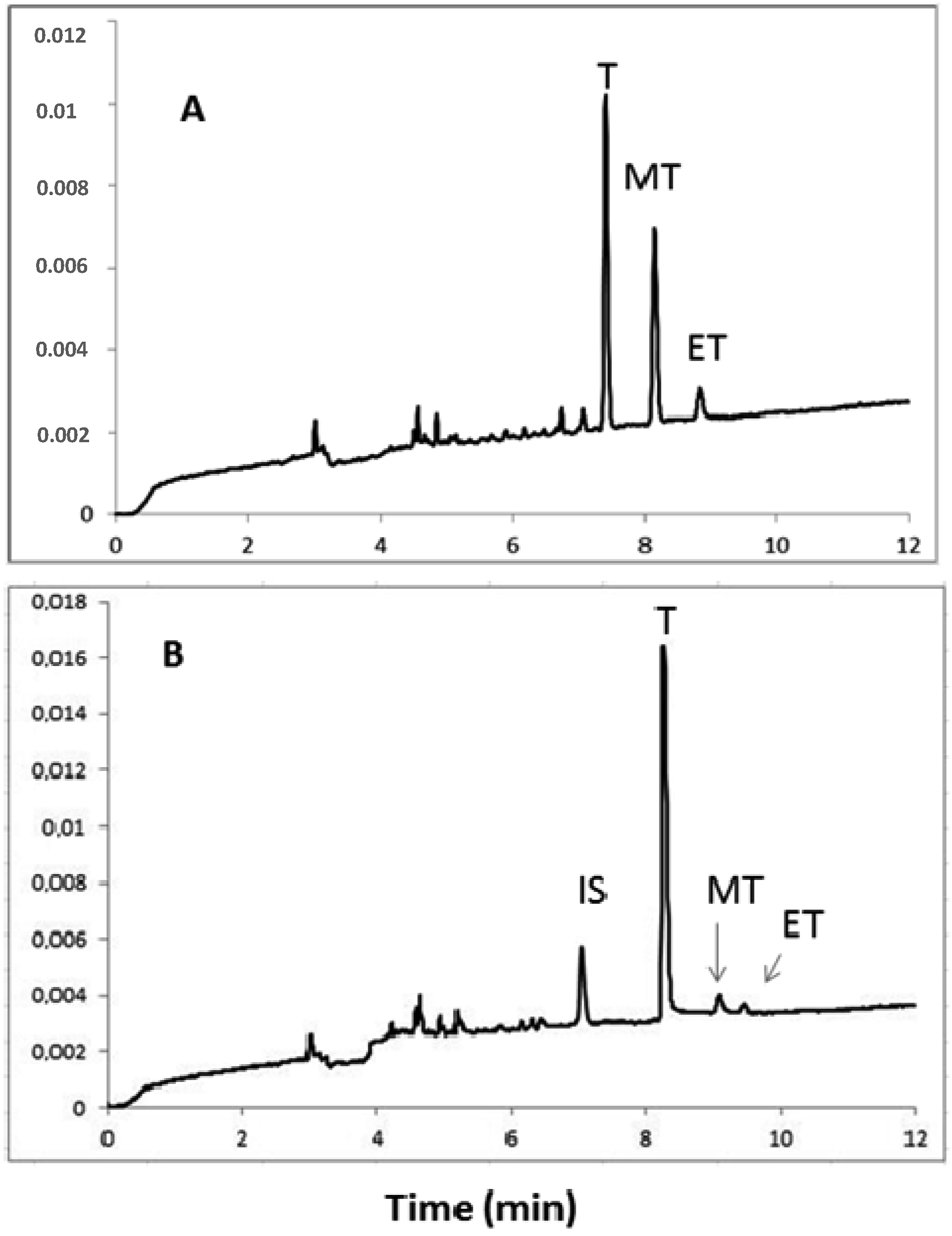
2.4. Application of Human Urine Samples
| Subject | Cortisone | Cortisol | Corticosterone | Testosterone | 17α-MT | Epitestosterone | Progesterone | T/ET |
|---|---|---|---|---|---|---|---|---|
| 1 | 181.8 | 170.7 | 57.3 | 1668.6 | 220.4 | 187.8 | n.a. | 8.88 |
| 2 | 222.4 | 228.3 | 90.2 | 1432.7 | 211.1 | 182.0 | 34.2 | 7.87 |
| 3 | 113.8 | 211.5 | 67.5 | 4940.9 | 211.1 | 355.2 | 91.1 | 13.9 |
| 4 | 167.9 | 269.7 | 64.1 | 2691.7 | 122.7 | 253.8 | n.a. | 10.6 |
| 5 | 153.0 | 260.1 | 47.2 | 1241.7 | 184.2 | 138.5 | n.a. | 8.96 |
| 6 | 145.6 | 234.1 | 65.0 | 2012.9 | 144.6 | 215.5 | 69.8 | 9.34 |
| 7 | 181.6 | 349.9 | 26.5 | 2371.3 | 108.4 | 223.3 | 144.5 | 10.61 |
| 8 | 151.3 | 548.3 | 75.5 | 3377.2 | 209.8 | 259.2 | n.a. | 13.03 |
| 9 | 231.9 | 553.1 | 100.4 | 3652.7 | 109.3 | 331.3 | 58.1 | 11.02 |
| 10 | 197.9 | 476.7 | 121.3 | 2879.1 | 210.4 | 281.2 | n.a. | 10.23 |
| 11 | 210.8 | 320.1 | 374.2 | 1401.5 | 181.9 | 209.9 | 160.3 | 6.67 |
| 12 | 209.3 | 318.0 | 129.2 | 1006.7 | 197.7 | 199.3 | n.a. | 5.05 |
| 13 | 267.1 | 501.8 | 100.5 | 2624.2 | 164.9 | 184.4 | 103.8 | 14.23 |
| Subject | Cortisone | Cortisol | Corticosterone | Testosterone | 17α-MT | Epitestosterone | Progesterone | T/ET |
