Theoretical Analysis on the Kinetic Isotope Effects of Bimolecular Nucleophilic Substitution (SN2) Reactions and Their Temperature Dependence

Abstract
:
1. Introduction
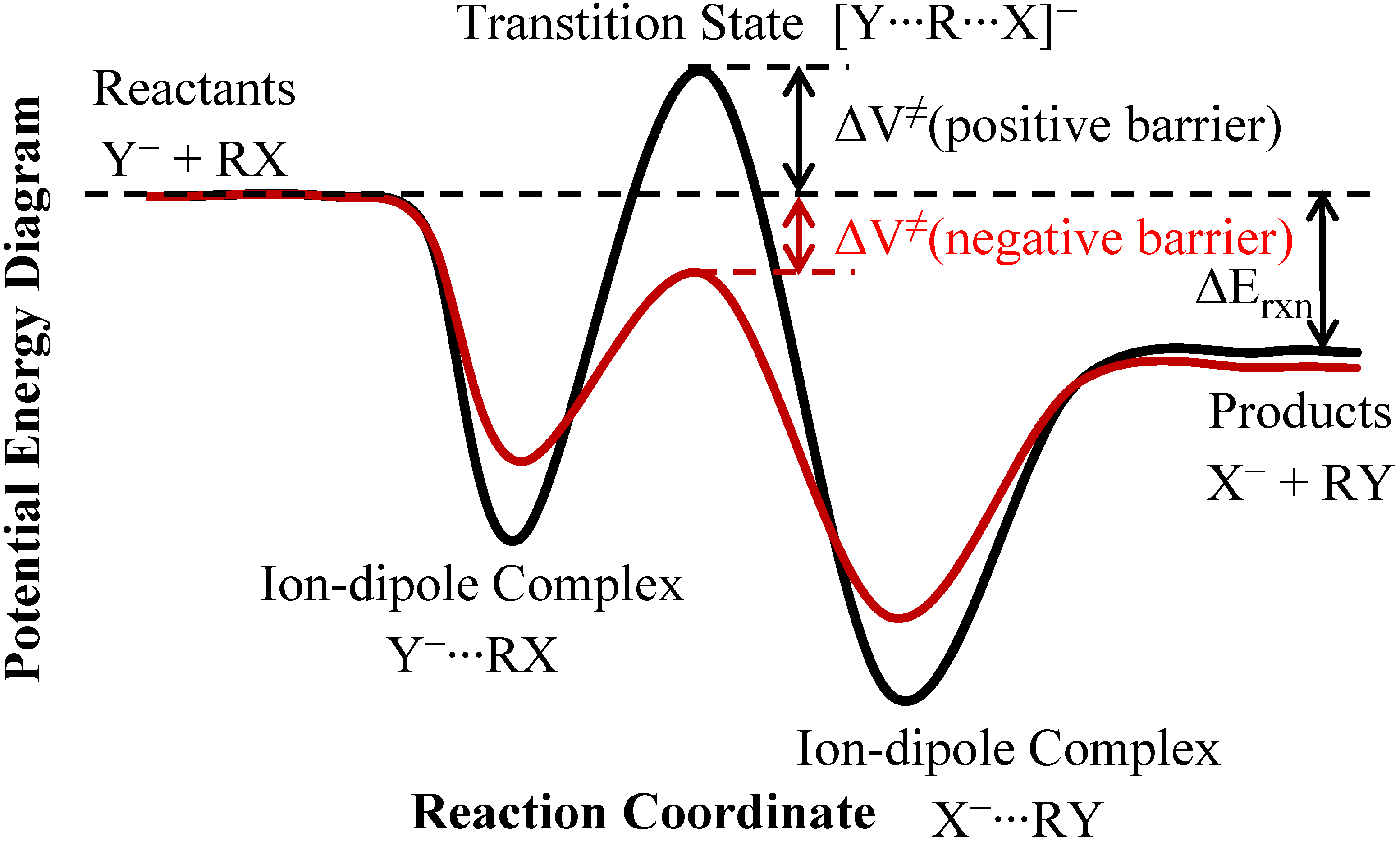
2. Theory and Methods
2.1. KIE Definition
2.2. KIEs Calculation from the Capture Rate Constants




2.3. KIEs Calculation Using the Canonical Unified Statistical (CUS) Model


2.4. KIEs Calculation Using the Transition State Theory (TST)







2.5. KIEs Calculation with Tunneling Correction



2.6. Electronic Structure Calculation
2.7. Rate Constant and KIEs Calculation
3. Results and Discussion
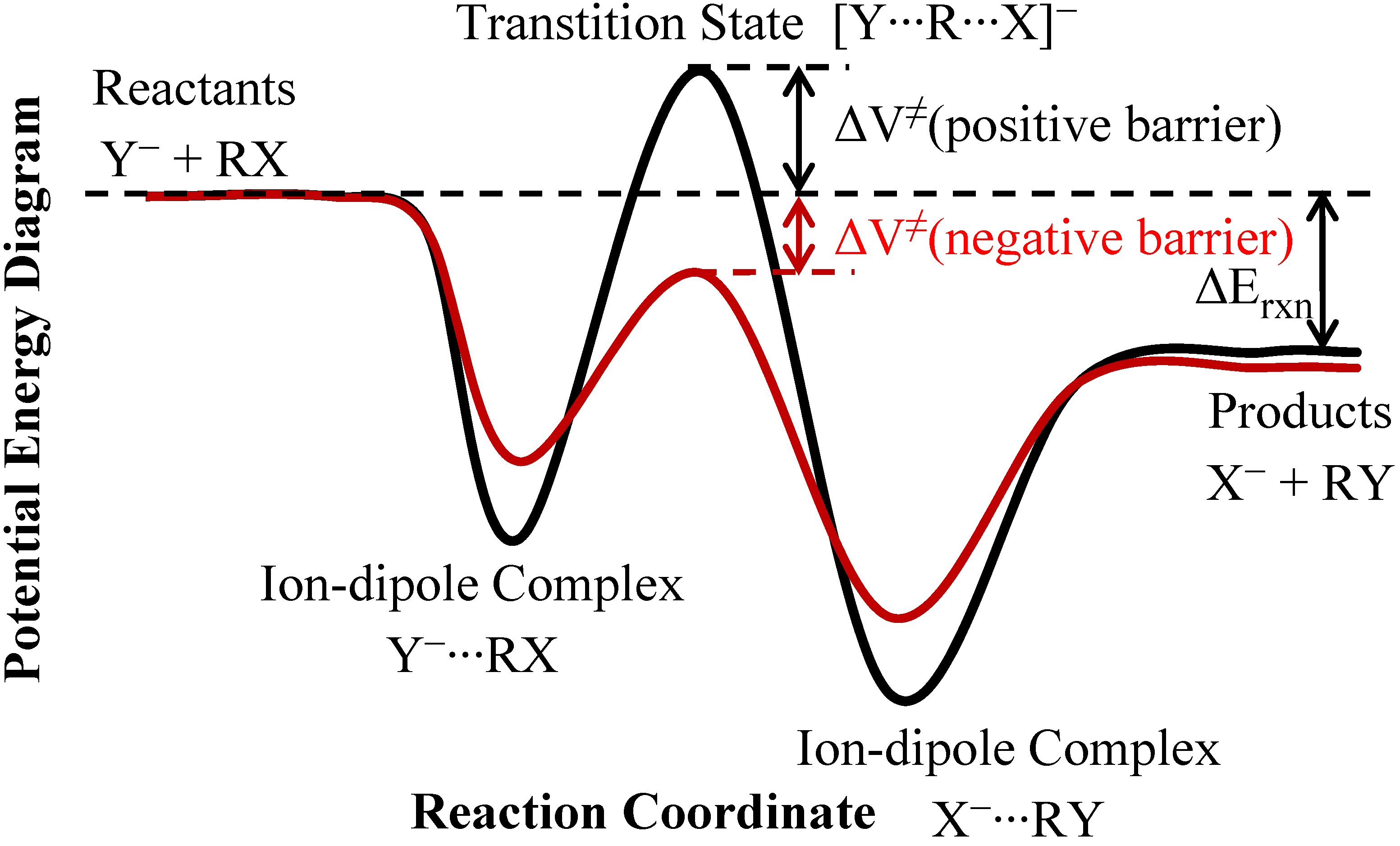
3.1. Systems with Very Low Barriers (Collision-Controlled Reactions)

| kcap | kexpt | Efficiency | KIEcap | KIEexpt | |
|---|---|---|---|---|---|
| F− + CH3Br a | 2.97(−09) c | 1.88(−09) | 0.631 | 1.003 | 0.98 ± 0.02 |
| F− + CH3I a | 2.79(−09) | 1.94(−09) | 0.695 | 1.001 | 0.98 ± 0.05 |
| CF3CH2O− + CH3CH2Br b | 1.87(−09) | 1.24(−09) | 0.665 | 1.011 | 1.10 ± 0.06 |
| CF3CH2O− + (CH3)2CHBr b | 1.98(−09) | 1.39(−09) | 0.703 | 1.012 | 1.20 ± 0.05 |
| H2NS− + CH3Br b | 2.10(−09) | 7.04(−10) | 0.336 | 1.005 | 1.04 ± 0.03 |
| H2NS− + CH3CH2Br b | 2.33(−09) | 9.05(−10) | 0.389 | 1.007 | 1.00 ± 0.04 |
| CF3CF2CH2O− + CH3Br b | 1.56(−09) | 8.48(−10) | 0.545 | 1.010 | 0.99 ± 0.04 |
| CF3CF2CH2O− + CH3CH2Br b | 1.69(−09) | 9.86(−10) | 0.582 | 1.013 | 1.16 ± 0.06 |
3.2. Systems with Low Barriers (both Collision- and TS-Controlled Reactions)
| F−(H2O) + CH3Br/F−(H2O) + CD3Br a | ||||||
|---|---|---|---|---|---|---|
| ∆V≠ | ∆Erxn | kTST | kCUS | KIETST | KIECUS | |
| M06-2X/6-31+G(d,p) | −7.8 | −28.2 | 4.11(−09) b | 1.47(−09) | 0.795 | 0.929 |
| M06-2X/6-311+G(d,p) | −8.6 | −30.4 | 2.87(−08) | 2.12(−09) | 0.628 | 0.977 |
| M06-2X/aug-cc-pVDZ | −8.9 | −31.0 | 1.40(−08) | 1.97(−09) | 0.763 | 0.970 |
| B3LYP/6-31+G(d,p) | −9.7 | −23.8 | 1.39(−06) | 2.29(−09) | 0.832 | 1.004 |
| B3LYP/6-311+G(d,p) | −10.5 | −25.7 | 2.76(−05) | 2.29(−09) | 0.836 | 1.004 |
| MP2/6-31+G(d,p) | −5.8 | −23.6 | 7.38(−09) | 1.75(−09) | 0.868 | 0.972 |
| MP2/6-311+G(d,p) | −2.2 | −23.2 | 4.02(−11) | 3.95(−11) | 0.833 | 0.836 |
| MP2/aug-cc-pVDZ | −6.3 | −22.8 | 1.48(−09) | 8.97(−10) | 0.820 | 0.892 |
| MP2/aug-cc-pVTZ | −4.0 | −21.7 | 6.61(−11) | 6.43(−11) | 0.857 | 0.861 |
| CCSD(T)/aug-cc-pVTZ c | −6.6 | −24.0 | 2.52(−09) | 1.20(−09) | 0.820 | 0.917 |
3.3. Systems with Small Barriers (TS Controlled Reactions)
3.3.1. Benchmark of Electronic Structure Methods
| kcap | kexpt | Efficiency | KIEcap | KIEexpt | |
|---|---|---|---|---|---|
| ClO− + CH3Cl a | 2.37(−09) f | 2.01(−10) | 0.085 | 1.015 | 0.85 ± 0.01 |
| ClO− + CH3CH2Cl a | 2.46(−09) | 2.25(−10) | 0.091 | 1.016 | 0.99 ± 0.01 |
| BrO− + CH3Cl a | 2.08(−09) | 1.08(−10) | 0.052 | 1.019 | 0.82 ± 0.03 |
| BrO− + CH3CH2Cl a | 2.12(−09) | 1.07(−10) | 0.050 | 1.022 | 0.96 ± 0.03 |
| HS− + CH3CH2Br b | 2.67(−09) | 1.95(−10) | 0.073 | 1.005 | 1.02 ± 0.07 |
| Cl− + CH3I b | 2.15(−09) | 1.66(−10) | 0.077 | 1.002 | 0.84 ± 0.02 |
| Br− + CH3I b | 1.60(−09) | 2.89(−11) | 0.018 | 1.004 | 0.76 ± 0.03 |
| CN− + CH3I c | 2.44(−09) | 1.28(−10) | 0.052 | 1.002 | 0.84 ± 0.03 |
| CN− + CH3CH2I c | 2.81(−09) | 2.99(−11) | 0.011 | 1.002 | 0.89 ± 0.02 |
| Cl− + CH3Br d | 2.33(−09) | 2.37(−11) | 0.010 | 1.004 | 0.88 ± 0.45 |
