3.1.2. Chemical Transformations
The pertinent literature contains very few publications regarding diosgenone synthesis; these have dealt with the Oppenauer reaction with aluminum alkoxides [
20], or ruthenium complexes [
21]. However, yields have been poor or absent. Enzymatic transformations have also been attempted with structurally-similar molecules [
22,
23]. Obtaining diosgenone via Swern oxidation with subsequent double bond isomerization with oxalic acid in refluxing ethanol is reported here (84% overall yield).
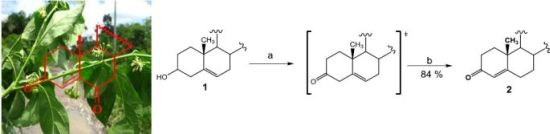
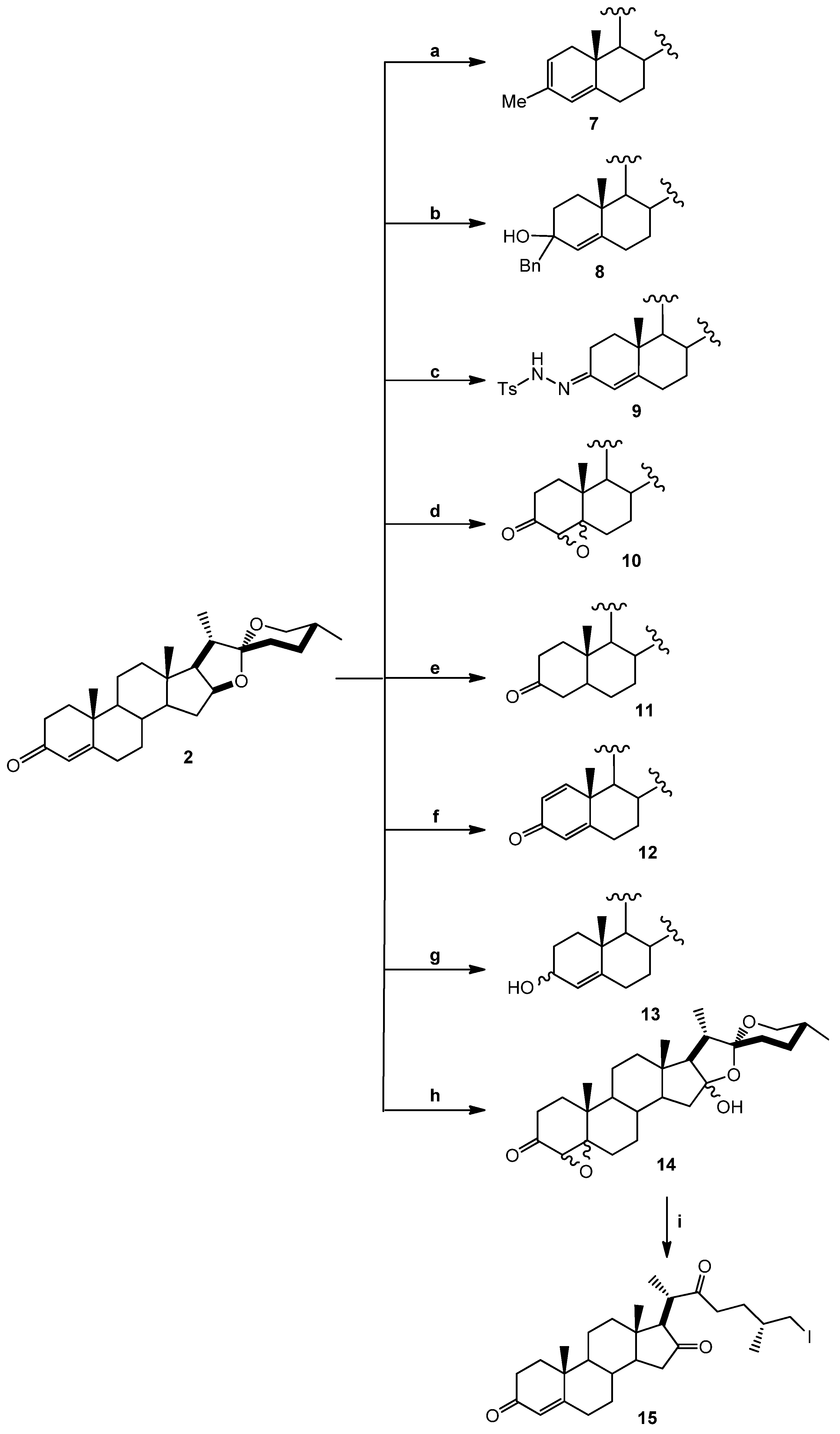
(25R)-4-Spirosten-3-one, diosgenone (2). A mixture of oxalyl chloride (1.08 mmol, 136 mg, 2 eq.) in dichloromethane (DCM, 5 mL) was treated with dimethyl sulfoxide (DMSO, 0.23 mL, 1.08 mmol, 84 mg, 2 eq.) in DCM (5 mL) and stirred at −60 °C for 5 min. (25R)-5-spirosten-3β-ol (3β-hydroxy-5-spirostene, diosgenin 1, 300 mg, 0.72 mmol, 1 eq.) in DCM (20 mL) was then added to the reaction mixture. Temperature was allowed to reach −10 °C after 20 min and then 0.65 mL N,N-diisopropyl ethylamine (3.73 mmol, 482 mg, 5.2 eq.) were added; stirring was continuous for 1 h to reach room temperature (RT) and HCl (20 mL, 5%) was added. The combined organic layer was washed with an aqueous solution (aq) of NaHCO3 (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was dissolved in an oxalic acid solution (1.4 g, 15.5 mmol, 21.5 eq.) in ethanol (20 mL) and heated to reflux for 5 h, extracted with DCM (3 × 15 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on silica and eluted with 4:1:1 hexane/DCM/Ethyl acetate (EtOAc), yielding diosgenone (2), as a white solid (255 mg, 84%), Rf = 0.6 (hexane/EtOAc 4:1). Melting point (m.p.): 188–190 °C. IR (KBr): 2950, 1680, 1470. 1H- NMR (CDCl3) δ: 5.6 (s, 1H, H-4), 4.3 (dd, J1 = 14.0 Hz, J2 = 7.0 Hz, 1H, H-16), 3.4 (dd, J1 = 11.0 Hz, J2 = 3.0 Hz, 1H, H-26), 2.5 (dt, J1 = 17.0 Hz, J2 = 7.0 Hz 1H, H-2), 2.4 (dt, J1 = 15.0 Hz, J2 = 12.0 Hz, 1H, H-2), 1.12 (s, 3H, H-19), 0.88 (d, J = 7.0 Hz, 3H, H-21), 0.76 (s, 3H, H-18), 0.72 (d, J = 6.3, 3H, H-27); 13C-NMR (CDCl3) δ: 199.3 (C-3), 171.0 (C-5), 123.7 (C-4), 109.1 (C-22), 80.4 (C-16), 66.7 (C-26), 61.9 (C-17), 55.5 (C-14), 53.6 (C-9), 41.5 (C-20), 40.2 (C-13), 39.5 (C-12), 38.5 (C-10), 35.5 (C-1), 35.0 (C-8), 33.8 (C-2), 32.6 (C-6), 32.0 (C-7), 31.6 (C-15), 31.2 (C-23), 30.1 (C-25), 28.6 (C-24), 20.7 (C-11), 17.2 (C-19), 17.0 (C-27). 16.2 (C-18), 14.4 (C-21). MS (m/z) (%): 413.0 (M+, 100).