|---|---|---|---|---|---|---|---|---|
| 1 | 128.4 | 185.1 | 79.8 | 87.6 | 14.2 | 31.5 | n.a. | 2.77 |
| 2 | 134.9 | 181.9 | 94.2 | 77.6 | 19.8 | 48.0 | n.a. | 1.61 |
| 3 | 129.7 | 167.6 | 89.1 | 79.9 | n.a. | 44.4 | n.a. | 1.78 |
| Subject (sex) | Cortisone | Cortisol | Corticosterone | Testosterone | 17α-MT | Epitestosterone | Progesterone | T/ET |
|---|---|---|---|---|---|---|---|---|
| 1 (M) | 197.8 | 349.6 | 57.3 | 18.5 | 5.5 | 38.6 | n.a. | 0.47 |
| 2 (M) | 199.5 | 288.3 | 90.2 | 21.3 | n.a. | 32.1 | n.a. | 0.66 |
| 3 (M) | 179.8 | 296.8 | 67.5 | 19.6 | n.a. | 49.3 | n.a | 0.4 |
| 4 (F) | 186.4 | 327.0 | 64.1 | 9.4 | n.a. | 21.2 | 29.2 | 0.44 |
| 5 (F) | 213.4 | 279.5 | 47.2 | 11.9 | n.a. | 18.7 | 25.6 | 0.63 |

3. Experimental
3.1. Material
3.2. Preparation of the Standard Solution of Steroids
3.3. Isolation Procedure of Steroid Hormones from Biological Samples
- Procedure 3. SPE using C18 cartridges and the elution of steroids with 2 mL of methanol.
- Procedure 4. SPE using C18 cartridges and the elution of steroids with 2 mL of dichloromethane.
- Procedure 5. SPE using HLB cartridges and an elution of steroids with 2 mL of methanol.
- Procedure 6. SPE using HLB cartridges and an elution of steroids with 2 mL of dichloromethane.