| F−(H2O) + CH3Cl e | 2.59(−09) | 1.49(−11) | 0.006 | 1.012 | 0.85 ± 0.03 |
| MUE | SD | |
|---|---|---|
| M06-2X/6-31+G(d,p) | 0.153 | 0.231 |
| M06-2X/6-311+G(d,p) | 0.087 | 0.055 |
| M06-2X/aug-cc-pVDZ | 0.079 | 0.058 |
| B3LYP/6-31+G(d,p) | 0.129 | 0.070 |
| B3LYP/6-311+G(d,p) | 0.172 | 0.094 |
| B3LYP/aug-cc-pVDZ | 0.099 | 0.056 |
| MP2/6-31+G(d,p) | 0.068 | 0.038 |
| MP2/6-311+G(d,p) | 0.121 | 0.073 |
| MP2/aug-cc-pVDZ | 0.049 | 0.039 |
| MP2/aug-cc-pVTZ | 0.075 | 0.047 |
3.3.2. Factor Analysis of KIE
3.3.2.1. Translational Contribution
3.3.2.2. Rotational Contribution
3.3.2.3. Vibrational Contribution
 |  |  | KIETST a | KIEexpt b | |
|---|---|---|---|---|---|
| ClO− + CH3Cl | 1.045 | 1.617 | 0.527 | 0.890 | 0.85 ± 0.01 |
| ClO− + CH3CH2Cl | 1.050 | 1.310 | 0.717 | 0.987 | 0.99 ± 0.01 |
| BrO− + CH3Cl | 1.059 | 1.625 | 0.519 | 0.893 | 0.82 ± 0.03 |
| BrO− + CH3CH2Cl | 1.069 | 1.312 | 0.710 | 0.995 | 0.96 ± 0.03 |
| HS− + CH3CH2Br | 1.016 | 1.271 | 0.831 | 1.073 | 1.02 ± 0.07 |
| Cl− + CH3I | 1.006 | 1.231 | 0.717 | 0.889 | 0.84 ± 0.02 |
| Br− + CH3I | 1.011 | 1.241 | 0.731 | 0.918 | 0.76 ± 0.03 |
| CN− + CH3I | 1.005 | 1.229 | 0.714 | 0.881 | 0.84 ± 0.03 |
| CN− + CH3CH2I | 1.007 | 1.264 | 0.724 | 0.921 | 0.89 ± 0.02 |
| Cl− + CH3Br | 1.013 | 1.234 | 0.732 | 0.915 | 0.88 ± 0.45 |
| F−(H2O) + CH3Cl | 1.037 | 1.660 | 0.481 | 0.828 | 0.85 ± 0.03 |
| CN− + (CH3)2CHI | 1.008 | 1.170 | 0.787 | 0.928 | |
| CN− + (CH3)3CI | 1.009 | 1.129 | 0.767 | 0.874 |
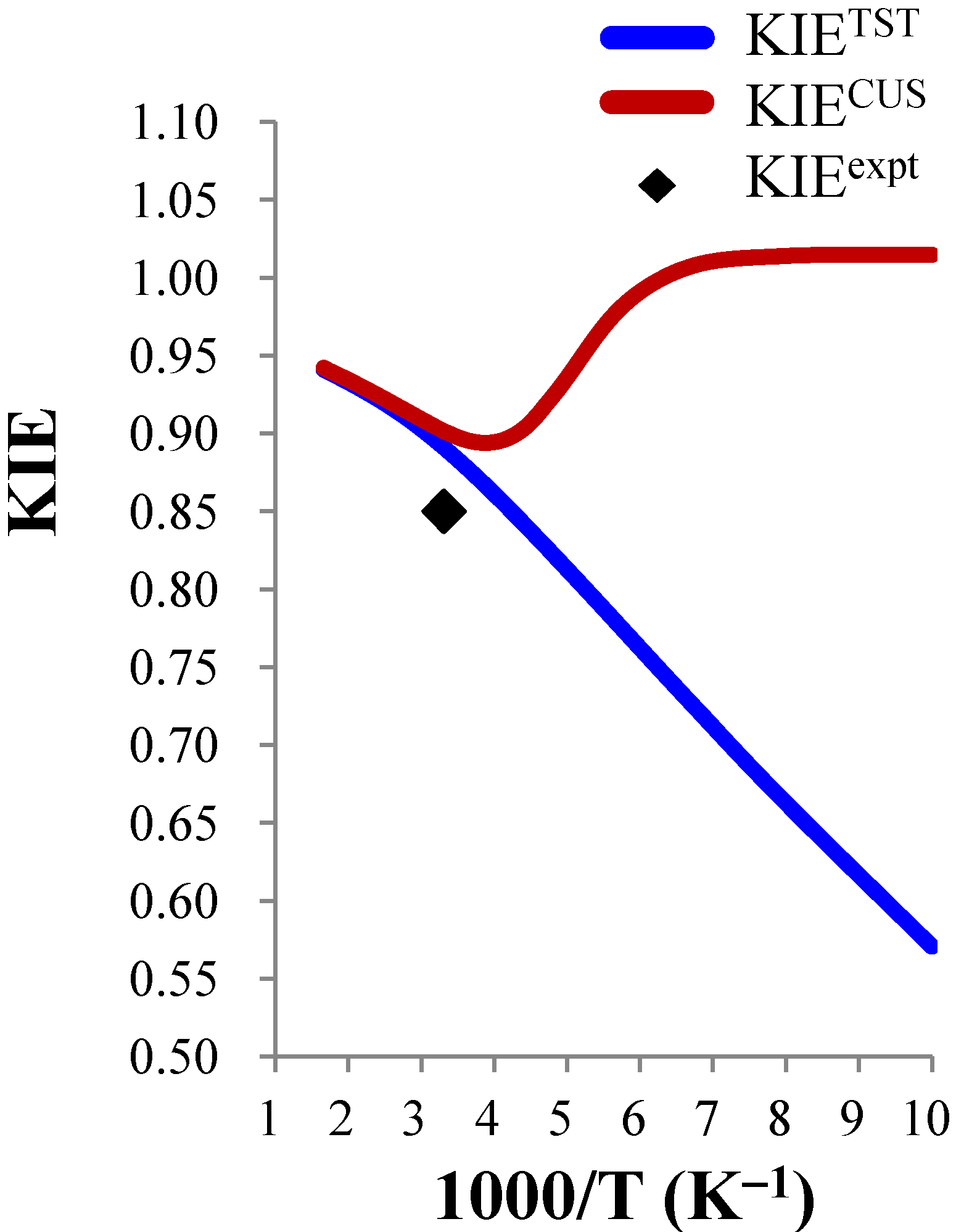
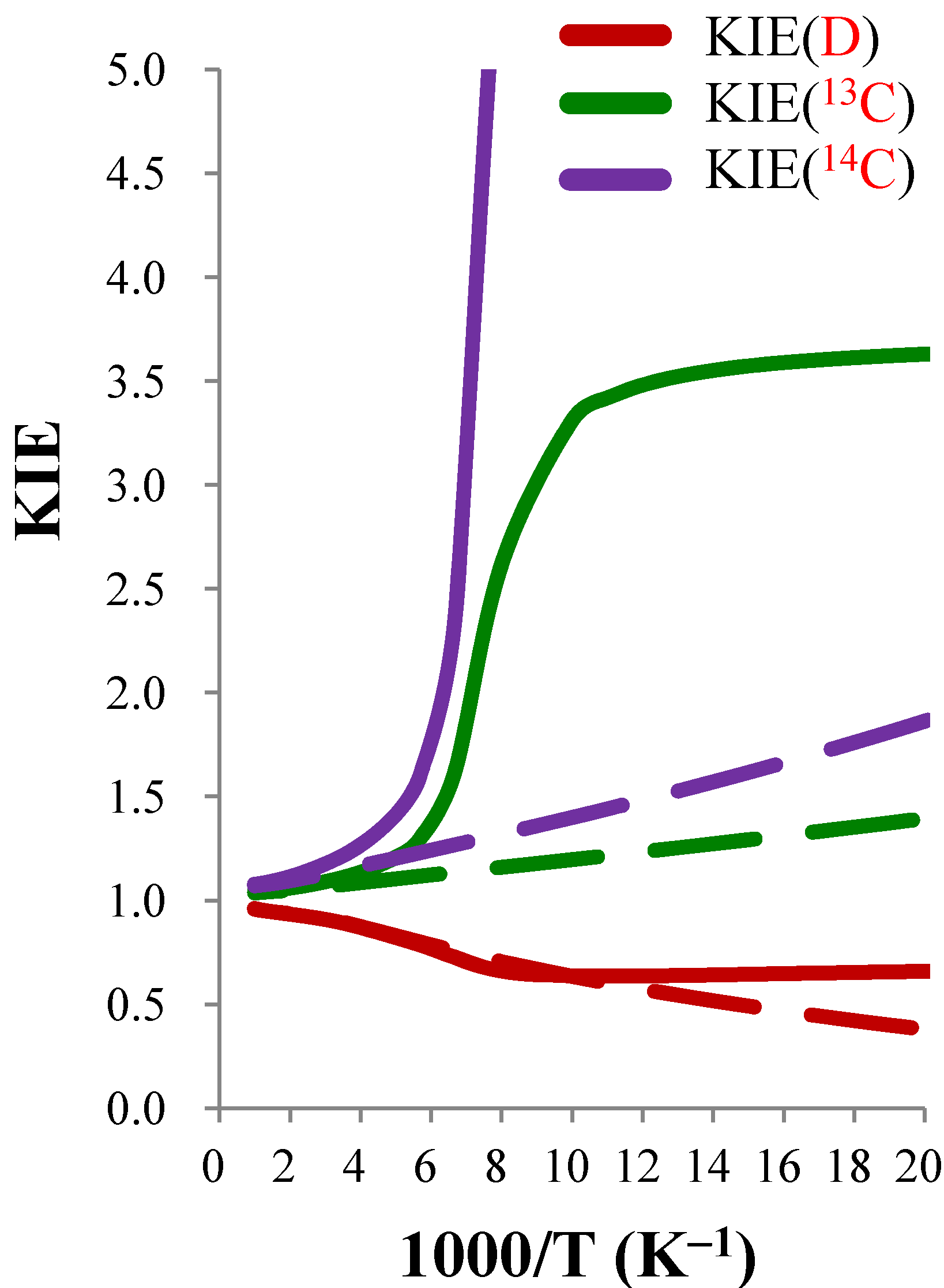
3.3.3. Temperature Dependence of the KIEs
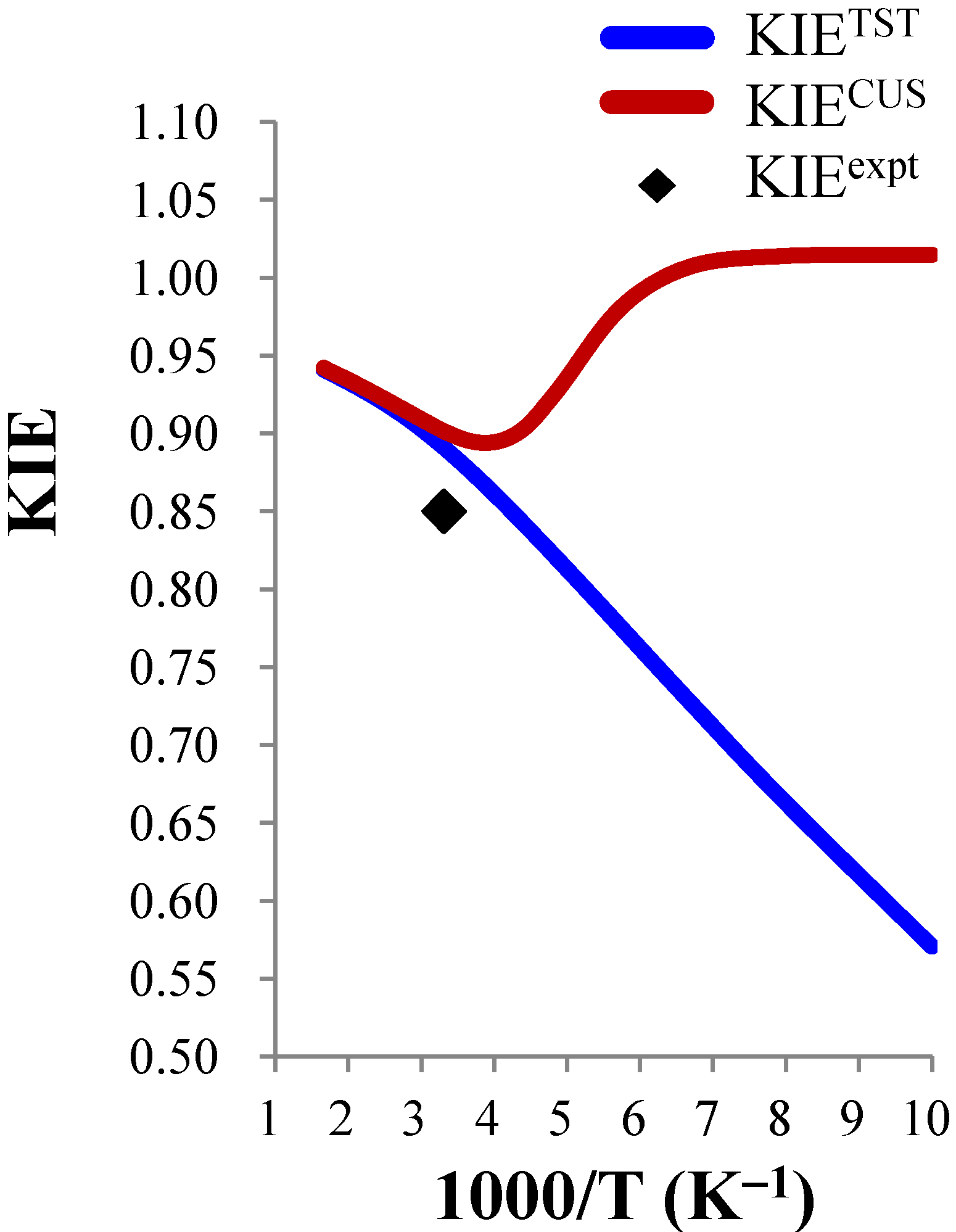
close to 1.00), the temperature dependence is expected to be small. For example, the ClO− + CH3CH2Cl and BrO− + CH3CH2Cl systems were predicted to show negligible temperature dependence of the deuterium KIE in the 300–600 K range. | ClO− + CH3Cl (0.85) b | ClO− + CH3CH2Cl (0.99) b | BrO− + CH3CH2Cl (0.96) b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T(K) | | KIETST | KIECUS | | KIETST | KIECUS | | KIETST | KIECUS |
| 100 | 0.338 | 0.571 | 1.015 | 0.636 | 0.875 | 1.016 | 0.639 | 0.896 | 1.022 |
| 200 | 0.481 | 0.813 | 0.936 | 0.703 | 0.966 | 1.007 | 0.698 | 0.978 | 1.005 |
| 300 | 0.527 | 0.890 | 0.901 | 0.717 | 0.987 | 0.990 | 0.710 | 0.995 | 0.996 |
| 400 | 0.544 | 0.919 | 0.922 | 0.719 | 0.989 | 0.990 | 0.710 | 0.995 | 0.995 |
| 500 | 0.552 | 0.933 | 0.934 | 0.718 | 0.988 | 0.988 | 0.708 | 0.992 | 0.992 |
| 600 | 0.557 | 0.941 | 0.942 | 0.718 | 0.987 | 0.987 | 0.707 | 0.990 | 0.991 |
| Cl− + CH3Br (0.88) b | F−(H2O) + CH3Cl (0.85) b | HS− + CH3CH2Br (1.02) b | |||||||
| T(K) | | KIETST | KIECUS | | KIETST | KIECUS | | KIETST | KIECUS |
| 100 | 0.553 | 0.691 | 0.762 | 0.273 | 0.470 | 0.642 | 0.957 | 1.235 | 1.005 |
| 200 | 0.695 | 0.868 | 0.871 | 0.425 | 0.732 | 0.735 | 0.868 | 1.122 | 1.066 |
| 300 | 0.732 | 0.915 | 0.916 | 0.481 | 0.828 | 0.829 | 0.831 | 1.073 | 1.068 |
| 400 | 0.742 | 0.928 | 0.929 | 0.505 | 0.870 | 0.871 | 0.807 | 1.042 | 1.041 |
| 500 | 0.746 | 0.933 | 0.934 | 0.518 | 0.892 | 0.893 | 0.791 | 1.022 | 1.022 |
| 600 | 0.749 | 0.937 | 0.937 | 0.527 | 0.907 | 0.908 | 0.782 | 1.010 | 1.010 |
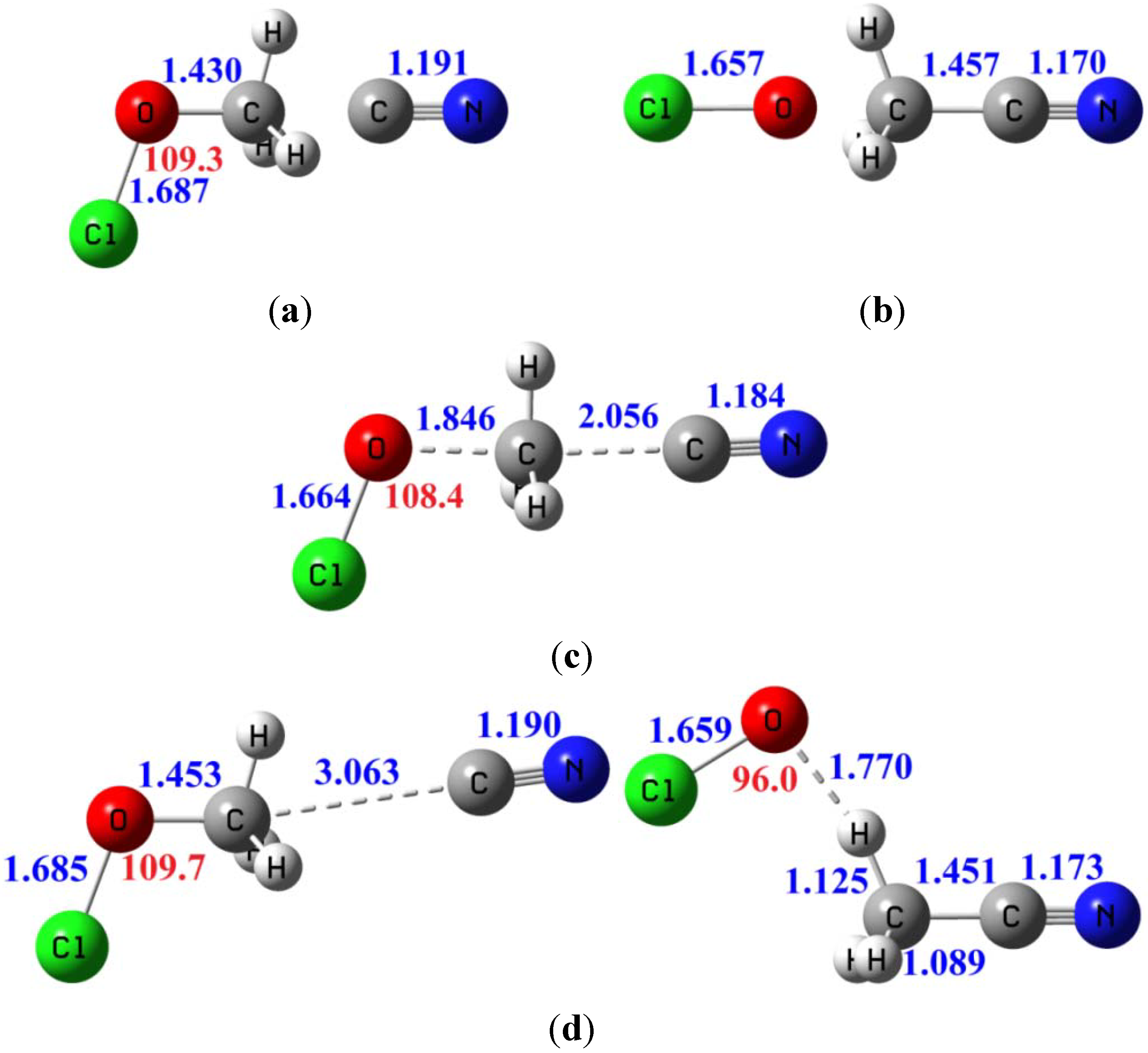
3.3.4. Tunneling Effects on Rate Constants and KIEs