(25R)-3-Methyl-spirostan-2,4-diene (7). Diosgenone (2, 25 mg, 0.6 mmol, 1 eq.) in diethyl ether (5 mL) was treated with methyl magnesium bromide solution (0.44 mL, 0.62 mmol, 1.1 eq., 1.4 M toluene/THF 3:1) and stirred at −15 °C (one hour) followed by 2 h at RT. The reaction mixture was slowly treated with 5% aq NH4Cl (20 mL) and extracted with three × 20 mL portions of DCM. The combined organic extract was washed with water (20 mL) and brine (20 mL). The dried (Na2SO4) organic phase was concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column (DCM/hexane 8:1) yielding compound 7 (14 mg, 54%): Rf = 0.4 (DCM). M.p.: 172–174 °C. IR (KBr): 2928. 1H-NMR (CDCl3) δ: 5.8 (s, 1H, H-4), 5.4 (dd, J = 5.0 Hz, 1H, H-2), 4.4 (dt, J1 = 7.9 Hz, J2 = 1.8 Hz, 1H, H-16), 3.5 (d, J = 10.7 Hz, 2H, H-26), 1.7 (s, 3H, Me), 1.0 (s, 3H, H-19), 0.9 (d, J = 7.1 Hz, 3H, H-21), 0.8 (s, 3H, H-18), 0.72 (d, J = 6.3, 3H, H-27);13C-NMR (CDCl3) δ: 142.0 (C-5), 133.4 (C-3), 130.4 (C-2), 124.8 (C-4), 120.9 (C-6), 109.7 (C-22), 81.3 (C-16), 67.3 (C-26), 62.5 (C-17), 57.1 (C-14), 48.7 (C-9), 40.8 (C-20), 40.3 (C-13), 32.1 (C-6), 32.0 (C-15), 31.9 (C-23), 31.8 (C-25), 28.9 (C-24), 28.3 (C-11), 24.3 (Me), 21.3 (C-18), 19.4 (C-27), 16.8 (C-18), 14.9 (C-21). MS (m/z) (%): 411.2 (M+, 10).
(25R)-3-Benzyl-spirostan-4-en-3-ol (8). Diosgenone (2, 50 mg, 0.12 mmol, 1 eq.) in diethyl ether (5 mL) was treated with benzylmagnesium bromide solution (0.14 mL, 0.14 mmol, 1.1 eq., 1.0 M THF) and treated as above. The residue was purified by chromatography on a silica gel column (DCM/hexane 8:1) yielding compound 8 as a white solid (56 mg, 92%): Rf = 0.4 (hexane/EtOAc 8:1). M.p.: 203–205 °C. IR (KBr): 2930, 2880, 1600–1700. 1H-NMR (CDCl3) δ: 7.40–7.09 (m, 5H, H-Ar), 5.1 (s, 1H, H-4), 4.4 (dt, J1 = 7.9, J2 1.7 Hz, 1H, H-16), 3.4 (d, J = 10.7 Hz, 2H, H-26), 2.8 (d, J = 14.7 Hz, 2H, Ar-CH2), 1.1 (s, 3H, H-19), 1.0 (d, 7.1 Hz, 3H, H-21), 0.8 (s, 3H, H-18), 0.8 (d, J = 6.2 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 146.8 (C-1'), 137.7 (C-5), 131.1 (C-4'), 129.3 (C-3', C-5'), 126.8 (C-2', C-4'), 126 (C-4), 109.7 (C-22), 81.2 (C-16), 71.7 (C-3), 67.3 (C-26), 62.5 (C-17), 56.4 (C-14), 54.8 (C-9), 47.2 (C-20), 40.3(C-13), 32.2 (C-6), 32.1 (C-15), 31.2 (C-22), 30.1 (C-25), 29.2 (C-26), 29.1 (C-10), 28.6 (C-23), 28.4 (C-24), 21.4 (C-19), 20.6 (C-11), 17.5 (C-27), 16.8 (C-18), 14.9 (C-21). MS (m/z) (%): 506.2 (M+ +1, 5), 487.2 (86).
(25R)-3-p-Toluenehydrazine-spirostan-4-ene (9). A mixture of diosgenone (2, 102 mg, 0.25 mmol, 1 eq.) and pTsNHNH2 (118 mg, 0.65 mmol, 2.6 eq.) in ethanol (5 mL) was heated to reflux for 5 h. The solvent was evaporated under vacuum and the residue was purified by chromatographic column on silica gel (hexane/EtOAc 5:1) yielding compound 9 as a white solid (122 mg, 84%): Rf = 0.45 (hexane/EtOAc 5:1). M.p.: 148–150 °C. IR (KBr): 3508, 2930. 1H-NMR (CDCl3) δ: 7.9 (d, J = 8.1 Hz, 2H, H-Ar), 7.3 (d, J = 8.1 Hz, 2H, H-Ar), 5.8 (s, 1H, H-4), 4.4 (dt, J1 = 7.9 Hz, J2 = 1.8 Hz, 1H, H-16), 3.4 (d, J = 10.7 Hz, 2H, H-26), 2.4 (s, 3H, Ar-CH3), 1.0 (s, 3H, H-19), 0.9 (d, J = 7.2 Hz, 3H, H-21), 0.76 (s, 3H, H-18), 0.7 (d, J = 6.3 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 145.0 (C-5), 142.6 (C-3), 139.8 (C-1'), 136.4 (C-4'), 129.9 (C-3', C-5'), 129.1 (C-4), 128.5 (C-3', C-5'), 120.8 (C-2', C-6'), 109.7 (C-22), 81,6 (C-16), 62.5 (C-26), 56.2 (C-17), 53.9 (C-14), 42,3 (C-20), 35.7 (C-8), 29.2 (C-23), 29.0 (C-10), 28.8 (C-1), 28.7 (C-2), 28.5 (C-15), 28.4 (C-6), 28.2 (C-25), 28.1 (C-24), 20.8 (C-11), 20.6 (C-Me) 17.5 (C-19), 16.7 (C-27), 14.9 (C-21), 14.5 (C-18), 14.1 (C-21). MS (m/z) (%): 581.3 (M+, 5); 539.1 (36).
