Comparing the Suitability of Autodock, Gold and Glide for the Docking and Predicting the Possible Targets of Ru(II)-Based Complexes as Anticancer Agents

Abstract
:1. Introduction
2. Results and Discussion

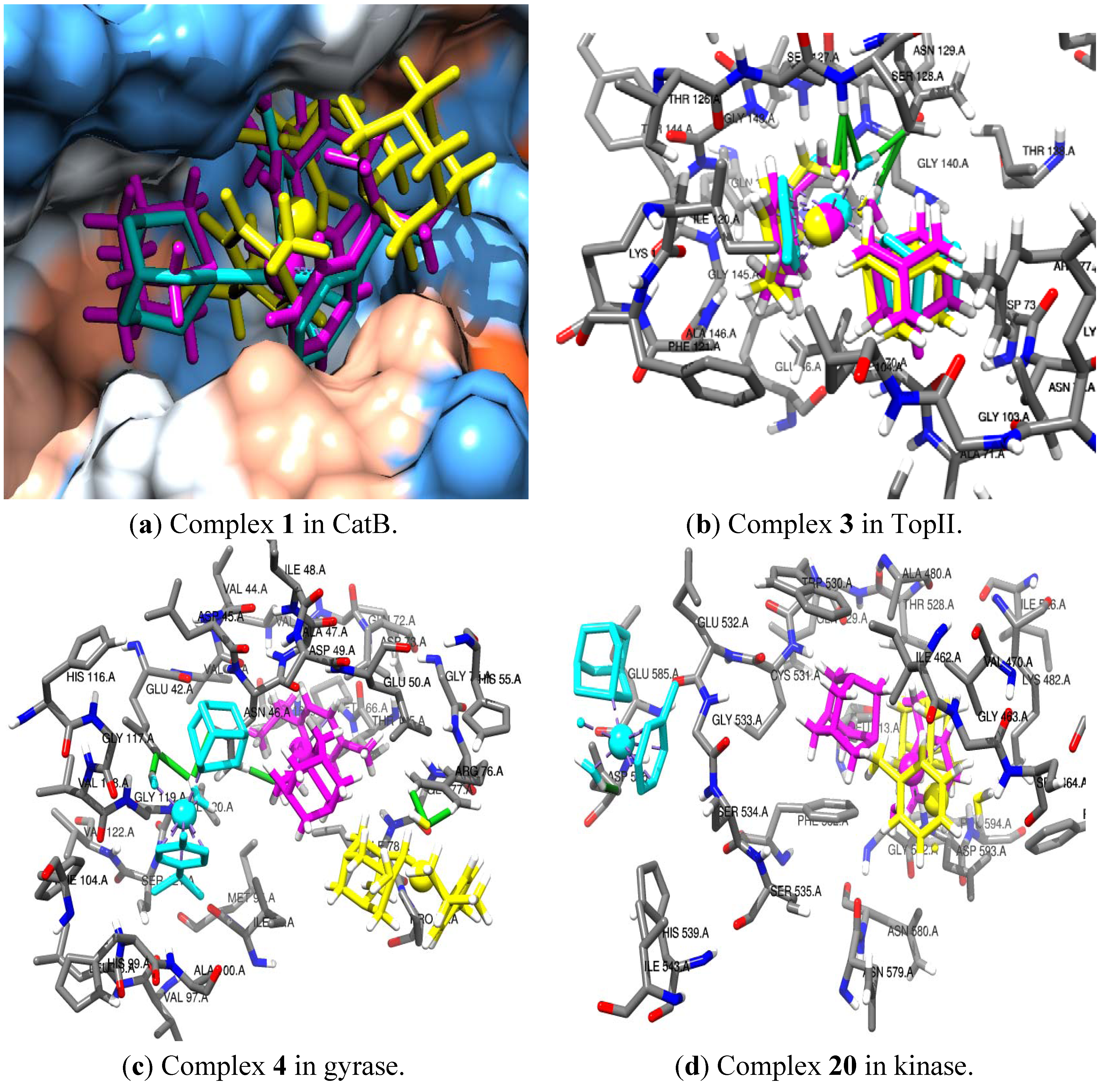{kind=link}
| No | Structure/Name | No | Structure/Name | No | Structure/Name | No | Structure/Name |
|---|---|---|---|---|---|---|---|
| 1 | 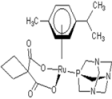 Carbo-rapta-C Carbo-rapta-C | 7 | 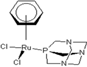 rapta-B rapta-B | 12 | 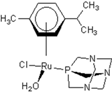 rapta-C-H2O rapta-C-H2O | 17 | 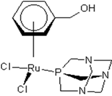 rapta-Ta-OH rapta-Ta-OH |
| 2 | 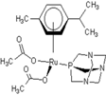 rapta-C-COOH rapta-C-COOH | 8 | 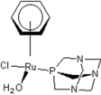 rapta-B-H2O rapta-B-H2O | 13 |  rapta-C-H rapta-C-H | 18 | 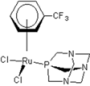 rapta-T-CF3 rapta-T-CF3 |
| 3 | 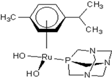 rapta-C-(OH)2 rapta-C-(OH)2 | 9 | 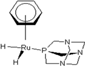 rapta-B-H rapta-B-H | 14 | 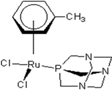 rapta-T rapta-T | 19 | 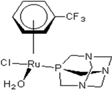 rapta-T-CF3(H2O) rapta-T-CF3(H2O) |
| 4 | 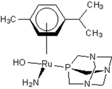 rapta-C-NH2(OH) rapta-C-NH2(OH) | 10 |  rapta-B-NH2 rapta-B-NH2 | 15 |  rapta-Ta-CH3 rapta-Ta-CH3 | 20 | 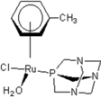 rapta-T-H2O rapta-T-H2O |
| 5 |  oxalo-rapta-C oxalo-rapta-C | 11 | 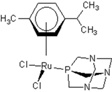 rapta-C rapta-C | 16 | 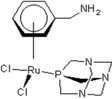 rapta-Ta-NH2 rapta-Ta-NH2 | 21 | 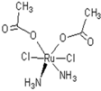 rClCOO-NH3 rClCOO-NH3 |
| 6 | 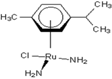 raC-NH2 raC-NH2 |
| CatB | DNA gyrase | HDAC7 | HP−NCP | KINASE | rHA | RNR | Top11 | TrxR | TS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | carbo-rapta-C | −2.14 | −1.68 | −1.41 | −2.67 | −3.45 | −0.97 | −1.51 | −1.70 | −1.00 | |
| 2 | rapta-C-COOH | −1.24 | −1.67 | −1.35 | −0.88 | −2.12 | −1.22 | ||||
| 3 | rapta-C-(OH)2 | −2.50 | −2.14 | −1.38 | −3.02 | −3.26 | −1.49 | −2.52 | −3.12 | −2.84 | −1.91 |
| 4 | rapta-C-NH2(OH) | −2.12 | −1.88 | −1.63 | −1.99 | −3.49 | −1.16 | −3.03 | −2.44 | −1.65 | |
| 5 | oxalo-rapta-C | −2.19 | −2.19 | −2.38 | −2.15 | −4.44 | −2.12 | −0.55 | −2.69 | −3.71 | |
| 6 | raC-NH2 | −2.68 | −2.51 | −1.93 | −3.56 | −5.17 | −2.91 | −1.80 | −3.44 | −2.92 | −2.93 |
| 7 | rapta-B | −3.59 | −2.53 | −2.30 | −2.84 | −3.20 | −2.49 | −2.88 | −3.75 | −2.54 | −1.74 |
| 8 | rapta-B-H2O | −4.04 | −3.18 | −2.16 | −3.41 | −3.10 | −2.40 | −3.52 | −5.68 | −4.10 | |