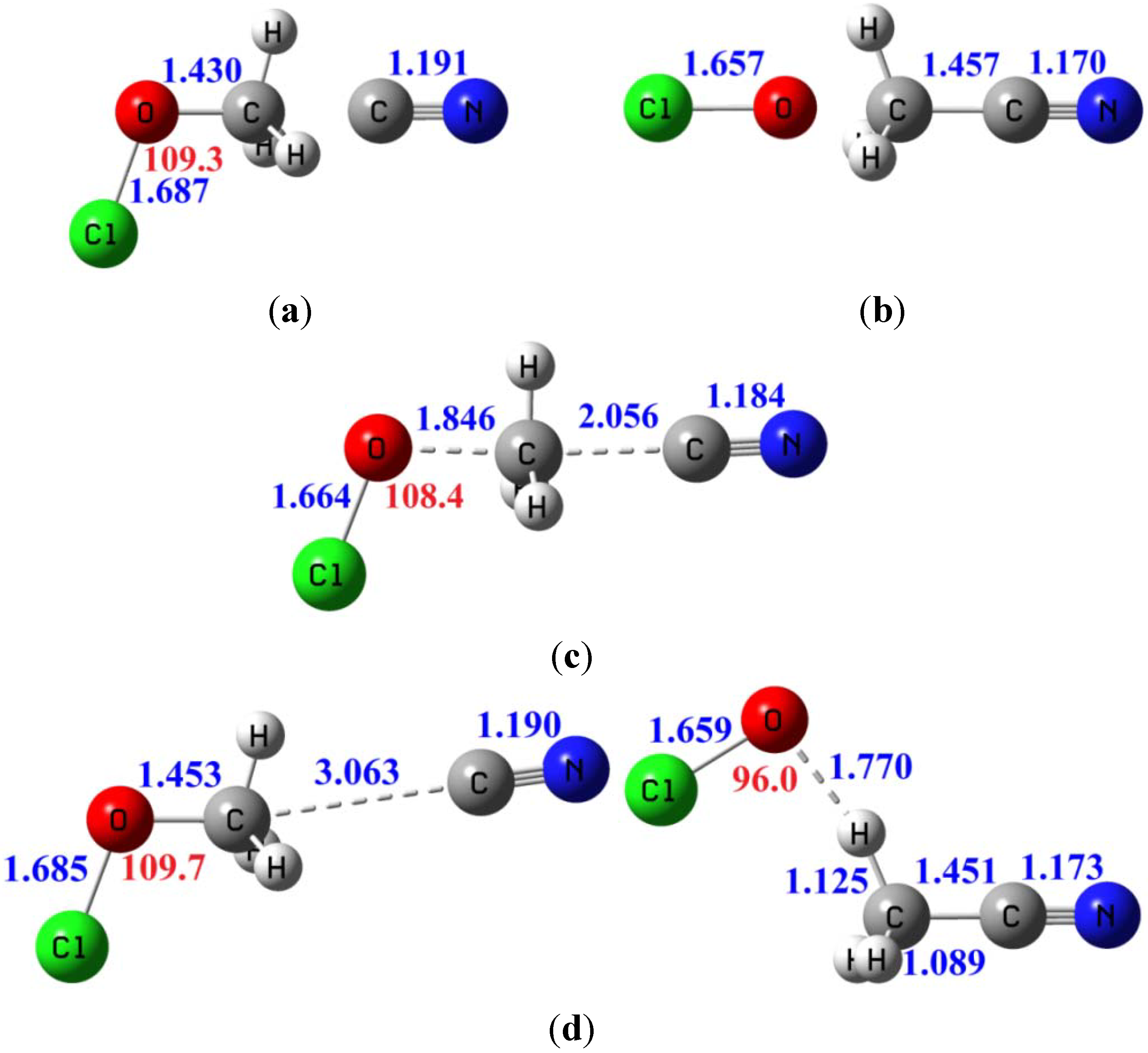
| Erxn b | ion-dipole complex CN−...CH3OCl | ion-dipole complex OCl−...CH3CN | Barrier height | |
|---|---|---|---|---|
| B3LYP/6-311+G(d,p) | −11.4 | −9.8 | −29.6 | 8.8 |
| MP2/aug-cc-pVDZ | −10.4 | −10.0 | −29.9 | 9.8 |
| MP2/aug-cc-pVTZ | −11.9 | −9.8 | −31.0 | 10.1 |
| CCSD(T)/aug-cc-pVTZ c | −9.9 | −9.8 | −28.7 | 9.8 |
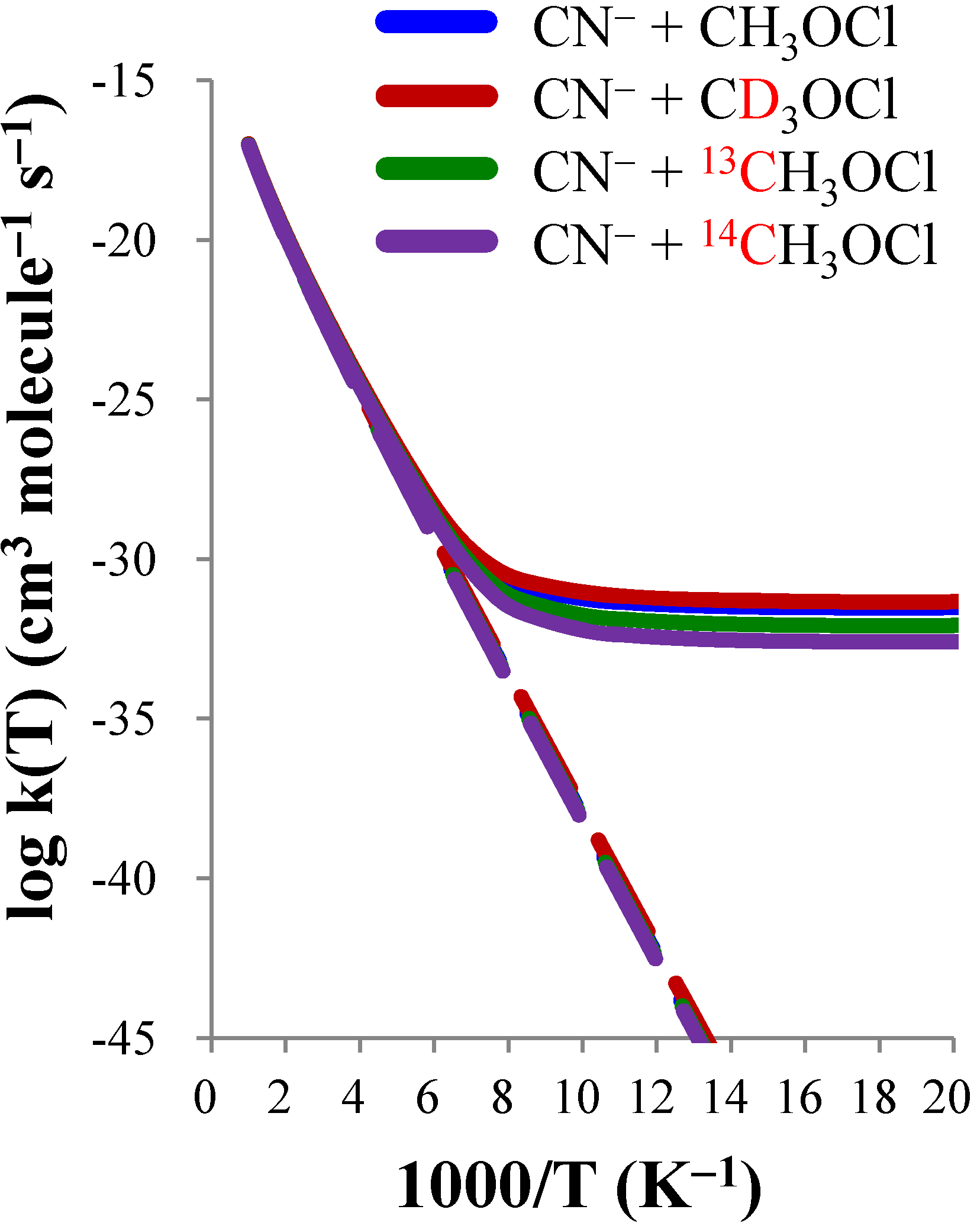
| CN− + CH3OCl | CN− + CD3OCl | CN− + 13CH3OCl | CN− + 14CH3OCl | |||||
|---|---|---|---|---|---|---|---|---|
| T(K) | TST | CVT/SCT | TST | CVT/SCT | TST | CVT/SCT | TST | CVT/SCT |
| 50 | 2.16(−60) a | 3.01(−32) | 5.70(−60) | 4.57(−32) | 1.55(−60) | 8.30(−33) | 1.16(−60) | 2.54(−33) |
| 100 | 8.22(−39) | 5.82(−32) | 1.30(−38) | 9.14(−32) | 6.87(−39) | 1.76(−32) | 5.88(−39) | 5.94(−33) |
| 150 | 1.64(−31) | 3.32(−30) | 2.16(−31) | 4.56(−30) | 1.44(−31) | 2.06(−30) | 1.29(−31) | 1.42(−30) |
| 200 | 8.72(−28) | 2.79(−27) | 1.05(−27) | 3.39(−27) | 7.90(−28) | 2.31(−27) | 7.25(−28) | 1.97(−27) |
| 250 | 1.70(−25) | 3.30(−25) | 1.94(−25) | 3.80(−25) | 1.56(−25) | 2.91(−25) | 1.46(−25) | 2.61(−25) |
| 300 | 6.23(−24) | 9.65(−24) | 6.92(−24) | 1.07(−23) | 5.81(−24) | 8.74(−24) | 5.47(−24) | 8.03(−24) |
| 400 | 6.69(−22) | 8.40(−22) | 7.22(−22) | 9.09(−22) | 6.33(−22) | 7.83(−22) | 6.02(−22) | 7.36(−22) |
| 500 | 1.28(−20) | 1.47(−20) | 1.37(−20) | 1.57(−20) | 1.22(−20) | 1.39(−20) | 1.17(−20) | 1.32(−20) |
| 600 | 1.02(−19) | 1.11(−19) | 1.08(−19) | 1.18(−19) | 9.73(−20) | 1.05(−19) | 9.36(−20) | 1.01(−19) |
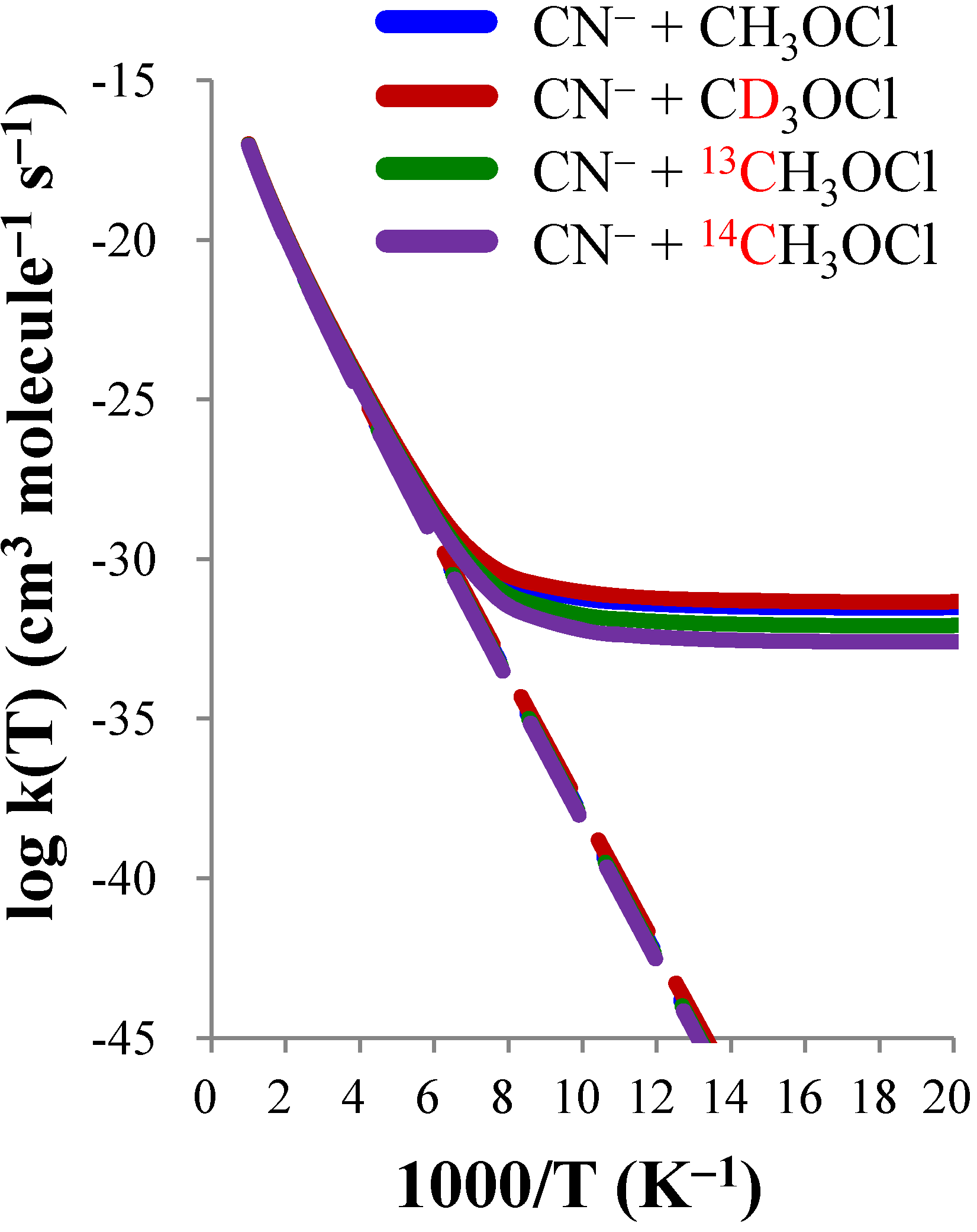
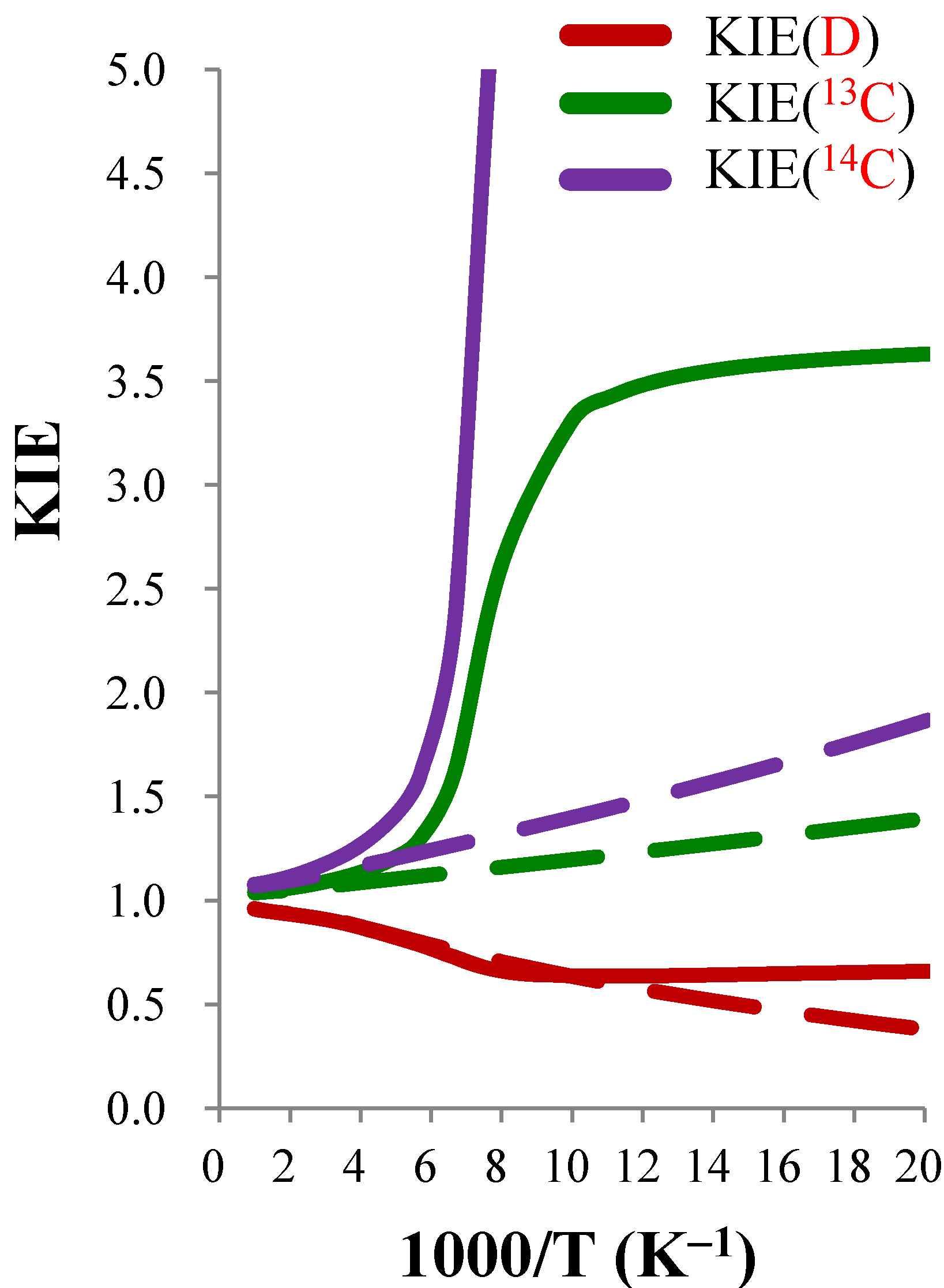
| KIE(D) | KIE(13C) | KIE(14C) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T(K) | TST |  | CVT/SCT | TST | | CVT/SCT | TST | | CVT/SCT |
| 50 | 0.380 | 1.734 | 0.659 | 1.394 | 2.604 | 3.629 | 1.863 | 6.356 | 11.842 |
| 100 | 0.635 | 1.003 | 0.637 | 1.196 | 2.768 | 3.310 | 1.399 | 7.003 | 9.797 |
| 150 | 0.758 | 0.959 | 0.727 | 1.134 | 1.422 | 1.613 | 1.267 | 1.848 | 2.342 |
| 200 | 0.831 | 0.990 | 0.822 | 1.103 | 1.095 | 1.208 | 1.203 | 1.175 | 1.413 |
| 250 | 0.874 | 0.995 | 0.870 | 1.084 | 1.047 | 1.136 | 1.165 | 1.087 | 1.266 |
| 300 | 0.901 | 0.996 | 0.897 | 1.072 | 1.030 | 1.104 | 1.140 | 1.054 | 1.201 |
| 400 | 0.926 | 0.997 | 0.924 | 1.057 | 1.016 | 1.073 | 1.110 | 1.028 | 1.142 |
| 500 | 0.937 | 0.997 | 0.934 | 1.049 | 1.010 | 1.059 | 1.094 | 1.018 | 1.114 |
| 600 | 0.943 | 0.997 | 0.940 | 1.044 | 1.007 | 1.051 | 1.085 | 1.013 | 1.099 |
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Wladkiwski, B.D.; Wilbur, J.L.; Brauman, J.I. Intrinsic structure-reactivity relationships in gas-phase SN2 reactions: Identity exchange of substituted benzyl chlorides with chloride ion. J. Am. Chem. Soc. 1994, 116, 2471–2480. [Google Scholar] [CrossRef]
- Meng, Q.; Gogoll, A.; Thibblin, A. Concerted and stepwise solvolytic elimination and substitution reactions: Stereochemistry and substituent effects. J. Am. Chem. Soc. 1997, 119, 1217–1223. [Google Scholar] [CrossRef]
- Gronert, S.; Fagin, A.E.; Wong, L. Direct measurements of deuterium kinetic isotope effects in anionic, gas-phase substitution and elimination reactions. J. Am. Chem. Soc. 2007, 129, 5330–5331. [Google Scholar] [CrossRef]