(25R)-4,5-Epoxy-spirostan-3-one (10). A mixture of diosgenone (2, 30 mg, 0.073 mmol, 1 eq.) and hydrogen peroxide (0.5 mL, 30%, 4.9 mmol, 67 eq.) in DCM/acetone 1:1 (20 mL) was treated with NaOH (1 mL, 2%) and stirred at RT for 48 h; then water was added to the reaction mixture and extracted with ethyl acetate (2 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc 10:1) yielding compound 10 as a white solid corresponding to the mixture of epoxides α (70%) y β (30%) (21 mg, 65%): Rf = 0.6 (hexane/EtOAc 9:1). M.p.: 184–186 °C. IR (KBr): 2899, 1715. 1H-NMR (CDCl3) δ: 4.4 (dt, J1 = 7.9 Hz, J2 = 1.8 Hz, 1H, H-16), 3.3 (d, J = 10.7 Hz, 2H, H-26), 3.0 (s, 1H, H-4β), 2.9 (s, 1H, H-4α), 1.2 (s, 3H, H-19), 1.0 (d, J = 7.1 Hz, 3H, H-21), 0.8 (s, 3H, H-18), 0.8 (d, J = 6.2 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 207.2 (C-3), 109.7 (C-22), 82.0 (C-16), 70.5 (C-5), 67.3 (C-26), 63.3 (C-4), 63.2 (C-17), 60.4 (C-17), 55.8 (C-14), 48.1 (C-9), 40.6 (C-12), 35.2 (C-20), 35.0 (C-17), 34.5 (C-8), 34.5 (C-10), 34.0 (C-2), 32.4 (C-1), 31.5 (C-15), 30.7 (C-6), 28.9 (C-23), 28.5 (C-7), 27.4 (C-24), 21.1 (C-11), 16.9 (C-27), 16.2 (C-19), 15.1 (C-21), 14.6 (C-18). MS (m/z) (%): 429.2 (M+, 82), 427.2 (12).
(25R)-Spirostan-3-one (11). A mixture of diosgenone (2, 40.8 mg, 0.1 mmol, 1 eq.) and palladium on carbon (Pd/C) 10% (2.0 mg, 10% mol, 0.01 mmol) in EtOAc (5 mL) was treated with hydrogen and stirred at RT overnight. The reaction mixture was filtered through a pad of celite and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc 10:1) yielding compound 11 as a white solid (35 mg, 85%): Rf = 0.4 (hexane/AcOEt 10:1). M.p.: 153–155 °C. IR (KBr): 2929, 1712. 1H-NMR (CDCl3) δ: 4.4 (dt, J1 = 7.9 Hz, J2 = 1.8 Hz, 1H, H-16), 3.4 (d, J = 10.7 Hz, 2H, H-26), 2.6 (d, J = 15.1 Hz, 2H, H-4), 1.1 (dt, J1 = 7.6 Hz, J2 3.6 Hz, 2H, H-6), 1.0 (s 3H, H-18), 0.9 (d, J = 7.0 Hz, 3H, H-21), 0.7 (d, J = 6.3 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 210.0 (C-3), 109.7 (C-22), 81.2 (C-16), 67.3 (C-26), 62.6 (C-17), 56.7 (C-14), 44.6 (C-4), 41.1 (C-20), 40.3 (C-13), 38.9 (C-12), 38.2 (C-1), 38.1 (C-2), 35.8 (C-10), 35.0 (C-8), 31.6 (C-15), 30.7 (C-23), 28.6 (C-24, C-6), 20.8 (C-11), 19.9 (C-27), 17.5 (C-21), 16.8 (C-18), 14.9 (C-19). MS (m/z) (%): 415.2 (M+, 86), 397.1 (6).
(25R)-1,4-Spirostadien-3-one (12). A mixture of diosgenone (2, 50.0 mg, 0.1 mmol, 1 eq.), benzoic acid (14.8 mg, 0.12 mmol, 1.1 eq.) and 2,3-Dichloro-5,6-dicyano-p-benzoquinone (DDQ, 40.9 mg, 0.18 mmol, 1.8 eq.) was dissolved in 10.0 mL toluene and then heated to reflux for 12 h. The reaction mixture was filtered through celite and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/DCM/EtOAc 4:1:1) yielding compound 12 as a white solid (30 mg, 60%): Rf = 0.3 (hexane/AcOEt 10:1). M.p.: 181–183 °C. IR (KBr): 2933, 2854, 1664, 1458. 1H-NMR (CDCl3) δ: 7.1 (d, J = 10.0, 1H, H-1), 6.3 (d, J = 10.0, 1H, H-2), 6.1 (s, 1H, H-4), 4.4 (dt, J1 = 7.9 Hz, J2 = 1.8 Hz, 1H, H-16), 3.6(d, J = 10.7 Hz, 2H, H-26), 2.6 (dt, J1 = 14.4, Hz J2 = 12.0 Hz, 2H, H-6), 1.3 (s, 3H, H-19), 0.9 (d, J = 7.1 Hz, 3H, H-21), 0.8 (d, J = 6.3 Hz, 3H, H-27), 0.7 (s, 3H, H-18); 13C-NMR (CDCl3) δ: 187.1 (C-3), 170.2 (C-5), 156.7 (C-1), 127.8 (C-2), 124.2 (C-4), 109.8 (C-22), 80.9 (C-16), 67.0 (C-26), 62.4 (C-17), 55.2 (C-14), 52.8 (C-9), 44.2 (C-10), 42.1 (C-20), 41.1 (C-13), 39.9 (C-12), 35.6 (C-8), 34.2 (C-7), 33.5 (C-6), 33.1 (C-15), 32.3 (C-23), 31.1 (C-25), 30.7 (C-24), 23.2 (C-11), 19.1 (C-19), 17.6 (C-27), 16.9 (C-18), 14.9 (C-21). MS (m/z) (%): 411.1 (M+, 100).
(25R)-4-Spirosten-3-ol (13). Diosgenone (2, 24 mg, 0.06 mmol, 1 eq.) in methanol (5 mL) was treated with NaBH4, (80 mg, 0.2 mmol, 3.3 eq.) and stirred at RT for 2 h overnight; water was then added to the reaction mixture and extracted with DCM (2 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc 5:1) yielding compound 13 as a white solid (21 mg, 65%): Rf = 0.4 (hexane/AcOEt 5:1). M.p.: 116–118 °C. IR (KBr): 3264, 2930, 2846, 1448. 1H-NMR (CDCl3) δ: 5.3 (d, J = 6.5 Hz, 1H, H-4), 4.4 (dt, J1 = 15.0, J2 = 7.5 Hz, 1H, H-16), 4.1 (dd, J1 = 18.0 Hz, J2 = 6.5 Hz, 1H, H-3), 3.5–3.3 (d, J = 10.7 Hz, 2H, H-26), 2.3 (dd, J1 = 14.0 Hz, J2 = 12.0 Hz, 1H, H-6), 1.0 (s, 3H, H-18), 0.9 (d, J = 6.9 Hz, 3H, H-21), 0.8 (s, 3H, H-19), 0.7 (d, J = 6.2 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 147.8 (C-5), 123.9 (C-4), 109.7 (C-22), 81.2 (C-16), 68.3 (C-3), 67.2 (C-26), 62.5 (C-17), 56.4 (C-14), 54.8 (C-9), 40.8 (C-20), 39.8 (C-13), 39.6 (C-12), 38.4 (C-10), 37.7 (C-1), 35.9 (C-8), 30.7 (C-25), 29.9 (C-2), 29.2 (C-24), 21.2 (C-11), 17.2 (C-19), 16.8 (C-27), 16.2 (C-18), 14.9 (C-21). MS (m/z) (%): 415.2 (M+, 80), 413.2 (21).