| 9 | rapta-B-H | −4.01 | −2.63 | −2.83 | −3.03 | −4.06 | −2.96 | −2.81 | −4.67 | −3.16 | −2.56 |
| 11 | rapta-C | −3.36 | −2.34 | −2.62 | −2.46 | −3.31 | −1.88 | −3.32 | −4.18 | −2.49 | −3.09 |
| 12 | rapta-C-H2O | −3.56 | −2.49 | −3.35 | −2.99 | −2.61 | −2.61 | −3.64 | −3.97 | −2.37 | −2.82 |
| 13 | rapta-C-H | −3.68 | −3.08 | −3.44 | −2.79 | −4.34 | −2.66 | −3.82 | −5.09 | −3.00 | −3.06 |
| 14 | rapta-T | −3.25 | −2.65 | −2.71 | −2.39 | −2.01 | −2.53 | −3.91 | −2.56 | −2.74 | |
| 15 | rapta-Ta-CH3 | −3.21 | −2.19 | −2.44 | −2.64 | −2.25 | −2.34 | −3.41 | −3.74 | −2.84 | −3.21 |
| 16 | rapta-Ta-NH2 | −3.05 | −2.56 | −3.06 | −2.97 | −3.96 | −2.68 | −4.15 | −4.76 | −2.73 | −3.22 |
| 17 | rapta-Ta-OH | −3.83 | −3.16 | −3.19 | −3.55 | −3.24 | −3.35 | −4.75 | −5.02 | −2.87 | −4.16 |
| 18 | rapta-T-CF3 | −3.81 | −2.21 | −2.58 | −2.33 | −2.13 | −2.31 | −3.99 | −4.19 | −1.91 | −2.73 |
| 19 | rapta-T-CF3(H2O) | −3.92 | −2.35 | −2.43 | −2.65 | −2.47 | −2.08 | −4.21 | −3.95 | −2.56 | −3.36 |
| 20 | rapta-T-H2O | −3.98 | −2.48 | −2.61 | −2.87 | −3.38 | −2.23 | −3.75 | −4.75 | −2.57 | −3.76 |
| 21 | rClCOO-NH3 | −3.18 | −2.20 | −1.92 | −2.95 | −3.79 | −3.82 | −1.96 | −2.76 | −3.79 | −3.19 |
| CatB | DNA-Gyrase | HDAC7 | HP-NCP | Kinase | rHA | RNR | Top11 | TrxR | TS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | carbo-rapta-C | 50.37 | 40.79 | −39.09 | 21.56 | 39 | 36.51 | 51.46 | 41.13 | 34.48 | 42.45 |
| 2 | rapta-C-COOH | 40.53 | 47.59 | 30.82 | 30.46 | 35.37 | 33.26 | 43.02 | 45.21 | 38.67 | 45.48 |
| 3 | rapta-C-(OH)2 | 45.53 | 39.29 | 37.14 | 21.63 | 42.04 | 39.06 | 38.54 | 60.73 | 41.92 | 52.63 |
| 4 | rapta-C-NH2(OH) | 53.29 | 46.78 | 39.97 | 32.2 | 39.68 | 43.75 | 40.18 | 64.48 | 40.31 | 51.22 |
| 5 | oxalo-rapta-C | 40.94 | 42.48 | 18.01 | 25.28 | 42.51 | 46.79 | 46.62 | 49.95 | 40.75 | 40.28 |
| 6 | raC-NH2 | 32.39 | 26.92 | 30.46 | 25.81 | 27.59 | 29.22 | 24.97 | 30.63 | 35.56 | 30.02 |
| 7 | rapta-B | 45.14 | 31.96 | 32.91 | 22.01 | 34.92 | 29.49 | 28.93 | 41.53 | 37.1 | 34.2 |
| 8 | rapta-B-H2O | 45.94 | 32.76 | 32.4 | 26.36 | 40.14 | 31.73 | 29.32 | 42.14 | 32.71 | 31.72 |
| 9 | rapta-B-H | 45.08 | 32.04 | 38.21 | 40.34 | 33.89 | 41.4 | 40.08 | |||
| 10 | rapta-B-NH2 | 47.66 | 37.76 | 40 | 28.61 | 42.27 | 42.57 | 36.13 | 47.9 | 48.6 | 38.03 |
| 11 | rapta-C | 44.84 | 41.05 | 1.63 | 24.02 | 46.02 | 35.18 | 27.07 | 53.56 | 33.47 | 38.49 |
| 12 | rapta-C-H2O | 40.34 | 39.56 | 10.84 | 26.19 | 42.19 | 36.96 | 36.94 | 52.18 | 37.26 | 37.39 |