3.4. Analytical Equipment
3.5. Validation Study
3.6. Application of the MEKC Method for Steroid Determination in Human Urine Samples
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Silva, C.A.; Pereira, E.A.; Micke, G.A.; Farah, J.P.S.; Tavares, M.F.M. Mixture-designed electrolytes for the MEKC separation of natural and synthetic steroids. Electrophoresis 2007, 28, 3722–3730. [Google Scholar] [CrossRef]
- Liu, Z.; Kanjo, Y.; Mizutani, S. Urinary excretion rates of natural estrogens and androgens from humans, and their occurrence and fate in the environment: A review. Sci. Total. Environ. 2009, 407, 4975–4985. [Google Scholar] [CrossRef]
- Tan, R.S. Measuring testosterone biomarkers in serum and saliva. Int. J. Pharm. Comp. 2004, 8, 419–423. [Google Scholar]
- Kataoka, H.; Matsuura, E.; Mitani, K. Determination of cortisol in human saliva by automated in-tube solid-phase microextraction coupled with liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2007, 44, 160–165. [Google Scholar] [CrossRef]
- Kotłowska, A.; Sworczak, K.; Stepnowski, P. Urine metabolomics analysis for adrenal incidentaloma activity detection and biomarker discovery. J. Chromatogr. B 2011, 879, 359–363. [Google Scholar] [CrossRef]
- Parr, M.K.; Schänzer, W. Detection of the misuse of steroids in doping control. J. Steroid Biochem. Mol. Biol. 2010, 121, 528–537. [Google Scholar] [CrossRef]
- Ai-Dujaili, E.A.S. Development and validation of a simple and direct ELISA method for the determination of conjugated (glucuronide) and non-conjugated testosterone excretion in urine. Clin. Chim. Acta 2006, 364, 172–179. [Google Scholar] [CrossRef]
- Isobe, N.; Nakao, T.; Yamashiro, H.; Shimada, M. Enzyme immunoassay of progesterone in the feces from beef cattle to monitor the ovarian cycle. Animal Reprod. Sci. 2005, 87, 1–10. [Google Scholar] [CrossRef]
- Furuta, T.; Mori, Ch.; Suzuki, A.; Shibasaki, H.; Yokokawa, A.; Kasuya, Y. Simultaneous determination of 6β-hydroxycortisol and cortisol in human urine by liquid chromatography with ultrafiolet absorbance detection for phenotyping the CYP3A activity determined by the cortisol 6β-hydroxylation clearance. J. Chromatogr. B 2004, 801, 165–171. [Google Scholar] [CrossRef]
- Antignac, J.P.; Monteau, F.; Negriolli, J.; Andre, F.; le Bizec, B. Application of hyphenated mass spectrometric techniques to the determination of corticosteroid residues in biological matrices. Chromatographia 2004, 59, 13–22. [Google Scholar] [CrossRef]
- Olędzka, I.; Plenis, A.; Konieczna, L.; Kowalski, P.; Bączek, T. Micellar electrokinetic chromatography for the determination of cortisol in urine samples in view of biomedical studies. Electrophoresis 2010, 31, 2356–2364. [Google Scholar] [CrossRef]
- Hagey, L.R.; Czekala, N.M. Comparative urinary androstanes in the great apes. Gen. Comp. Endocrinol. 2003, 130, 64–69. [Google Scholar] [CrossRef]
- De la Torre, X.; Colamonici, C.; Curcio, D.; Molaioni, F.; Pizzardi, M.; Botre, F. A simplified procedure for GC/C/IRMS analysis of underivatized 19-norandrosterone in urine following HPLC purification. Steroids 2011, 76, 471–477. [Google Scholar] [CrossRef]
- Sharef, O.A.; Feely, J.; Kavanagh, P.V.; Scott, K.R.; Sharma, S.C. An HPLC method for the determination of the free cortisol/cortisone ratio in human urine. Biomed. Chromatogr. 2007, 21, 1201–1206. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, H.L.; Ding, Y.J.; Xia, A.L.; Cui, H.; Yu, R.Q. Simultaneous determination of cortisol and prednisolone in body fluids by using HPLC-DAD coupled with second-order calibration based on alternating trilinear decomposition. J. Chromatogr. B 2006, 840, 116–123. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, J.; He, Y.; Chi, Y.; Chen, G. A new mixed micellar electrokinetic chromatography method for analysis of natural and synthetic anabolic steroids. Talanta 2009, 77, 1002–1008. [Google Scholar] [CrossRef]
- Kartsova, A.A.; Velikanova, L.I.; Pavlova, E.G.; Bessonova, E.A. Steroidogenesis in patients with various adrenal cortex diseases as studied by reversed-phase high-performance liquid chromatography. J. Anal. Chem. 2004, 59, 976–982. [Google Scholar] [CrossRef]
- Rouits, E.; Boisdron-Celle, M.; Morel, A.; Gamelin, E. Simple and sensitive high-performance liquid chromatography method for simultaneous determination of urinary free cortisol and 6β-hydrocortisol in routine practice For CYP 3A4 activity evaluation in basal conditions and after grapefruit juice intake. J. Chromatogr. B 2003, 793, 357–366. [Google Scholar] [CrossRef]