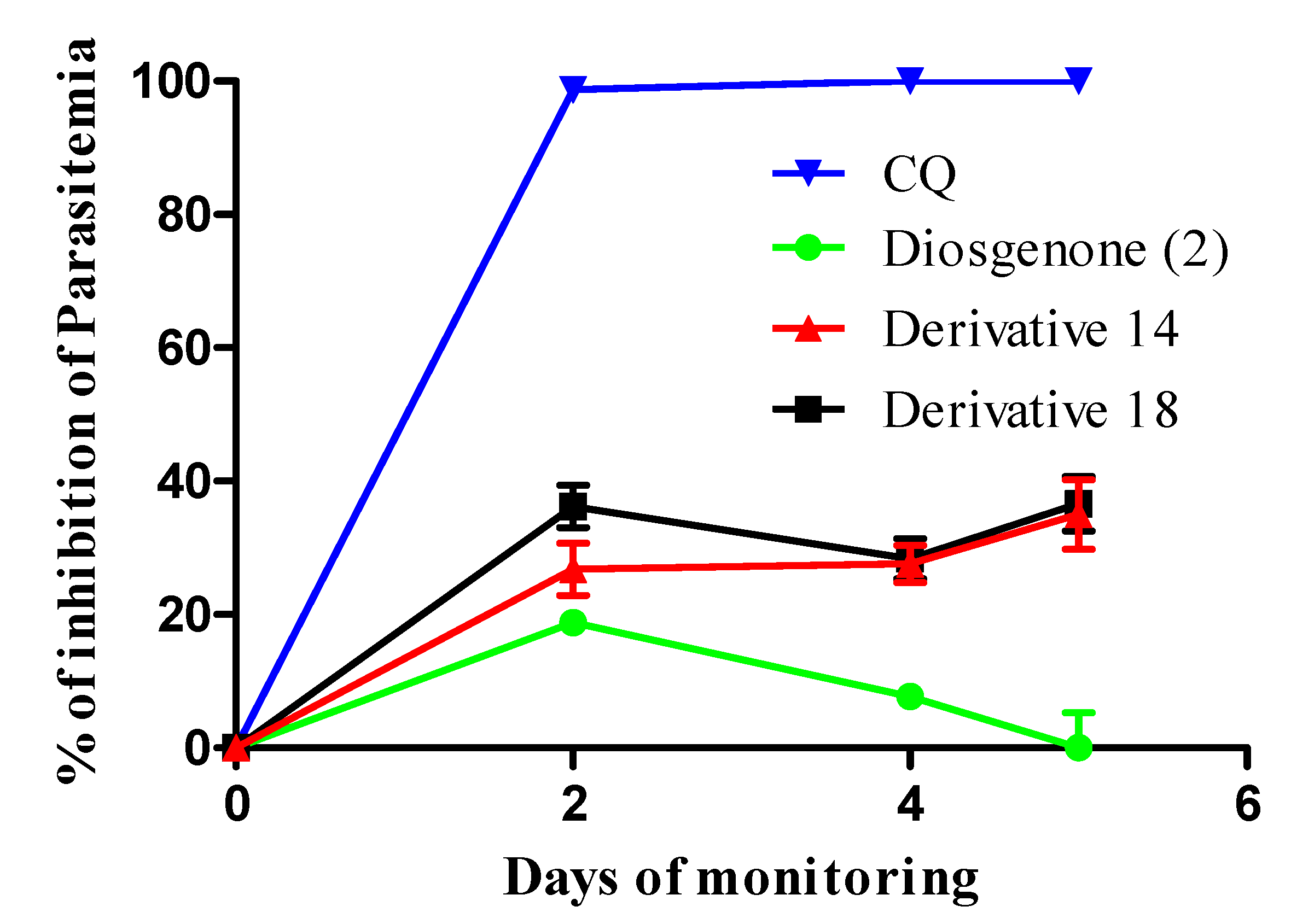
(25R)-4,5-Epoxy-spirostan-16α-hydroxy-3-one (14). A mixture of diosgenone (2, 200 mg, 0.48 mmol, 1 eq.) and NaHCO3 (1.89 g, 22.5 mmol, 47 eq.) in CHCl3-acetone-1.0 mM Na2EDTA (1:1:1, 37.5 mL, 4.65 g, 12.5 mmol, 26 eq.) was treated with oxone (6 g, 9.8 mmol, 20.4 eq.) in EDTA-Na2 (1 Mm) and stirred at RT for 48 h. The reaction mixture was extracted with DCM (2 × 15 mL) and the combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/DCM /EtOAc 3:1:1) yielding compound 14 as a white solid corresponding to the mixture of epoxides α (70%) y β (30%) (170 mg, 80%): Rf = 0.55 (hexane/AcOEt 3:1). M.p.: 145–147 °C. IR (KBr): 3450, 2940, 1710. 1H-NMR (CDCl3) δ: 3.6 (d, J1 = 10.7 Hz, J2 = 8.2 Hz, 2H, H-26), 3.0 (s, 1H, H-4α), 2.97 (s, 1H, H-4β), 1.8 (dd, J1 = 17.6 Hz, J2 = 2.4 Hz, 2H, H-2), 1.5 (d, J = 10.7 Hz, 2H, H-15), 1.0 (d, J = 7.1 Hz, 3H, H-21), 1.0 (s, 3H, H-18), 0.8 (s, 3H, H-19), 0.8 (d, J = 6.2 Hz, 3H, H-27). 13C-NMR (CDCl3) δ: 207.7 (C-3), 116.5 (C-22), 111.6 (C-16), 70.8 (C-5), 70.7 (C-3), 68.7 (C-26), 63.3 (C-4), 55.2 (C-17), 51.0 (C-15), 47.0 (C-9), 42.9 (C-15), 40.3 (C-12), 39.4 (C-20), 37.6 (C-8), 33.2 (C-1), 32.7 (C-6), 31.5 (C-23), 30.3 (C-25), 29.8 (C-24), 29.7 (C-1), 29.2 (C-7), 27.2 (C-2), 21.8 (C-11), 20.0 (C-11), 18.0 (C-27), 16.0 (C-19), 15.5 (C-18), 15.0 (C-21). MS (m/z) (%): 445.0 (M+, 3), 427.0 (96).
(25R)-26-Iodine-cholest-4-en-3,16,22-trione (15). A mixture of sodium iodide (NaI, 3 g, 20 mmol, 20 eq.), TMSCl (0.63 mL, 5 mmol, 5 eq.) and molecular sieve (5 g) in CH3CN (24 mL) was stirred at RT for 2 h. Compound 9 (446 mg, 1 mmol, 1 eq.) in DCM (15 mL) was added for 20 min. The reaction mixture was extracted with DCM (2 × 15 mL) and the combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/DCM/EtOAc 2:1:1) yielding compound 15 as a white solid (404 mg, 75%): Rf = 0.4 (hexane/EtOAc 2:1). M.p.: 148–150 °C. IR (KBr): 2942, 1733, 1717. 1H-NMR (CDCl3) δ: 5.7 (s, 1H, H-4), 3.3 (d, J = 4.9 Hz, 2H, H-26), 2.6 (d, J = 8.6 Hz, 1H, H-17), 1.6 (d, J = 16.4 Hz, 2H, H-23), 1.2 (d, J = 12.0 Hz, 3H, H-21), 1.1 (d, J = 6.2 Hz, 3H, H-27), 1.0 (s, 3H, H-18), 0.8 (s, 3H, H-19); 13C-NMR (CDCl3) δ: 218.2 (C-16), 213.7 (C-22), 199.9 (C-3), 170.8 (C-5), 124.9 (C-4), 66.9 (C-17), 53.9 (C-9), 51.2 (C-14), 44.0 (C-), 42.5 (C-13), 40.2 (C-20), 39.3 (C-12), 39.2 (C-15), 39.0 (C-10), 36.2 (C-23), 36.1 (C-8), 34.6 (C-1), 34.5 (C-24), 33.2 (C-2), 32.7 (C-6), 30.6 (C-7), 21.2 (C-25), 20.9 (C-27), 18.6 (C-11), 18.1 (C-26), 17.6 (C-19), 16.1 (C-21), 13.8 (C-18). MS (m/z) (%): 539.1 (M+, 96), 500.1 (5).