| 13 | rapta-C-H | 47.22 | 39.11 | 32.49 | 15.25 | 43.03 | 39.26 | 24.33 | 51.92 | 32.93 | 39.36 |
| 14 | rapta-T | 41.09 | 34.83 | 34.43 | 22.33 | 38.57 | 31.1 | 28.8 | 46.48 | 40.48 | 34.66 |
| 15 | rapta-Ta-CH3 | 50.09 | 38.46 | 38.59 | 25.11 | 39.04 | 35.87 | 29.64 | 48.55 | 30.9 | 38.84 |
| 16 | rapta-Ta-NH2 | 50.46 | 44.05 | 34.02 | 30.56 | 46.61 | 39.58 | 41.03 | 53.8 | 35.93 | 38.7 |
| 17 | rapta-Ta-OH | 49.51 | 43.87 | −4.38 | 28.01 | 38.15 | 34.39 | 42 | 49.75 | 36.91 | 40.75 |
| 18 | rapta-T-CF3 | 45.33 | 32.37 | 33.2 | 22.83 | 39.65 | 30.95 | 31.99 | 43.82 | 37.93 | 35.02 |
| 19 | rapta-T-CF3(H2O) | 46.03 | 27.52 | 29.51 | 26.64 | 43.79 | 29.8 | 34.39 | 45.61 | 30.34 | 31.96 |
| 20 | rapta-T-H2O | 45.13 | 33.69 | 26.55 | 26.86 | 44.12 | 29.71 | 37.95 | 46.1 | 28.8 | 34.72 |
| 21 | rClCOO-NH3 | 35.59 | 38.42 | 38.51 | 28.92 | 32.83 | 32.54 | 33.89 | 40.86 | 39.64 | 48.42 |
| CatB | DNA gyrase | HDAC7 | HP-NCP | Kinase | rHA | RNR | Top11 | TrxR | TS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | carbo-rapta-C | −9.29 | −8.92 | −8.33 | −3.93 | −6.57 | −6.49 | −5.49 | −4.94 | −3.04 | −8.05 |
| 2 | rapta-C-COOH | −8.33 | −6.63 | −7.68 | −3.06 | −5.51 | −5.02 | −4.12 | −2.63 | −5.32 | −6.66 |
| 3 | rapta-C-(OH)2 | −9.96 | −7.9 | −8.1 | −3.46 | −6.65 | −5.58 | −4 | −3.82 | −6.43 | −6.27 |
| 4 | rapta-C-NH2(OH) | −9.42 | −6.79 | −7.37 | −2.95 | −6.16 | −5.25 | −3.73 | −3.1 | −6.22 | −5.79 |
| 5 | oxalo-rapta-C | −9.11 | −8.35 | −7.93 | −3.66 | −6.44 | −5.52 | −5.16 | −3.8 | −7.26 | −7.15 |
| 6 | raC-NH2 | −3.22 | −2.72 | −3.69 | −2.11 | −3.41 | −2.96 | −2.94 | −1.84 | −2.78 | −2.73 |
| 7 | rapta-B | −5.01 | −3.95 | −4.09 | −2.61 | −3.82 | −4.11 | −2.65 | −2.38 | −2.84 | −3.89 |
| 8 | rapta-B-H2O | −7.16 | −6.21 | −6.95 | −2.98 | −6.3 | −4.72 | −3.79 | −3.22 | −5.69 | −5.46 |
| 9 | rapta-B-H | −4.83 | −4.77 | −5.07 | −2.71 | −4.81 | −4.4 | −2.83 | −2.42 | −3.03 | −4.01 |
| 10 | rapta-B-NH2 | −7.09 | −5.65 | −6.76 | −2.76 | −5.76 | −4.23 | −3.31 | −2.54 | −4.24 | −4.68 |
| 11 | rapta-C | −5.73 | −5.36 | −5.2 | −3.08 | −4.71 | −4.34 | −3.46 | −3.18 | −2.18 | −4.65 |
| 12 | rapta-C-H2O | −8.15 | −6.58 | −6.63 | −3.61 | −6.21 | −5.11 | −3.89 | −2.78 | −5.87 | −5.17 |
| 13 | rapta-C-H | −5.44 | −5.14 | −5.1 | −2.83 | −4.69 | −4.24 | −2.99 | −2.65 | −3.46 | −4.32 |
| 14 | rapta-T | −7.1 | −6.15 | −6.23 | −2.79 | −5.32 | −4.62 | −3.74 | −3.23 | −4.67 | −5.31 |
| 15 | rapta-Ta-CH3 | −5.42 | −4.94 | −4.49 | −3.04 | −4.18 | −4.48 | −3.68 | −2.86 | −2.64 | −4.13 |
| 16 | rapta-Ta-NH2 | −5.02 | −4.76 | −4.37 | −2.96 | −4 | −4.21 | −3.11 | −2.7 | −2.5 | −3.77 |