- Poirier, R.A.; Wang, Y.; Westaway, K.C. A theoretical study of the relationship between Secondary α-Deuterium kinetic isotope effects and the structure of SN2 transition states. J. Am. Chem. Soc. 1994, 116, 2526–2533. [Google Scholar] [CrossRef]
- Graul, S.T.; Bowers, M.T. Vibrational excitation in products of nucleophilic substitution: The dissociation of metastable X− (CH3Y) in the gas phase. J. Am. Chem. Soc. 1994, 116, 3875–3883. [Google Scholar] [CrossRef]
- Cyr, D.M.; Scarton, M.G.; Wiberg, K.B.; Johnson, M.A.; Nonose, S.; Hirokawa, J.; Tanaka, H.; Kondow, T.; Morris, R.A.; Viggiano, A.A. Observation of the XY− abstraction products in the ion-molecule reactions X− + RY → XY− + R: An alternative to the SN2 mechanism at suprathermal collision energies. J. Am. Chem. Soc. 1995, 117, 1828–1832. [Google Scholar] [CrossRef]
- Laerdahl, J.K.; Uggerud, E. Gas phase nucleophilic substitution. Int. J. Mass Spectrom. 2002, 214, 277–314. [Google Scholar] [CrossRef]
- Gronert, S.; Depuy, C.H.; Bierbaum, V.M. Deuterium isotope effects in gas-phase reactions of alkyl halides: Distinguishing E2 and SN2 pathways. J. Am. Chem. Soc. 1991, 113, 4009–4010. [Google Scholar] [CrossRef]
- Almerindo, G.I.; Pliego, R., Jr. Ab Initio Study of the SN2 and E2 Mechanisms in the reaction between the cyanide ion and ethyl chloride in dimethyl sulfoxide solution. Org. Lett. 2005, 7, 1821–1823. [Google Scholar] [CrossRef]
- Bento, A.P.; Solà, M.; Bickelhaupt, F.M. E2 and SN2 Reactions of X− + CH3CH2X (X = F, Cl); an ab Initio and DFT Benchmark Study. J. Chem. Theory Comput. 2008, 4, 929–940. [Google Scholar] [CrossRef]
- Matsson, O.; Dybala-Defratyka, A.; Rostkowski, M.; Paneth, P.; Westaway, K.C. A theoretical investigation of α-Carbon kinetic isotope effects and their relationship to the transition-state structure of SN2 reactions. J. Org. Chem. 2005, 70, 4022–4027. [Google Scholar] [CrossRef]
- Fang, Y.-R.; MacMillar, S.; Eriksson, J.; Kołodziejska-Huben, M.; Dybała-Defratyka, A.; Paneth, P.; Matsson, O.; Westaway, K.C. The effect of solvent on the structure of the transition state for the SN2 reaction between cyanide ion and ethyl chloride in DMSO and THF probed with six different kinetic isotope effects. J. Org. Chem. 2006, 71, 4742–4747. [Google Scholar] [CrossRef]
- Hu, W.-P.; Truhlar, D.G. Factors affecting competitive ion-molecule reactions: ClO− + C2H5Cl and C2D5Cl via E2 and Sn2channels. J. Am. Chem. Soc. 1996, 118, 860–869. [Google Scholar] [CrossRef]
- Villano, S.M.; Kato, S.; Bierbaum, V.M. Deuterium kinetic isotope effects in gas-phase SN2 and E2 reactions: Comparison of experiment and theory. J. Am. Chem. Soc. 2006, 128, 736–737. [Google Scholar] [CrossRef]
- Villano, S.M.; Eyet, N.; Lineberger, W.C.; Bierbaum, V.M. Reactions of α-nucleophiles with alkyl chlorides: Competition between SN2 and E2 mechanisms and the gas-phase α-effect. J. Am. Chem. Soc. 2009, 131, 8227–8233. [Google Scholar] [CrossRef]
- Garver, J.M.; Fang, Y.-R.; Eyet, N.; Villano, S.M.; Bierbaum, V.M.; Westaway, K.C. A Direct comparison of reactivity and mechanism in the gas phase and in solution. J. Am. Chem. Soc. 2010, 132, 3808–3814. [Google Scholar]
- Zhao, X.G.; Tucker, S.C.; Truhlar, D.G. Solvent and secondary kinetic isotope effects for the microhydrated SN2 reaction of C1−(H2O)n with CH3Cl. J. Am. Chem. Soc. 1991, 113, 826–832. [Google Scholar] [CrossRef]