(25R)-3-Acetoxy-spirostan-5-ene (16). A mixture of diosgenine (1, 500 mg, 1.2 mmol, 1 eq.), acetic anhydride (1 mL, 1.05 mmol, 8.8 eq.) and ZnCl2 (0.5 g, 3.6 mmol, 3 eq.) was stirred at RT for 3 h. An Na2CO3 solution was added to the reaction mixture and extracted with EtOAc (2 × 15 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc/DCM 5:1:1) yielding compound 16 as a white solid (452 mg, 82%) Rf = 0.5 (hexane/EtOAc 3:1). M.p.: 182–184 °C. 1H-NMR (CDCl3) δ: 5.4 (d, 1H, J = 4.7 Hz, H-6), 4.6 (m, 1H, H-3), 4.4 (dt, 1H, J1 = 15 Hz, J2 = 7 Hz, H-16), 3.5(d, J = 10.7 Hz, 2H, H-26), 2.1 (s, 3H, 3-OCOCH3), 1.6 (s 3H, H-18), 1.0 (s 3H, H-19), 0.9 (d, 3H, J = 7.1 Hz, H-21), 0.8 (d, 3H, J = 6.3 Hz, H-27); 13C-NMR (CDCl3) δ: 170.5 (3-OCOCH3), 139.6 (C-5), 122.3 (C-6), 109.2 (C-22), 80.8 (C-16), 73.8 (C-3), 66.8 (C-26), 62.0 (C-17), 56.4 (C-14), 49.9 (C-9), 41.6 (C-20), 40.2 (C-13), 39.7 (C-4), 38.0 (C-12), 36.9 (C-1), 36.7 (C-10), 32.0 (C-7), 31.8 (C-15), 31.4 (C-8), 31.3 (C-23), 30.3 (C-25), 28.8 (C-2), 27.7 (C-24), 21.4 (3-OCH3), 20.8 (C-11), 19.3 (C-19), 17.1 (C-27), 16.2 (C-18), 14.5 (C-21).
(25R)-5,6-Epoxy-spirostan-3-ol (17). A mixture of diosgenine (1, 100 mg, 2.0 mmol, 1 eq.) and NaHCO3 (1.89 g, 22.5 mmol, 11 eq.) in CHCl3-acetone-1.0 mM Na2EDTA (1:1:1, 37.5 mL) was treated with oxone (6.0 g, 9.8 mmol) in Na2EDTA 1mM and stirred at RT for 16 h. The reaction mixture was extracted with DCM (2 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/DCM/EtOAc 1:1:1) yielding compound 17 as a white solid corresponding to the mixture of epoxides α (70%) y β (30%) (85.1 mg, 70.1%): Rf = 0.4 (hexane/EtOAc 2:3). M.p.: 97–99 °C. 1H-NMR (CDCl3) δ: 4.38 (dd, 1 H, J = 7.4 Hz, H-16), 3.9 (m, 1H, H-3), 3.5 (dd, 1H, J = 11, 3 Hz, H-26), 3.36 (dd, 1H, J = 11 Hz, H-26), 3.10 (d, 1H, J = 4Hz, H-6β), 2.92 (d, 1H, J = 4 Hz, H-6α), 1.09 (s, 3H, H-19), 0.97 (d, 3H, J = 7 Hz, H-21), 0.80 (d,3H, J = 6 Hz, H-27), 0.74 (s, 3H, H-18); 13C-NMR (CDCl3) δ: 110.0 (C-22), 81.3 (C-16), 69.3 (C-3), 67.5 (C-26), 66.4 (C-5), 62.6 (C-17), 59.8 (C-6), 55.7 (C-14), 43.3 (C-4), 42.3 (C-12), 41.0 (C-13), 40.5 (C-20), 35.7 (C-10), 32.4 (C-1), 31.8 (C-15), 31.7 (C-7), 31.0 (C-23), 30.1 (C-9), 30.0 (C-26), 29.7 (C-2), 29.5 (C-24), 22.5 (C-11), 17.0 (C-27), 16.9 (C-19), 16.7 (C-21), 15.2 (C-18).
(25R)-4-Spirosten-3,6-dione (18). A mixture of diosgenine (1, 50 mg, 0.12 mmol, 1 eq.) and pyridinium chlorochromate (PCC, 100 mg, 0.46 mmol, 3.8 eq.) in DCM (30 mL) was stirred at RT for 5 h. The reaction mixture was extracted with DCM (2 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/DCM/EtOAc 2:1:1) yielding compound 18 as a yellow solid (28 mg, 55%): Rf = 0.3 (hexane/AcOEt/DCM 7:1:1). M.p.: 200–202 °C. IR (KBr): 2951, 2925, 2850, 1700. 1H-NMR (CDCl3) δ: 6.2 (s, 1H, H-4), 4.42 (dd, J1 = 7.6 Hz, J2 = 13.8 Hz, 1H, H-16), 3.5(d, J = 10.7 Hz, 2H, H-26), 2.8 (d, J = 14.7 Hz, 2H, H-7), 1.2 (s, 3H, H-19), 1.0 (d, J = 7.1 Hz, 3H, H-21), 0.8 (d, J = 6.2 Hz, 3H, H-27), 0.7 (s, 3H, H-18); 13C-NMR (CDCl3) δ: 202.1 (C-7), 199.6 (C-3), 160.8 (C-5), 125.9 (C-4), 109.6 (C-22), 80.5 (C-16), 67.2 (C-26), 62.1 (C-17), 56.5 (C-14), 51.1 (C-9), 47.0 (C-7), 41.9 (C-20), 40.7 (C-12), 39.9 (C-13), 39.4 (C-10), 35.7 (C-1), 34.2 (C-2), 33.9 (C-8), 31.7 (C-15), 31.5 (C-23), 30.4 (C-25), 29.0 (C-24), 20.9 (C-11), 17.8 (C-19), 17.3 (C-27), 16.5 (C-18), 14.7 (C-21). MS (m/z) (%): 427.0 (M+, 100).