| 17 | rapta-Ta-OH | −7.15 | −7.03 | −7.51 | −5.14 | −6.48 | −5.83 | −5.32 | −3.98 | −4.58 | −5.99 |
| 18 | rapta-T-CF3 | −5.02 | −4.59 | −4.98 | −1.86 | −4.33 | −3.89 | −2.8 | −2.04 | −1.77 | −3.91 |
| 19 | rapta-T-CF3(H2O) | −6.57 | −5.86 | −5.91 | −2.31 | −5.87 | −4.34 | −3.07 | −2.53 | −4.41 | −4.76 |
| 20 | rapta-T-H2O | −7.27 | −6.26 | −6.61 | −2.9 | −6.32 | −5.04 | −3.62 | −3.23 | −4.89 | −5.52 |
| 21 | rClCOO-NH3 | −5.74 | −4.88 | −5.57 | −2.78 | −5.32 | −4.81 | −3.37 | −3.09 | −4.25 | −3.85 |
| No | Method | Receptor Interactions |
|---|---|---|
| 1 | Autodock | CatBb{[HB: 1.92 Å (O@COO)-(H@imHIS 111E)], MR:4.07Å (arTRP 221E)}; |
| Gyrasea{[HB:1.74Å (O@COO)-(H@NHVAL 120A)]; [MR:4.35Å (NH2ASN 46A)}; | ||
| HDAC7a{[MR:3.52Å (COOH ASP 626A)]}; | ||
| Kinaseb{[HB:1.36 (O@COO)- (H@imHIS 584A)]}; | ||
| rHAa{[MR:4.05Å (NH2 ASN 109A)], [MR:4.21Å (COOH GLU 425A)]}; | ||
| RNRa {[HB:1.99Å (O@COO)- (H@NHTHR 209A)], [HB:2.55Å (N@PTA)- (H@COOHGLU 441A],} | ||
| TSa {[no HB and MR]} | ||
| Gold | CatBb{[MR: 4.32Å (arTRP 221E)]}; RNRa {[MR:3.83Å (HOTHR 209A)]} | |
| 3 | Autodock | CatBa{[HB:1.59Å (OH1)-(O@COOGLU 122E)], [HB:1.52Å (OH2)-(O@COOH GLU 122E)], [HB:2.52Å (HO2)-(H@NH2GLN 23D)], [MR:3.88Å (CH2GLY 121E)}; HDAC7b{[HB:1.69 (OH)-(O@COOASP 626A)],[MR:4.37Å (arPHE 738A)], [MR:4.39Å (COOH ASP 626A)]; HP-NCPa{[HB:2.22 (OH)-(O@COOGLU 64G)],[MR:2.83Å (COOGLU 64G)], [MR:4.19Å (COOGLU 61G)]} Kinasea{[HB:1.96Å (H@OH1)-(O@COOH ASP 586A)], [HB:2.54Å (H@HO2)-(O@COOH ASP 586A)], [HB:3.02Å (O@HO2)-(H@COOH ASP 586A)],} |
| Gold | Gyrasea{[MR:4.48Å (COO GLU 50A)], } | |
| Top11b { [HB:2.11Å (O@OH)- (H@NH SER 128A)], [MR:3.97Å (CHSER 127A)], [MR:4.23Å (CO ASN 71A)]} | ||
| TrxRb{ [HB:1.73Å (O@OH)-(H@NH2 ARG 166A)], [MR:3.67Å (NH2ARG 166A)]} | ||
| TSa {[HB:1.11Å (O@OH1)-(H@NH2ARG 218A], [HB:2.30Å (O@OH1)-(H@OH SER 219A], [HB:2.52Å (O@OH2)-(H@NH2ARG 23A)][MR:3.47Å (CH2ARG 23A)], [MR:3.20Å (NH2ARG 218A)]} | ||
| 4 | Autodock | CatBa{[HB: 1.44 Å (NH2)-(O@COOH GLU 122E)], MR:3.23Å (CH2 GLY 29D)}; |
| Gyraseb{HB: 1.87 Å (OH)-(H@NH2 ASN 46A)], [MR:2.07Å (CH2ASN 46A)], [MR:4.17Å (CH3ILE 78A)]} | ||
| HDAC7a{[MR:1.87Å (arPHE 679A)], [MR:3.73Å (im HIS 709A) ] } | ||
| TSb {[HB:2.57Å (N@PTA)-(H@OH SER 219A], [MR:2.90Å (SH CYS 198A)], [MR:3.48Å (CH3LEU 195A)]} | ||
| 5 | Autodock | HP-NCPa{[IT:2.90 (N@PTA)-(O@COOGLU 64G)],[MR:3.73Å (COOGLU 61G)]} |
| Gyraseb{[HB:1.78Å (O@COO)-(H@NHVAL 120A)], [MR:4.41Å (COGLY 117A)], [MR:4.48Å (CH2GLY 119A)]}; | ||
| Top11a{[HB:2.08Å (O@COO)-(H@OHSER 128A)], [MR:3.18Å (COOGLU 134A)]} | ||