- Viggiano, A.A.; Morris, R.A.; Paschkewitz, J.S.; Paulson, J. Kinetics of the gas-phase reactions of C1– with CH3Br and CD3Br: Experimental evidence for nonstatistical behavior? J. Am. Chem. Soc. 1992, 114, 10477–10482. [Google Scholar] [CrossRef]
- Boyd, R.J.; Kim, C.K.; Shi, Z.; Weinberg, N.; Wolfe, S. Secondary H/D Isotope effects and transition state looseness in nonidentity methyl transfer reactions. Implications for the concept of enzymatic catalysis via transition state compression. J. Am. Chem. Soc. 1993, 115, 10147–10152. [Google Scholar] [CrossRef]
- O’Hair, R.A.J.; Dang, T.T.; DePuy, C.H.; Bierbaum, V.M. Measurements of solvent and secondary kinetic isotope effects for the gas-phase SN2 reactions of fluoride with methyl halides. J. Am. Chem. Soc. 1994, 116, 3609–3610. [Google Scholar] [CrossRef]
- Hu, W.-P.; Truhlar, D.G. Modeling transition state solvation at the single-molecule level: Test of correlated ab Initio predictions against experiment for the gas-phase SN2 reaction of microhydrated fluoride with methyl chloride. J. Am. Chem. Soc. 1994, 116, 7797–7800. [Google Scholar] [CrossRef]
- Hu, W.-P.; Truhlar, D.G. Deuterium kinetic isotope effects and their temperature dependence in the gas-phase SN2 reactions X– + CH3Y → CH3X + Y– (X, Y = C1, Br, I). J. Am. Chem. Soc. 1995, 117, 10726–10734. [Google Scholar] [CrossRef]
- Viggiano, A.A.; Arnold, S.T.; Morris, R.A.; Ahrens, A.F.; Hierl, P.M. Temperature dependences of the rate constants and branching ratios for the reactions of OH−(H2O)0–4 + CH3Br. J. Phys. Chem. 1996, 100, 14397–14402. [Google Scholar] [CrossRef]
- Davico, G.E. Interpretation of the gas-phase solvent deuterium kinetic isotope effects in the SN2 reaction mechanism: Comparison of theoretical and experimental results in the reaction of microsolvated fluoride ions with methyl halides. J. Phys. Chem. A 2006, 110, 13112–13121. [Google Scholar] [CrossRef]
- Chen, J.-L.; Hu, W.-P. Theoretical study on the gas-phase SN2 reaction of microhydrated fluoride with methyl fluoride. J. Chin. Chem. Soc. 2012, 59, 1401–1408. [Google Scholar] [CrossRef]
- Wu, Y.-R.; Hu, W.-P. Reaction dynamics study on the tunneling effects of a microsolvated E2 reaction: FO−(H2O) + C2H5Cl → HOF(H2O) + C2H4 + Cl−. J. Am. Chem. Soc. 1999, 121, 10168–10177. [Google Scholar] [CrossRef]
- Eyet, N.; Villano, S.M.; Kato, S.; Bierbaum, V.M. Deuterium kinetic isotope effects in microsolvated gas-phase E2 reactions. J. Am. Soc. Mass Spectrom. 2007, 18, 1046–1051. [Google Scholar] [CrossRef]
- Eyet, N.; Villano, S.M.; Bierbaum, V.M. Gas-Phase reactions of microsolvated fluoride ions: An investigation of different solvents. J. Phys. Chem. A 2013, 117, 1136–1143. [Google Scholar] [CrossRef]
- Pabis, A.; Paluch, P.; Szala, J.; Paneth, P. A DFT study of the kinetic isotope effects on the competing SN2 and E2 reactions between hypochlorite anion and ethyl chloride. J. Chem. Theory. Comput. 2009, 5, 33–36. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Laidler, K.J.; King, M.C. The development of transition-state theory. J. Phys. Chem. 1983, 87, 2657–2664. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Hase, W.L.; Hynes, J.T. Current status of transition-state theory. J. Phys. Chem. 1983, 87, 2664–2682. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Hirschfelder, J.O.; Wigner, E. Some quantum mechanical considerations in the theory of reactions involving an activation energy. J. Chem. Phys. 1939, 7, 616–628. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Isaacson, A.D.; Garrett, B.C. Generalized Transition State Theory. In Theory of Chemical Reaction Dynamics; Baer, M., Ed.; CRC Press: Boca Raton, FL, USA, 1985; Volume 4, pp. 65–137. [Google Scholar]