(25R)-3,5-Spirostadiene (19). HCl (2.5 mL, 36.5%, 31.8 mmol, 635 eq.) was added to a solution of diosgenine (1, 20 mg, 0.05 mmol, 1 eq.), zinc powder (250 mg, 3.8 mmol, 76 eq.) in ethanol (5 mL) and stirred at RT for 2 h. NaOH (15 mL, 10%) was added to the reaction mixture and extracted with DCM (2 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (DCM/methanol (MeOH) 100:1) generating 19 as white solid (13 mg, 71%): Rf = 0.5 (DCM/MeOH 20:1). M.p.: 116–118 °C. 1H-NMR (CDCl3) δ 5.9: (d, J = 4.0 Hz, 1H, H-3), 5.8 (d, J = 4.0 Hz, 1H, H-4), 5.3 (dd, J = 6.0, 1.3 Hz, 1H, H-6, 4.39 (dt, J1 = 7.6 Hz, J2 = 13.8 Hz, 1H, H-16), 3.4 (d, J = 10.7 Hz, 2H, H-26), 1.2 (s, 3H, H-19), 1.0 (d J = 7.1 Hz, 3H, H-21), 0.9 (d, J = 6.2 Hz, 3H, H-27), 0.8 (s, 3H, H-18); 13C-NMR (CDCl3) δ: 141.9 (C-5), 129.3 (C-3), 125.4 (C-4), 123.1 (C-6), 109.7 (C-22), 81.2 (C-16), 67.2 (C-26), 62.5 (C-17), 57.2 (C-14), 48.7 (C-9), 41.8 (C-20), 40.3 (C-12), 35.3 (C-10), 34.6 (C-1), 34.0 (C-8), 33.7 (C-15), 33.4 (C-23), 32.0 (C-8), 30.6 (C-25), 28.9 (C-24), 23.3 (C-2), 21.1 (C-11), 19.1 (C-19), 18.2 (C-27), 17.5 (C-18), 16.6 (C-21).
(25R)-23-Acetyl-3,16-diacetoxy-22,23-pyran-cholesta-5,22-diene (20). ZnCl2 (1.0 g, 7.2 mmol, 6 eq.) was added to a solution of diosgenine (1, 500 mg, 1.2 mmol, 1 eq.) in acetic anhydride (5 mL, 0.5 mmol, 0.4 eq.) and stirred at RT for 72 h. Water was added to the reaction mixture and extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc/DCM, 9:1:1) yielding compound 20 as a white solid (620 mg, 96%): Rf = 0.6 (hexane/EtOAc 4:1).1H-NMR (CDCl3) δ: 5.3 (d, J = 4.3Hz, 1H, H-6), 5.1 (dt, J1 = 5.9 Hz, J2 = 4.9 Hz, 1H, H-16), 4.6 (dt, J = 12.9, 8.1 Hz, 1H, H-3), 4.0 (dd, J1 = 10.7 Hz, J2 = 4.7 Hz, 2H, H-26), 2.9 (dd, J = 6.5 Hz, 1H, H-20,), 2.2 (s, 3H, 28-COCH3), 2.0 (s, 3H, 3-OCOCH3), 1.9 (s, 3H, 16-OCOCH3), 1.2 (d, J = 7.0 Hz, 3H, H-21,), 1.0 (s, 3H, H-19), 0.9 (s, 3H, H-18,), 0,8 (d, J = 6.3, 