| TrxRa{[HB:1.83Å (O@COO)-(H@NHSER 386A)], [MR:3.96Å (COGLY 38A)]} | ||
| TSb {[HB:1.86Å (O@COO)-(H@SHCYS 198A], [HB:2.22Å (CO)-(H@NH ASP 221A], [MR:4.14Å (CH2GLY 225A)]} | ||
| Gold | rHAa {[HB:1.68Å (O@COO)-(H@NH2ASN 111A], [HB:2.08Å (O@COO)-(H@NH ASN 111A], [MR:3.20Å (CH2 GLN 33A)]} | |
| RNRb {[HB:1.86Å (O@COO1)- (H@NH GLU 623A)], [HB:1.98Å (O@COO1)- (H@NH THR 624A)], HB:2.56Å (O@COO1)- (H@OH THR 624A)], HB:1.98Å (O@COO2)- (H@OH SER 625A)], [IT:2.81Å (N@PTA)- (O@COPRO 621A], [MR:3.40Å (HOTHR 209A)]} | ||
| 6 | Glide | HP-NCPa{[MR:2.42Å (NH3 LYS 113H)]} |
| Kinasea{[MR:3.01Å (NH ASP 593A) ]} | ||
| 8 | Glide | CatBa{[HB:1.96Å (H@H2O)-(O@COOGLU 122E)]} |
| Gyrasea{[HB:1.67Å (H@H2O)-(O@COO ASP 49A)]} | ||
| Top11a{[HB:1.83Å (H@H2O)- (O@OHSER 128A)], [HB:2.04Å (H@H2O)- (O@CO ASN 129A)], [IT:2.97Å (N@PTA)- (O@CO ASN 70A)], [MR:3.66Å (NH SER 128A)]} TSb{[HB:1.69Å (H@H2O)- (H@OHASP 257A)], [MR:3.73Å (OH SER 219A)]} | ||
| 9 | Glide | CatBb{[MR:3.09Å (CH2 GLY121E)], [MR:4.29Å (COOH GLY122E)]} |
| TrxRb{[MR:3.42Å OH SER 199A]} | ||
| 10 | Gold | rHAb { [MR:1.55Å (CH3@S(CH3) MET 87A)]} |
| TrxRa{ [MR:4.18Å (ar@ph TYR 200A)]} | ||
| 12 | Autodock | |
| Glide | HDAC7b{[HB:1.64Å (H@H2O)-(O@COOASP 626A)]} | |
| 13 | Glide | HDAC7a{[MR:3.21Å (ar PHE 679A)], [MR:4.42Å (CH3 LEV 810A)]} |
| Kinasea{[MR:3.33Å (ar PHE 582A) ], [MR:3.30Å (CH3ILE 462A)]} | ||
| Top11b{[MR:4.10Å (CH2ASN 70A)], [MR:3.78Å (CH3 ILE 104A)]} | ||
| 15 | Gold | HDAC7b{[MR:2.80Å (arPHE 679A)], [MR:3.52Å (im HIS 709A) ] |
| 16 | Gold | HP-NCPb{[HB: 1.69Å(NH2@ar)-(O@COOH GLU 61G)]} |
| Kinasea{[MR:4.24Å (ar TRP 530A) ], [MR:4.34Å (ar PHE 582A) ] } | ||
| 17 | Autodock | HP-NCPa{[HB:2.044Å(OH@ar)-(O@COTHR 101G)]}; |
| rHAb {[HB:1.88Å (OH@ar)-(O@COPRO 113A], [HB:2.02Å (HO@ar)-(H@NH ARG 145A], [HB:2.11Å (HO@ar)-(H@NH LEU 115A], [HB:2.44Å (N@PTA)-(H@COOH GLU 425A]}; | ||
| RNRb {[HB:1.89Å (HO@ar)-(H@NH SER 625A]} | ||
| Top11b {[no HB and MR]} | ||
| Glide | Gyraseb{[MR:3.26Å (CH2 ILE 78A)], [MR:3.99Å (CH2 ASN 46A)]} | |
| Top11b{[HB:1.69Å (OH@ar)- (O@COASP 73A)], [MR:4.33Å (COO GLU 134A)]} | ||
| rHAb {[HB:1.77Å (OH@ar)-(O@CO PRO 110A]} | ||
| RNRa{[HB:1.80Å (OH@ar)- (O@OHSER 625A)], [HB:1.86Å (OH@ar)- (H@NHSER 625A)], [MR:4.08Å (H@OH THR 209A)]} | ||
| TSa{[HB:2.63Å (OH@ar)- (H@NH3ARG 23A)], [MR:4.39Å (NH2 ASP 221A)], [MR:3.43Å (SH CYS 198A)]} | ||
| 19 | Glide | RNRb{[HB andMR]} |
| 20 | Gold | Kinasea{[MR:2.83Å (ar PHE 582A) ], [MR:3.46Å (CH3VAL 470A)] } |
| 21 | Autodock | TrxRb {[HB:1.70Å (H@NH3)-(O1@COOGLU 341A], [HB:2.32Å (H@NH3)-(O2@COOGLU 341A)], [HB:1.70Å (H@NH3)-(O@COARG 293A], [MR: 3.26Å (COOGLU 341A)], [MR: 3.88Å (COOARG 166A)], [MR: 4.09Å (NH3LYS 315A)]} |