- Truhlar, D.G.; Garrett, B.C. Variational transition-state theory. Acc. Chem. Res. 1980, 13, 440–448. [Google Scholar] [CrossRef]
- Miller, W.H. Unified statistical model for “complex” and “direct” reaction mechanisms. J. Chem. Phys. 1976, 65, 2216–2223. [Google Scholar] [CrossRef]
- Garrett, B.C.; Truhlar, D.G. Canonical unified statistical model. Classical mechanical theory and applications to collinear reactions. J. Chem. Phys. 1982, 76, 1853–1858. [Google Scholar] [CrossRef]
- Chesnavich, W.J.; Su, T.; Bowers, M.T. Collisions in a noncentral field: A variational and trajectory investigation of ion-dipole capture. J. Chem. Phys. 1980, 72, 2641–2655. [Google Scholar] [CrossRef]
- Celli, F.; Weddle, G.; Ridge, D.P. On statistical and thermodynamic approaches to ion polar molecule collisions. J. Chem. Phys. 1980, 73, 801–812. [Google Scholar] [CrossRef]
- Garrett, B.C.; Truhlar, D.G.; Magnuson, A.W. New semiempirical method of modeling potential energy surfaces for generalized TST and application to the kinetic isotope effects in the Cl–H–H system. J. Chem. Phys. 1982, 76, 2321–2331. [Google Scholar] [CrossRef]
- Lu, D.-H.; Maurice, D.; Truhlar, D.G. What is the effect of variational optimization of the transition state on α-Deuterium secondary kinetic isotope effects? A prototype: CD3H + H
![Molecules 18 04816 i029]() CD3 + H2. J. Am. Chem. Soc. 1990, 112, 6206–6214. [Google Scholar] [CrossRef]
CD3 + H2. J. Am. Chem. Soc. 1990, 112, 6206–6214. [Google Scholar] [CrossRef] - Zhao, X.G.; Lu, D.-H.; Liu, Y.-P.; Lynch, G.C.; Truhlar, D.G. Use of an improved ion-solvent potential-energy function to calculate the reaction rate and α-deuterium and microsolvation kinetic isotope effects for the gas-phase SN2 reaction of Cl−(H2O) with CH3Cl. J. Chem. Phys. 1992, 97, 6369–6383. [Google Scholar] [CrossRef]
- Huang, C.-H.; Tsai, L.-C.; Hu, W.-P. Dual-Level direct dynamics study on the diels-alder reaction of ethylene and 1,3-Butadiene. J. Phys. Chem. A 2001, 105, 9945–9953. [Google Scholar] [CrossRef]
- Albu, T.V.; Lynch, B.J.; Truhlar, D.G.; Goren, A.C.; Hrovat, D.A.; Borden, W.T.; Moss, R.A. Dynamics of 1,2-Hydrogen migration in carbenes and ring expansion in cyclopropylcarbenes. J. Phys. Chem. A 2002, 106, 5323–5338. [Google Scholar] [CrossRef]
- Zuev, P.S.; Sheridan, R.S.; Albu, T.V.; Truhlar, D.G.; Hrovat, D.A.; Borden, W.T. Carbon tunneling from a single quantum state. Science 2003, 299, 867–870. [Google Scholar] [CrossRef]
- Datta, A.; Hrovat, D.A.; Borden, W.T. Calculations predict rapid tunneling by carbon from the vibrational ground state in the ring opening of cyclopropylcarbinyl radical at cryogenic temperatures. J. Am. Chem. Soc. 2008, 130, 6684–6685. [Google Scholar] [CrossRef]
- Gonzalez-James, O.M.; Zhang, X.; Datta, A.; Hrovat, D.A.; Borden, W.T.; Singleton, D.A. Experimental evidence for heavy-atom tunneling in the ring-opening of cyclopropylcarbinyl radical from intramolecular 12C/13C kinetic isotope effects. J. Am. Chem. Soc. 2010, 132, 12548–12549. [Google Scholar]