- Rao, L.V.; Petersen, J.R.; Bissell, M.G.; Okorodudu, A.O.; Mohammad, A.A. Development of urinary free cortisol assay using solid-phase extraction-capillary electrophoresis. J. Chromatogr. B 1999, 730, 123–128. [Google Scholar] [CrossRef]
- Amundsen, L.K.; Nevanen, T.K.; Takkinen, K.; Rovio, S.; Sirén, H. Microscale immunoaffinity SPE and MEKC in fast determination of testosterone in male urine. Electrophoresis 2007, 28, 3232–3241. [Google Scholar] [CrossRef]
- Amundsen, L.A. Analysis of anabolic steroids by partial filling micellar electrokinetic capillary chromatography and electrospray mass spectrometry. J. Chromatogr. A 2004, 1040, 123–131. [Google Scholar]
- AbuRuz, S.; Millership, J.; Heaney, L.; McElnay, J. Simple liquid chromatography method for the rapid simultaneous determination of prednisolone and cortisol in plasma and urine using hydrophilic lipophilic balanced solid chase extraction cartridges. J. Chromatogr. B 2003, 798, 193–201. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.D.; Lee, W.Y.; Chung, B.C.; Choi, M.H. Quantitative metabolic profiling of 21 endogenous corticosteroids in urine by liquid chromatography-triple quadrupole-mass spectrometry. Anal. Chim. Acta 2009, 632, 101–108. [Google Scholar] [CrossRef]
- Hu, Z.; Gong, Q.; Hu, X.; Wang, L.; Cao, Y.; Cao, W.; Yu, Q.; Cheng, Z. Simultaneous determination of 6β-hydroxycortisol and cortisol in human urine and plasma by liquid chromatography with ultraviolet absorbance detection for phenotyping the CYP3A activity. J. Chromatogr. B 2005, 826, 238–243. [Google Scholar] [CrossRef]
- Kartsova, L.A.; Strelnikova, G. Determination of Endo- and Exogenous Corticosteroids by Cyclodextrin-Modified Micellar Electrokinetic Chromatography with the Use of On-Line Preconcentration. J. Anal. Chem. 2007, 62, 716–720. [Google Scholar] [CrossRef]
- Izquierdo-Hornillos, R.; Gonzalo-Lumbreras, R.; Santos-Montes, A. Method development for cortisol and cortisone by micellar liquid chromatography using sodium dodecyl sulphate: Application to urine samples of rugby players. J. Chromatogr. Sci. 2005, 43, 235–240. [Google Scholar] [CrossRef]
- Chen, M.C.; Chou, S.H.; Lin, C.H. Determination of corticosterone and 17-hydroxycorticosterone in plasma and urine samples by sleeping techniques using micellar electrokinetic chromatography. J. Chromatogr. B 2004, 801, 347–353. [Google Scholar] [CrossRef]
- Abubaker, M.A.; Petersen, J.R.; Bissell, M.G. Micellar electrokinetic capillary chromatographic separation of steroids in urine by trioctylphosphine oxide and cationic surfaktant. J. Chromatogr. B 1995, 674, 31–38. [Google Scholar] [CrossRef]
- Pomponio, R.; Gotti, R.; Fiori, J.; Cavrini, V. Microemulsion electrokinetic chromatography of corticosteroids effect of surfactants and cyclodextrins on the separation selectivity. J. Chromatogr. A 2005, 1081, 24–30. [Google Scholar]
- Sirén, H.; Seppänen-Laakso, T.; Orešič, T. Capillary electrophoresis with UV detection and mass spectrometry in method development for profiling metabolites of steroid hormone metabolism. J. Chromatogr. B 2008, 871, 375–382. [Google Scholar] [CrossRef]
- Qi, X.H. Simultaneous determination of nandrolone, testosterone, and methyltestosterone by multi-immunoaffinity column and capillary electrophoresis. Electrophoresis 2008, 29, 3398–3405. [Google Scholar] [CrossRef]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef]
- Goyal, R.N.; Gupta, V.K.; Chatterjee, S. Electrochemical investigations of corticosteroid isomers-testosterone and epitestosterone and their simultaneous determination in human urine. Anal. Chim. Acta 2010, 657, 147–153. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olędzka, I.; Kowalski, P.; Dziomba, S.; Szmudanowski, P.; Bączek, T. Optimization of a Pre-MEKC Separation SPE Procedure for Steroid Molecules in Human Urine Samples. Molecules 2013, 18, 14013-14032. https://doi.org/10.3390/molecules181114013
Olędzka I, Kowalski P, Dziomba S, Szmudanowski P, Bączek T. Optimization of a Pre-MEKC Separation SPE Procedure for Steroid Molecules in Human Urine Samples. Molecules. 2013; 18(11):14013-14032. https://doi.org/10.3390/molecules181114013
Chicago/Turabian StyleOlędzka, Ilona, Piotr Kowalski, Szymon Dziomba, Piotr Szmudanowski, and Tomasz Bączek. 2013. "Optimization of a Pre-MEKC Separation SPE Procedure for Steroid Molecules in Human Urine Samples" Molecules 18, no. 11: 14013-14032. https://doi.org/10.3390/molecules181114013