| Glide | rHAa {[HB:1.74Å (O@COO1)-(H@NH2ARG 144A], [HB:2.03Å (O@COO2)-(H@NH2ARG 145A], [HB:1.95Å (O@COO)-(H@OH GLU 141A]} | |
| TrxRa {[HB:2.62Å (H@NH3)-(O@CO VAL 291A], [HB:1.85Å (O@COO)-(H@NH ALA 198A], [MR:3.27Å NH2ARG 221A], [MR:3.81Å CH2ARG 226A]} |

| Autodock vs. Gold | Autodock vs. Glide | Gold vs. Glide | |
|---|---|---|---|
| CatB | 0.27 | −0.50 | 0.10 |
| DNA-Gyrase | 0.54 | −0.32 | −0.33 |
| HDAC7 | −0.13 | −0.33 | −0.38 |
| HP-NCP | 0.24 | −0.17 | −0.18 |
| KINASE | 0.32 | −0.17 | −0.26 |
| rHA | 0.45 | −0.37 | −0.20 |
| RNR | 0.76 | −0.32 | −0.36 |
| topoII | 0.41 | −0.06 | −0.11 |
| TrxR | 0.32 | 0.13 | 0.07 |
| TS | 0.42 | −0.28 | −0.46 |
| Factors | gauss_s | gauss_e | gauss_h | gauss_a | gauss_d | S.D | R^2 | R^2-CV | R^2-Scramble | Stability | F | P | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CatB | 1 | 0.53 | 0.15 | 0.22 | 0.06 | 0.05 | 0.33 | 0.85 | 0.41 | 0.39 | 0.66 | 138.8 | 1.83 E-011 |
| DNA gyrase | 1 | 0.52 | 0.17 | 0.19 | 0.08 | 0.04 | 0.53 | 0.53 | 0.19 | 0.39 | 0.86 | 27.2 | 2.41 E-005 |
| HDAC7 | 1 | 0.5 | 0.14 | 0.19 | 0.07 | 0.1 | 0.42 | 0.65 | 0.04 | 0.45 | 0.58 | 41.3 | 1.82 E-006 |
| HP-NCP | 1 | 0.59 | 0.14 | 0.19 | 0.04 | 0.03 | 0.42 | 0.71 | 0.35 | 0.38 | 0.77 | 58.5 | 6.88 E-008 |
| Kinase | 1 | 0.63 | 0.08 | 0.16 | 0.07 | 0.06 | 0.68 | 0.49 | 0.25 | 0.35 | 0.94 | 21.8 | 1.07 E-004 |
| rHA | 1 | 0.53 | 0.16 | 0.22 | 0.04 | 0.05 | 0.41 | 0.72 | 0.02 | 0.55 | 0.48 | 52.9 | 3.68 E-007 |
| RNR | 1 | 0.63 | 0.13 | 0.15 | 0.05 | 0.04 | 1 | 0.62 | 0.39 | 0.45 | 0.93 | 38.7 | 2.00 E-006 |
| TopoII | 1 | 0.43 | 0.18 | 0.18 | 0.07 | 0.14 | 0.45 | 0.65 | 0.27 | 0.45 | 0.72 | 38.4 | 3.78 E-006 |
| TrxR | 1 | 0.48 | 0.15 | 0.23 | 0.08 | 0.06 | 0.32 | 0.8 | 0.03 | 0.55 | 0.31 | 92.6 | 1.57 E-009 |
| TS | 1 | 0.6 | 0.16 | 0.15 | 0.05 | 0.03 | 0.7 | 0.49 | 0.27 | 0.36 | 0.94 | 23.4 | 6.35 E-005 |
3. Experimental
Computational Methods
4. Conclusions
Acknowledgments
References
- Sava, G.; Bergamoa, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dorcier, A.; Scolaro, C.; Dyson, P.J. Development of organometallic (organo-transition metal) pharmaceuticals. Appl. Organometal. Chem. 2005, 19, 1–10. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging protein targets for anticancer metallodrugs: inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar] [CrossRef]
- Page, S. Ruthenium compounds as anticancer agents. Education in Chemistry, 2012. Available online: http://www.rsc.org/eic (accessed on 1 August 2012).