- Chen, J-.L.; Hu, W.-P. Theoretical Prediction on the Thermal Stability of Cyclic Ozone and Strong Oxygen Tunneling. J. Am. Chem. Soc. 2011, 133, 16045–16053. [Google Scholar] [CrossRef]
- Schreiner, P.R.; Reisenauer, H.P.; Ley, D.; Gerbig, D.; Wu, C.-H.; Allen, W.D. Methylhydroxycarbene: Tunneling control of a chemical reaction. Science 2011, 332, 1300–1303. [Google Scholar] [CrossRef]
- Wigner, E. On the quantum correction for thermodynamic equilibrium. Phys. Rev. 1932, 40, 749–459. [Google Scholar] [CrossRef]
- Wigner, E. Über das Überschreiten von Potentialschwellen bei chemischen Reaktionen. Z. Physik. Chem. 1932, B19, 203–216. [Google Scholar]
- Lu, D.-H.; Truong, T.N.; Melissas, V.S.; Lynch, G.C.; Liu, Y.-P.; Garrett, B.C.; Steckler, R.; Isaacson, A.D.; Rai, S.N.; Hancock, G.C.; et al. POLYRATE 4: A new version of a computer program for the calculation of chemical reaction rates for polyatomics. Comput. Phys. Commun. 1992, 71, 235–262. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Lynch, G.C.; Truong, T.N.; Lu, D.-H.; Truhlar, D.G.; Garrett, B.C. Molecular modeling of the kinetic isotope effect for the [1,5] sigmatropic rearrangement of cis-1,3-pentadiene. J. Am. Chem. Soc. 1993, 115, 2408–2415. [Google Scholar]
- Liu, Y.-P.; Lu, D.-H.; Gonzalez-Lafont, A.; Truhlar, D.G.; Garrett, B.C. Direct dynamics calculation of the kinetic isotope effect for an organic hydrogen-transfer reaction, including corner-cutting tunneling in 21 dimensions. J. Am. Chem. Soc. 1993, 115, 7806–7817. [Google Scholar] [CrossRef]
- Truong, T.N.; Lu, D.-H.; Lynch, G.C.; Liu, Y.-P.; Melissas, V.S.; Gonzalez-Lafont, A.; Rai, S.N.; Steckler, R.; Garrett, B.C.; Joseph, T.; et al. MORATE: A program for direct dynamics calculations of chemical reaction rates by semiempirical molecular orbital theory. Comput. Phys. Commun. 1993, 75, 143–159. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasrkhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Pross, A.; McGrath, M.P.; Radom, L. Extension of Gaussian-2 (G2) theory to bromine- and iodine-containing molecules: Use of effective core potentials. J. Chem. Phys. 1995, 103, 1878–1885. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Pople, J.A.; HeadGordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Miller, W.H.; Handy, N.C.; Adams, J.E. Reaction path Hamiltonian for polyatomic molecules. J. Chem. Phys. 1980, 72, 99–112. [Google Scholar] [CrossRef]
- Minichino, C.; Barone, V. From concepts to algorithms for the characterization of reaction mechanisms. H2CS as a case study. J. Chem. Phys. 1994, 100, 3717–3741. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 2005, 122, 1–10. [Google Scholar]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the spectroscopic and thermochemical properties of ClO, BrO, IO, and their anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef]
- Dunning, T.H.; Peterson, K.A.; Wilson, A.K. Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited. J. Chem. Phys. 2001, 114, 9244–9253. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- CRC Handbook of Chemistry and Physics, Internet Version 2007, 87th; Lide, D.R. (Ed.) Taylor and Francis: Boca Raton, FL, USA, 2007.