3H, H-27); 13C-NMR (CDCl3) δ: 198.1 (C-27), 171.3 (C-22), 170.6 (16-OCOCH3), 170.4 (3-OCOCH3), 139.7 (C-5), 122.2 (C-6), 106.9 (C-23), 75.1 (C-16), 73.8 (C-3), 71.5 (C-26), 55.9 (C-17), 54.2 (C-14), 49.9 (C-9), 42.2 (C-13), 39.7 (C-12), 38.0 (C-4), 36.8 (C-1), 36.5 (C-10), 34.8 (C-15), 32.8 (C-20), 31.6 (C-24), 31.5 (C-7), 31.3 (C-8), 29.7 (COCH3), 28.4 (C-28), 27.8 (C-2), 22.2 (C-11), 21.2 (C-25), 21.3 (COCH3), 20.7 (C-19 C24), 19.3 (C-21), 19.2 (C-18).
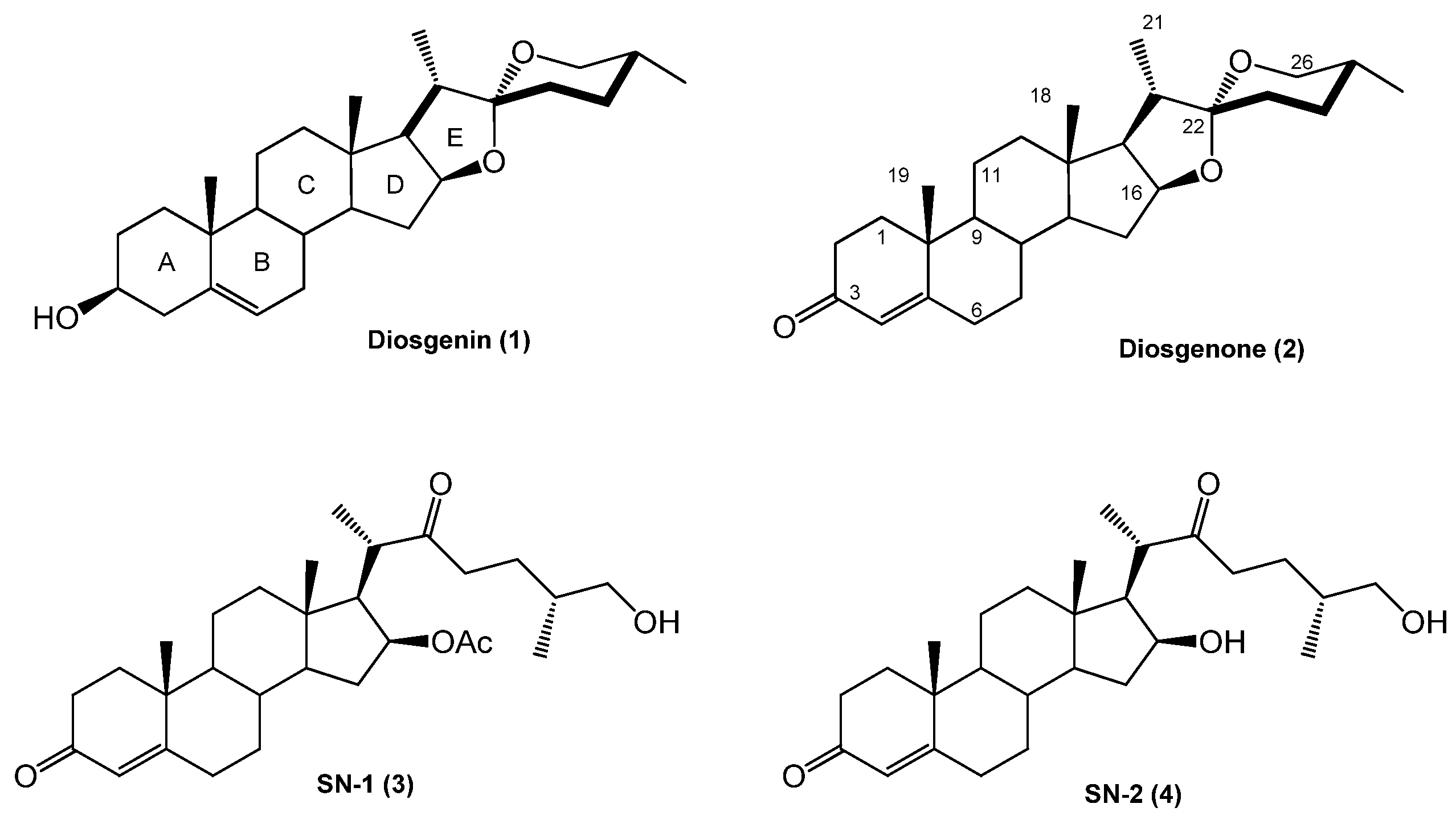
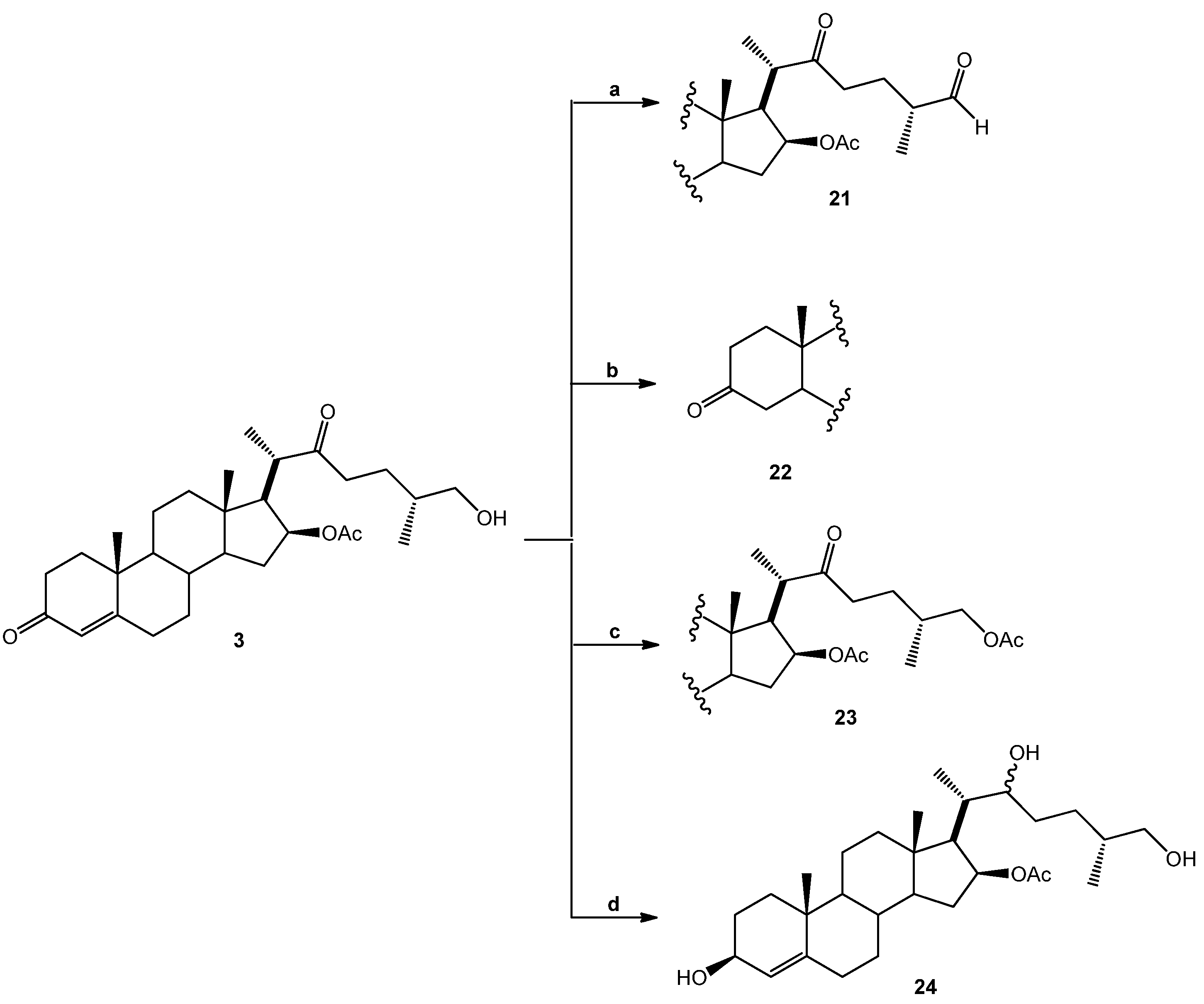
(25R)-16-Acetoxy-4-cholesten-3,22-dione-26-al (21). DMSO (0.2 mL, 2.8 mmol, 93 eq.) was added dropwise to a solution of (COCl)2 (0.1 mL, 2.3 mmol, 29 eq.) in DMC (5 mL) at −60 °C and the resulting solution was stirred for 15 min. A solution of SN-1 (14.2 mg, 0.03 mmol, 1 eq.) in DMC (3 mL) was added dropwise to this solution and stirred for 30 min. (iPr)2NEt (0.62 g, 4.8 mmol, 162 eq.) was added dropwise, and the solution was warmed at RT for 1 h. The solution was diluted with water (10 mL) and extracted with DCM (4 × 15 mL). The organic extracts were combined, washed with water (30 mL), brine (30 mL), dried over Na2SO4, and concentrated in vacuo to yield 21 as a white solid. (8.2 mg, 58.1%): Rf = 0.6 (hexane/EtOAc 1:2). 1H-NMR (CDCl3) δ: 9.6 (s, 1H, CHO), 5.7 (s, 1H, H-4), 4.9 (dt, J = 10.7, 5.9 Hz, 1H, H-16), 2.6 (dd, J = 16.4, 86 Hz, 2H, H-23), 2.0 (s, 3H, 16-OCOCH3), 1.2 (s, 3H, H-19), 1.1 (d, J = 7.1 Hz, 3H, H-27), 1.0 (d, J = 7.1 Hz, 3H, H-21), 0.9 (s, 3H, H-18); 13C-NMR (CDCl3) δ: 212.2 (C-22), 204.4 (C-26), 199.4 (C-3), 170.7 (16-OCOCH3), 170.6 (C-5), 124.0 (C-4), 78.4 (C-16), 57.7 (C-17), 53.4 (C-9), 52.8 (C-14), 47.9 (C-25), 45.6 (C-20), 43.6 (C-13), 39.3 (C-10), 38.7 (C-12), 38.5 (C-23), 35.6 (C-15), 34.9 (C-8), 34.1 (C-1), 33.9 (C-2), 32.6 (C-6), 31.7 (C-7), 23.9 (C-24), 21.2 (16-OCOCH3), 20.6 (C-11), 17.3 (C-19), 16.3 (C-18), 13.6 (C-21), 13.3 (C-27).