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platium Metal 2001, 45, 62–69. [Google Scholar]
- Heffeter, P.; Bock, K.; Atil, B.; Hoda, M.A.R.; Korner, W.; Bartel, C.; Jungwirth, U.; Keppler, B.K.; Micksche, M.; Berger, W.; et al. Intracellular protein binding patterns of the anticancer ruthenium drugs KP1019 and KP1339. J. Biol. Inorg. Chem. 2010, 15, 737–748. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium(II)-arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53–JNK pathways. J. Biol. Inorg. Chem. 2008, 13, 1149–1155. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A new redox-active anticancer agent—preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodiver. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Wu, B.; Ong, M.S.; Groessl, M.; Adhireksan, Z.; Hartinger, C.G.; Dyson, P.J.; Davey, C.A. A ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core. Chem. Eur. J. 2011, 17, 3562–3566. [Google Scholar]
- Ang, W.H.; Daldini, E.; Scolaro, C.; Scopelliti, R.; Juillerat-Jeannerat, L.; Dyson, P.J. Development of organometallic ruthenium−arene anticancer drugs that resist hydrolysis. Inorg. Chem. 2006, 45, 9006–9013. [Google Scholar]
- Egger, A.E.; Hartinger, C.G.; Renfrew, A.K.; Dyson, P.J. Metabolization of [Ru(ŋ6-C6H5CF3)(pta)Cl2]: A cytotoxic RAPTA-type complex with a strongly electron withdrawing arene ligand. J. Biol. Inorg. Chem. 2010, 15, 919–927. [Google Scholar] [CrossRef]
- Wu, A.; Dehestani, A.; Saganic, E.; Crevier, T.J.; Kaminsky, W.; Cohen, D.E.; Mayer, J.M. Reactions of Tp–Os nitrido complexes with the nucleophiles hydroxide and thiosulfate. Inorg. Chim. Acta 2006, 359, 2842–2849. [Google Scholar] [CrossRef]
- Richardson, D.E.; Lang, G.H.L.; Crestoni, E.; Ryan, M.F.; Eyler, J.R. Intrinsic reactivity of metal–hydroxide complexes: O-H bond activation and adduct formation in gas-phase reactions of Cp2ZrOH+. Int. J. Mass Spectrom. 2001, 204, 255–266. [Google Scholar] [CrossRef]
- Shubina, E.S.; Belkova, N.V.; Epstein, L.M. Novel types of hydrogen bonding with transition metal pi-complexes and hydrides. J. Organomet. Chem. 1997, 536–537, 17–29. [Google Scholar] [CrossRef]
- Hoskin, A.J.; Stephan, D.W. Early transition metal hydride complexes: Synthesis and reactivity. Coord. Chem. Rev. 2002, 233–234, 107–129. [Google Scholar] [CrossRef]
- Heinekey, D.M.; Mellows, H.; Pratum, T. Dynamic Processes in cis-dihydrogen/hydride complexes of ruthenium. J. Am. Chem. Soc. 2000, 122, 6498–6499. [Google Scholar] [CrossRef]
- Lee, D.; Lippard, S.J. Structural flexibility within a sterically hindered ligand platform: mononuclear iron(II) carboxylate complexes as subsite models for diiron(II) centers. Inorg. Chim. Acta 2002, 341, 1–11. [Google Scholar] [CrossRef]
- Kannan, S.; Venkatachalam, G.; Lee, H.; Min, B.K.; Kim, W.; Koo, E.; Do, Y.R.; Yoon, S. Mononuclear transition metal complexes with sterically hindered carboxylate ligands: Synthesis, structural and spectral properties. Polyhedron 2011, 30, 340–346. [Google Scholar] [CrossRef]
- Kaplan, A.W.; Ritter, J.C.M.; Bergman, R.G. Synthesis and structural characterization of late transition metal parent amido (LnM-NH2) complexes: An acid/conjugate base metathesis approach. J. Am. Chem. Soc. 1998, 120, 6828–6829. [Google Scholar] [CrossRef]
- Ciancetta, A.; Genheden, S.; Ryde, U. A QM/MM study of the binding of RAPTA ligands to cathepsin B. J. Comput. Aided Mol. Des. 2011, 25, 729–742. [Google Scholar] [CrossRef]
- Hanif, M.; Henke, H.; Meier, S.M.; Martic, S.; Labib, M.; Kandioller, W.; Jakupec, M.A.; Arion, V.B.; Kraatz, H.; Keppler, B.K.; et al. Is the reactivity of M(II)-arene complexes of 3-hydroxy-2(1H)-pyridones to biomolecules the anticancer activity determining parameter? Inorg. Chem. 2010, 49, 7953–7963. [Google Scholar] [CrossRef]
- Turel, I.; Kljun, J.; Perdih, F.; Morozova, E.; Bakulev, V.; Kasyanenko, N.; Byl, J.A.W.; Osheroff, N. First ruthenium organometallic complex of antibacterial agent ofloxacin. Crystal structure and interactions with DNA. Inorg. Chem. 2010, 49, 10750–10752. [Google Scholar] [CrossRef]
- Xie, P.; Streu, C.; Qin, J.; Bregman, H.; Pagano, N.; Meggers, E.; Marmorstein, R. The crystal structure of BRAF in complex with an organoruthenium inhibitor reveals a mechanism for inhibition of an active form of BRAF kinase. Biochemistry 2009, 48, 5187–5198. [Google Scholar] [CrossRef]