- Hu, W.-P.; Liu, Y.-P.; Truhlar, D.G. Variational transition-state theory and semiclassical tunneling calculations with interpolated corrections: A new approach to interfacing electronic structure theory and dynamics for organic reactions. J. Chem. Soc. Faraday Trans. 1994, 90, 1715–1725. [Google Scholar] [CrossRef]
- Corchado, J.C.; Espinosa-Garcia, J.; Hu, W.-P.; Rossi, I.; Truhlar, D.G. Dual-Level Reaction-Path Dynamics (the /// Approach to VTST with Semiclassical Tunneling). Application to OH + NH3 → H2O + NH2. J. Phys. Chem. 1995, 99, 687–694. [Google Scholar] [CrossRef]
- Page, M.; McIver, J.W. On evaluating the reaction path Hamiltonian. J. Chem. Phys. 1988, 88, 922–935. [Google Scholar] [CrossRef]
- Page, M.; Doubleday, C.; Mclver, J.M. Following steepest descent reaction paths. The use of higher energy derivatives with ab initio electronic structure methods. J. Chem. Phys. 1990, 93, 5634–5642. [Google Scholar] [CrossRef]
- Pulay, P.; Fogarasi, G. Geometry optimization in redundant internal coordinates. J. Chem. Phys. 1992, 96, 2856–2860. [Google Scholar] [CrossRef]
- Jackels, C.F.; Gu, Z.; Truhlar, D.G. Reaction-path potential and vibrational frequencies in terms of curvilinear internal coordinates. J. Chem. Phys. 1995, 102, 3188–3201. [Google Scholar] [CrossRef]
- Nguyen, K.A.; Jackels, C.F.; Truhlar, D.G. Reaction-path dynamics in curvilinear internal coordinates including torsions. J. Chem. Phys. 1996, 104, 6491–6496. [Google Scholar] [CrossRef]
- Chuang, Y.-Y.; Truhlar, D.G. Reaction-Path dynamics in redundant internal coordinates. J. Phys. Chem. A 1998, 102, 242–247. [Google Scholar] [CrossRef]
- Huang, C.-H.; You, R.-M.; Lian, P.-Y.; Hu, W.-P. Improved interpolated correction schemes for dual-level direct dynamics calculation. J. Phys. Chem. A 2000, 104, 7200–7208. [Google Scholar] [CrossRef]
- Corchado, J.C.; Chunag, Y.-Y.; Coitino, E.L.; Truhlar, D.G. Gaussrate, version 8.2, University of Minnesota: Minneapolis, MN, USA, 1999.
- Chuang, Y.-Y.; Corchado, J.C.; Fast, P.L.; Villa, J.; Hu, W.-P.; Liu, Y.-P.; Lynch, G.C.; Nguyen, K.A.; Jackels, C.F.; Gu, M.Z.; et al. Polyrate—Version 8.2, University of Minnesota: Minneapolis, MN, USA, 1999.
- Zhao, Y.; González-Garciá, N.; Truhlar, D.G. Benchmark database of barrier heights for heavy atom transfer, nucleophilic substitution, association, and unimolecular reactions and its use to test theoretical methods. J. Phys. Chem. A 2005, 109, 2012–2018. [Google Scholar] [CrossRef]
- Chen, J.-L.; Hong, J.-T.; Wu, K.-J.; Hu, W.-P. The MC-DFT approach to the M06-2X, B2K-PLYP, and B2T-PLYP functionals. Chem. Phys. Lett. 2009, 468, 307–312. [Google Scholar] [CrossRef]
- Sun, Y.-L.; Li, T.-H.; Chen, J.-L.; Hu, W.-P. Accurate multi-coefficient electronic structure methods MLSE(Cn)-DFT for thermochemical kinetics. Chem. Phys. Lett. 2009, 475, 141–145. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functional Calculations of E2 and SN2 Reactions: Effects of the Choice of Density Functional, Basis set, and Self-consistent Iterations. J. Chem. Theory. Comput. 2010, 6, 1104–1108. [Google Scholar] [CrossRef]
- Thornton, E.R. Physical organic chemistry. Annu. Rev. Phys. Chem. 1996, 17, 349–372. [Google Scholar] [CrossRef]
- Davico, G.E. Distinguishing the SN2 and SN2’ Mechanisms in the Gas Phase. Org. Lett. 1999, 1, 1675–1678. [Google Scholar] [CrossRef]
- Davico, G.E.; Bierbaum, V.M. Reactivity and secondary kinetic isotope effects in the SN2 reaction mechanism: Dioxygen radical anion and related nucleophilies. J. Am. Chem. Soc. 2000, 122, 1740–1748. [Google Scholar] [CrossRef]
- Meyer, M.P. New applications of isotope effects in the determination of organic reaction mechanism. Adv. Phys. Org. Chem. 2012, 46, 57–120. [Google Scholar] [CrossRef]
- Handy, N.C.; Lee, A.M. The adiabatic approximation. Chem. Phys. Lett. 1996, 252, 425–430. [Google Scholar] [CrossRef]
- Harding, M.E.; Vazquez, J.; Ruscic, B.; Wilson, A.K.; Gauss, J.; Stanton, J.F. High-accuracy extrapolated ab initio thermochemistry. III. Additional improvements and overview. J. Chem. Phys. 2008, 128, 114111–114115. [Google Scholar] [CrossRef]
- Mielke, S.L.; Schwenke, D.W.; Schatz, G.C.; Garrett, B.C.; Peterson, K.A. Functional representation for the born-oppenheimer diagonal correction and born-huang adiabatic potential energy surfaces for isotopomers of H3. J. Phys. Chem. A 2009, 113, 4479–4488. [Google Scholar] [CrossRef]
- Fleming, D.G.; Arseneau, D.J.; Sukhorukov, O.; Brewer, J.H.; Mielke, S.L.; Schatz, G.C.; Garrett, B.C.; Peterson, K.A.; Truhlar, D.G. Kinetic isotope effects for the reactions of muonic helium and muonium with H2. Science 2011, 331, 448–450. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsai, W.-C.; Hu, W.-P. Theoretical Analysis on the Kinetic Isotope Effects of Bimolecular Nucleophilic Substitution (SN2) Reactions and Their Temperature Dependence. Molecules 2013, 18, 4816-4843. https://doi.org/10.3390/molecules18044816
Tsai W-C, Hu W-P. Theoretical Analysis on the Kinetic Isotope Effects of Bimolecular Nucleophilic Substitution (SN2) Reactions and Their Temperature Dependence. Molecules. 2013; 18(4):4816-4843. https://doi.org/10.3390/molecules18044816
Chicago/Turabian StyleTsai, Wan-Chen, and Wei-Ping Hu. 2013. "Theoretical Analysis on the Kinetic Isotope Effects of Bimolecular Nucleophilic Substitution (SN2) Reactions and Their Temperature Dependence" Molecules 18, no. 4: 4816-4843. https://doi.org/10.3390/molecules18044816