(25R)-16-Acetoxy-26-hydroxy-cholestan-3,22-dione (22). A mixture of SN-1 (20.9 mg, 0.04 mmol, 1 eq.) and Pd/C 10% (1.0 mg, 10% mol, 0.1 eq.) in EtOAc (5 mL) was treated with hydrogen and stirred at RT overnight. The reaction mixture was filtered through celite and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc 10:1) yielding compound 22 as a white solid (18.1 mg, 96.1%): Rf = 0.4 (hexane/AcOEt 2:1). 1H-NMR (CDCl3) δ: 4.83 (dd, J1 =10.7, J2 = 5.9 Hz, 1H, H-16), 3.33 (d, , 2H, H-26), 2.4 (dd, , 2H, H-24), 2.0 (s, 3H, 28-COCH3), 1.1 (d, J = 7.1 Hz, 3H, H-21), 1.0 (s, 3H, H-19), 0.9 (s, 3H, H-18), 0.8 (d, J = 6.4 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 212.8 (C-22), 212.1 (C-3), 170.0 (16-OCOCH3), 77.4 (C-16), 65.7 (C-26), 56.7 (C-17), 52.2 (C-14), 51.9 (C-9), 51.7 (C-5), 46.4 (C-20), 42.6 (C-4), 42.5 (C-13), 38.4 (C-1), 38.2 (C-12), 38.1 (C-23), 38.0 (C-2), 37.0 (C-15), 36.7 (C-10), 33.8 (C-25), 33.5 (C-8), 30.1 (C-6), 29.6 (C-7), 25.0 (C-24), 21.2 (16-OCOCH3), 19.8 (C-19), 19.6 (C-18), 19.4 (C-11), 15.9 (C-27), 15.1 (C-21).
(25R)-16,26-Diacetoxy-4-cholesten-3,22-dione (23). Acetyl chloride (0.15 mL, 1.8 mmol, 450 eq.) was added to a mixture of SN-1 (20.1 mg, 0.04 mmol, 1 eq.) in pyridine (2 mL) and the resulting solution was stirred for 30 min at RT. Water was added to the reaction mixture and extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc, 5:2) yielding compound 23 as a white solid (22.1 mg, 100%): Rf = 0.6 (hexane/EtOAc 4:1). 1H-NMR (CDCl3) δ: 5.68 (s, 1H, H-4), 4.92 (dd, J1 = 10.7 Hz, J2 = 5.9 Hz, 1H, H-16), 3.55 (d, J = 10.7 Hz, 2H, H-26), 2.4 (dd, J = 16.4 Hz, 2H, H-23), 2.0 (s, 6H, COCH3), 1.2 (s, 3H, H-19), 1.1 (d, J = 7.1 Hz, 3H, H-21), 0.9 (s, 3H, H-18), 0.8 (d, J = 6.3 Hz, 3H, H-27). 13C-NMR (CDCl3) δ: 212.8 (C-22), 199.4 (C-3), 171.2 (24-OCOCH3), 170.6 (16-OCOCH3), 124.0 (C-4), 78.6 (C-16), 68.9 (C-26), 57.8 (C-17), 53.5 (C-14), 53.4 (C-9), 52.8 (C-5), 47.9 (C-20), 43.6 (C-4), 39.3 (C-13), 39.1 (C-1), 38.5 (C-12), 35.6 (C-23), 34.9 (C-2), 34.1 (C-15), 33.9 (C-10), 32.6 (C-25), 32.1 (C-8), 31.7 (C-6), 26.7 (C-7), 21.2 (C-24), 21.2 (16-OCOCH3), 20.8 (24-OCOCH3), 17.8 (C-19), 15.9 (C-27), 15.1 (C-21), 13.1 (C-18).
(25R)-16-Acetoxy-4-cholesten-3,22,26-triol (24). SN-1 (19.1 mg, 0.04 mmol, 1 eq.) in MeOH (5 mL) was treated with NaBH4 (38.2 mg, 1 mmol, 25 eq.) and stirred at −15 °C for 2 h and then at RT overnight. Water was added to the reaction mixture and extracted with DCM (2 × 15 mL). The combined organic layer was washed with an aq NaHCO3 solution (30 mL), dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by chromatography on a silica gel column (hexane/EtOAc 1:1) yielding compound 24 as a white solid (12 mg, 63%): Rf = 0.3 (hexane/AcOEt 1:1). M.p.: 144–146 °C. 1H-NMR (CDCl3) δ: 5.3 (d, J = 6.7 Hz, 1H, H-4), 5.0 (dt, J1 = 10.7 Hz, J2 = 5.9 Hz, 1H, H-16), 4.1 (q, J = 6.7 Hz, 1H, H-3,), 3.5 (tq, J = 8.0, 7.0 Hz, 2H, H-22), 3.4 (d, J = 10.4 Hz, 2H, H-26), 2.0 (s, 3H, 16-OCOCH3), 1.2 (d, J = 7.1 Hz, 2H, H-21), 1.0 (s, 3H, H-19), 0.8 (s, 3H, H-18); 0.7 (d, J = 7.1 Hz, 3H, H-27); 13C-NMR (CDCl3) δ: 170.7 (16-OCOCH3), 147.1 (C-5), 123.6 (C-4), 79.9 (C-16), 73.3 (C-22), 68.1 (C-26), 67.9 (C-3), 58.4 (C-17), 54.1 (C-9), 53.4 (C-14), 43.3 (C-13), 39.9 (C-12), 38.4 (C-20), 37.2 (C-10), 35.9 (C-25), 35.3 (C-8), 35.2 (C-15), 34.3 (C-1), 32.8 (C-6), 32.0 (C-7), 31.6 (C-24), 29.9 (C-23), 29.4 (C-2), 21.4 (C-18), 21.0 (16-OCOCH3), 20.7 (C-11), 18.8 (C-19), 16.6 (C-27), 14.2 (C-21).
(25R)-16-Hydroxy-4-cholesten-3,22-dione-26-al (25). DMSO (0.2 mL, 2.8 mmol, 93 eq.) was added dropwise to a solution of (COCl)2 (0.1 mL, 2.4 mmol, 47 eq.) in DCM (5 mL) at −60 °C and the resulting solution was stirred for 15 min. A solution of SN-2 (21.5 mg, 0.05 mmol, 1 eq.) in DMC (3 mL) was added dropwise to this solution and stirred for 30 min. (iPr)2NEt (0.62 g, 4.8 mmol, 162 eq.) was added dropwise, and the solution was warmed at RT for 1 h. The solution was diluted with water (10 mL) and extracted with DMC (4 × 15 mL). The organic extracts were combined, washed with water (30 mL), brine (30 mL), dried over Na2SO4, and concentrated in vacuo to yield compound 25 as a white solid (14.9 mg, 69.8%): Rf = 0.5 (hexane/EtOAc 1:2). 1H-NMR (CDCl3) δ: 9.6 (s, 1H, CHO), 5.7 (s, 1H, H4), 2.9 (s, 1H, H-17), 2.6 (dd, J = 16.4 Hz, 2H, H-23), 1.1 (d, J = 7.1 Hz, 3H, H-21), 1.0 (d, J = 6.5 Hz, 3H, H-27), 0.8 (s, 3H, H-18); 13C-NMR (CDCl3) δ: 216.8 (C-16), 213.0 (C-22), 204.4 (C-26), 199.7 (C-3), 169.9 (C-5), 124.2 (C-4), 66.3 (C-17), 53.2 (C-9), 50.5 (C-14), 45.6 (C-25), 43.2 (C-20), 41.7 (C-13), 39.4 (C-15), 38.9 (C-10), 38.4 (C-12), 37.1 (C-23), 35.4 (C-1), 34.6 (C-8), 33.9 (C-2), 32.5 (C-6), 31.9 (C-7), 24.0 (C-24), 20.5 (C-11), 17.3 (C-19), 15.4 (C-18), 14.1 (C-21), 13.1 (C-27).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





