- Hu, W.; Luo, Q.; Ma, X.; Wu, K.; Liu, J.; Chen, Y.; Xiong, S.; Wang, J.; Sadler, P.J.; Wang, F. Arene control over thiolate to sulfinate oxidation in albumin by organometallic ruthenium anticancer complexes. Chem. Eur. J. 2009, 15, 6586–6594. [Google Scholar]
- Stepanenko, I.N.; Casini, A.; Edafe, F.; Novak, M.S.; Arion, V.B.; Dyson, P.J.; Jakupec, M.A.; Keppler, B.K. Conjugation of organoruthenium(II) 3-(1H-benzimidazol-2-yl)pyrazolo[3,4-b]pyridines and Indolo[3,2-d]benzazepines to recombinant human serum albumin: A strategy to enhance cytotoxicity in cancer cells. Inorg. Chem. 2011, 50, 12669–12679. [Google Scholar]
- Zhuang, W.; Wu, X.; Zhou, Y.; Liu, G.; Wu, T.; Yao, X.; Du, L.; Wei, M. Polymorphisms of thymidylate synthase in the 5'- and 3' -untranslated regions and gastric cancer. Dig. Dis. Sci. 2009, 54, 1379–1385. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Malla, R.R.; Gopinath, S.; Gondi, C.S.; Alapati, K.; Dinh, D.H.; Tsung, A.J.; Rao, J.S. uPAR and cathepsin B down regulation induces apoptosis by targeting calcineurin A to BAD via Bcl-2 in glioma. J. Neurooncol. 2012, 107, 69–80. [Google Scholar] [CrossRef]
- Hu, X.; Shelver, W.H. Docking studies of matrix metalloproteinase inhibitors: zinc parameter optimization to improve the binding free energy prediction. J. Mol. Graphics Model. 2003, 22, 115–126. [Google Scholar] [CrossRef]
- Adeniyi, A.A.; Ajibade, P.A. Inhibitory activities and possible anticancer targets of Ru(II)-based complexes using computational docking method. J. Mol. Graph. Model. 2012, 38, 60–69. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly version 7.1.G. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 5 July 2012).
- Caballero, N.A.; Meléndez, F.J.; Niño, A.; Muñoz-Caro, C. Molecular docking study of the binding of aminopyridines within the K+ channel. J. Mol. Model. 2007, 13, 579–586. [Google Scholar] [CrossRef]
- Ma, D.-L.; Ma, V.P.-Y.; Chan, D.S.-H.; Leung, K.-H.; Zhong, H.-J.; Leung, C.-H. In silico screening of quadruplex-binding ligands. Methods 2012, 57, 106–114. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Wang, F.; Habtemariam, A.; van der Geer, E.P.L.; Fernandez, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.A.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. PNAS 2005, 102, 18269–18274. [Google Scholar] [CrossRef]
- Research collaboratory for structural bioinformatics (RCSB) online pdb database. Available online: http://www.rcsb.org (accessed on 1 May 2012).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Cole, J.C.; Nissink, J.W.M.; Taylor, R. Protein-ligand Docking and Virtual Screening with GOLD in Virtual Screening in Drug Discovery; Shoichet, B., Alvarez, J., Eds.; Taylor & Francis CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. “Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity”. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Autodock. Available online: http://autodock.1369657.n2.nabble.com/ADL-Parameters-for-docking-with-metal-ions-in-receptor-td2505649.html (accessed on 6 May 2012).
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, Accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- The R project for statistical computing. Available online: http://www.R-project.org (accessed on 1 May 2012).
- Sample Availability: These are modelled compounds.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adeniyi, A.A.; Ajibade, P.A. Comparing the Suitability of Autodock, Gold and Glide for the Docking and Predicting the Possible Targets of Ru(II)-Based Complexes as Anticancer Agents. Molecules 2013, 18, 3760-3778. https://doi.org/10.3390/molecules18043760
Adeniyi AA, Ajibade PA. Comparing the Suitability of Autodock, Gold and Glide for the Docking and Predicting the Possible Targets of Ru(II)-Based Complexes as Anticancer Agents. Molecules. 2013; 18(4):3760-3778. https://doi.org/10.3390/molecules18043760
Chicago/Turabian StyleAdeniyi, Adebayo A., and Peter A. Ajibade. 2013. "Comparing the Suitability of Autodock, Gold and Glide for the Docking and Predicting the Possible Targets of Ru(II)-Based Complexes as Anticancer Agents" Molecules 18, no. 4: 3760-3778. https://doi.org/10.3390/molecules18043760