Theoretical Study of H/D Isotope Effects on Nuclear Magnetic Shieldings Using an ab initio Multi-Component Molecular Orbital Method

Abstract
:1. Introduction
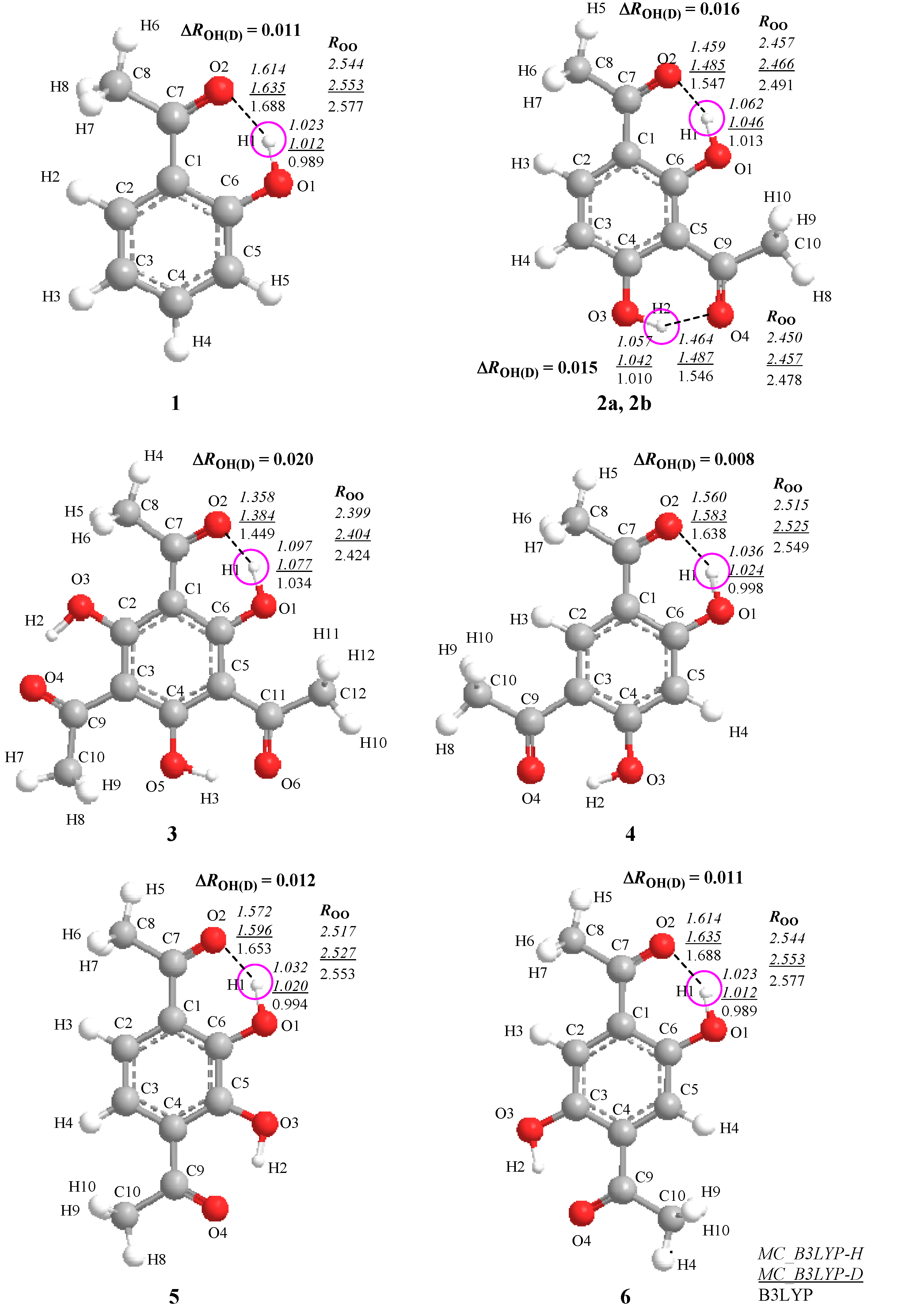
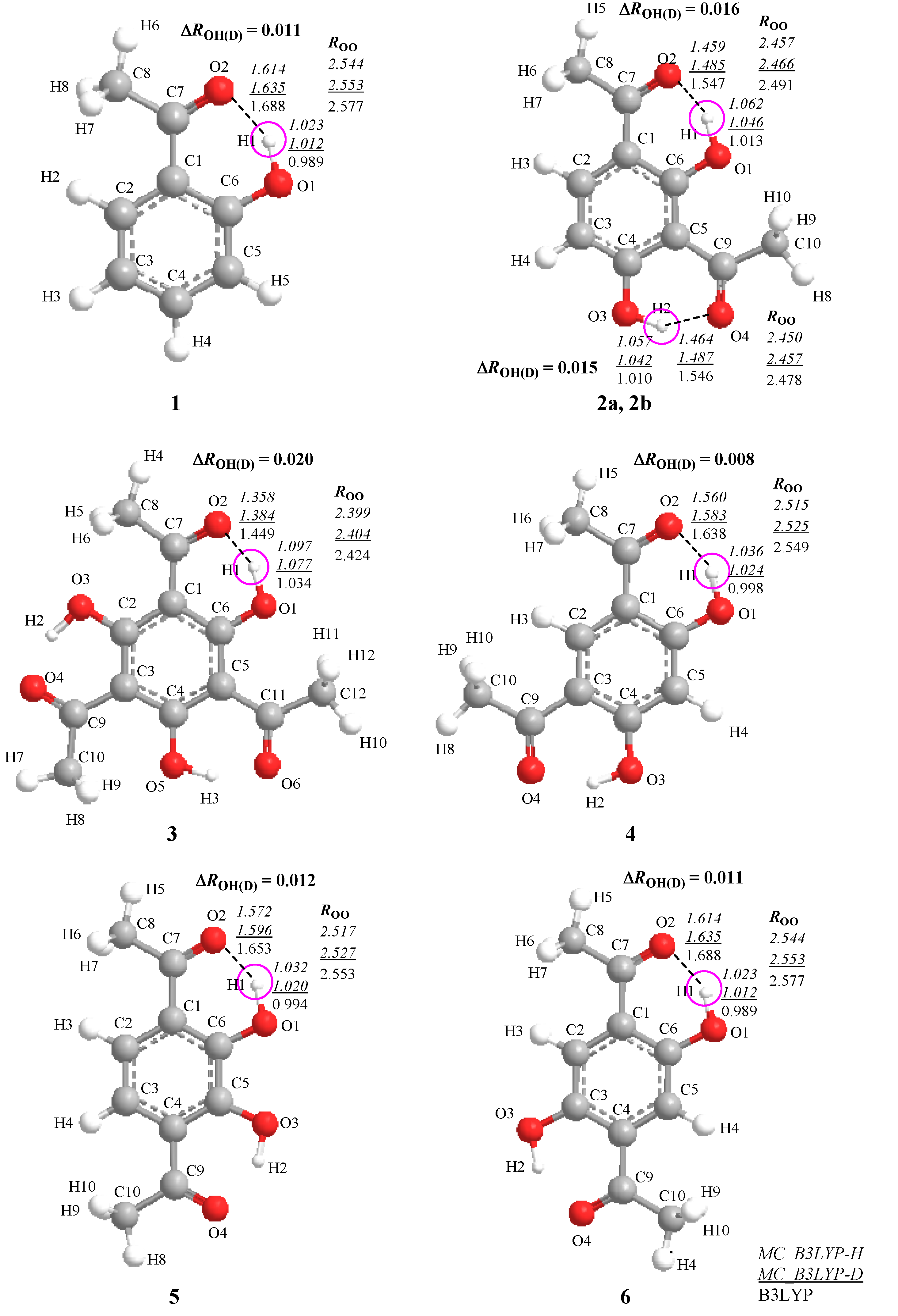
2. Results and Discussion
2.1. Geometrical Changes Induced by H/D Isotope Effect
 >
>  >
>  , where and are the covalent O–H and O–D bond lengths calculated by MC_B3LYP method, respectively, and is the equilibrium length with the conventional B3LYP method. This tendency is due to the direct inclusion of the anharmonicity of the potential by MC_DFT method. In addition, the contraction of O–H(D) bond, and the elongation of H(D)…O and O…O distances by deuterium substitution are clearly found. These changes can be interpreted by using the idea of Ubbelohde effect in hydrogen-bonded crystals [21]. Since these geometrical changes are induced by the difference between quantum nature of proton and deuteron, this geometrical H/D isotope effect is appeared in all the systems studied here.
, where and are the covalent O–H and O–D bond lengths calculated by MC_B3LYP method, respectively, and is the equilibrium length with the conventional B3LYP method. This tendency is due to the direct inclusion of the anharmonicity of the potential by MC_DFT method. In addition, the contraction of O–H(D) bond, and the elongation of H(D)…O and O…O distances by deuterium substitution are clearly found. These changes can be interpreted by using the idea of Ubbelohde effect in hydrogen-bonded crystals [21]. Since these geometrical changes are induced by the difference between quantum nature of proton and deuteron, this geometrical H/D isotope effect is appeared in all the systems studied here.
{kind=link}
{kind=link}
{kind=link}
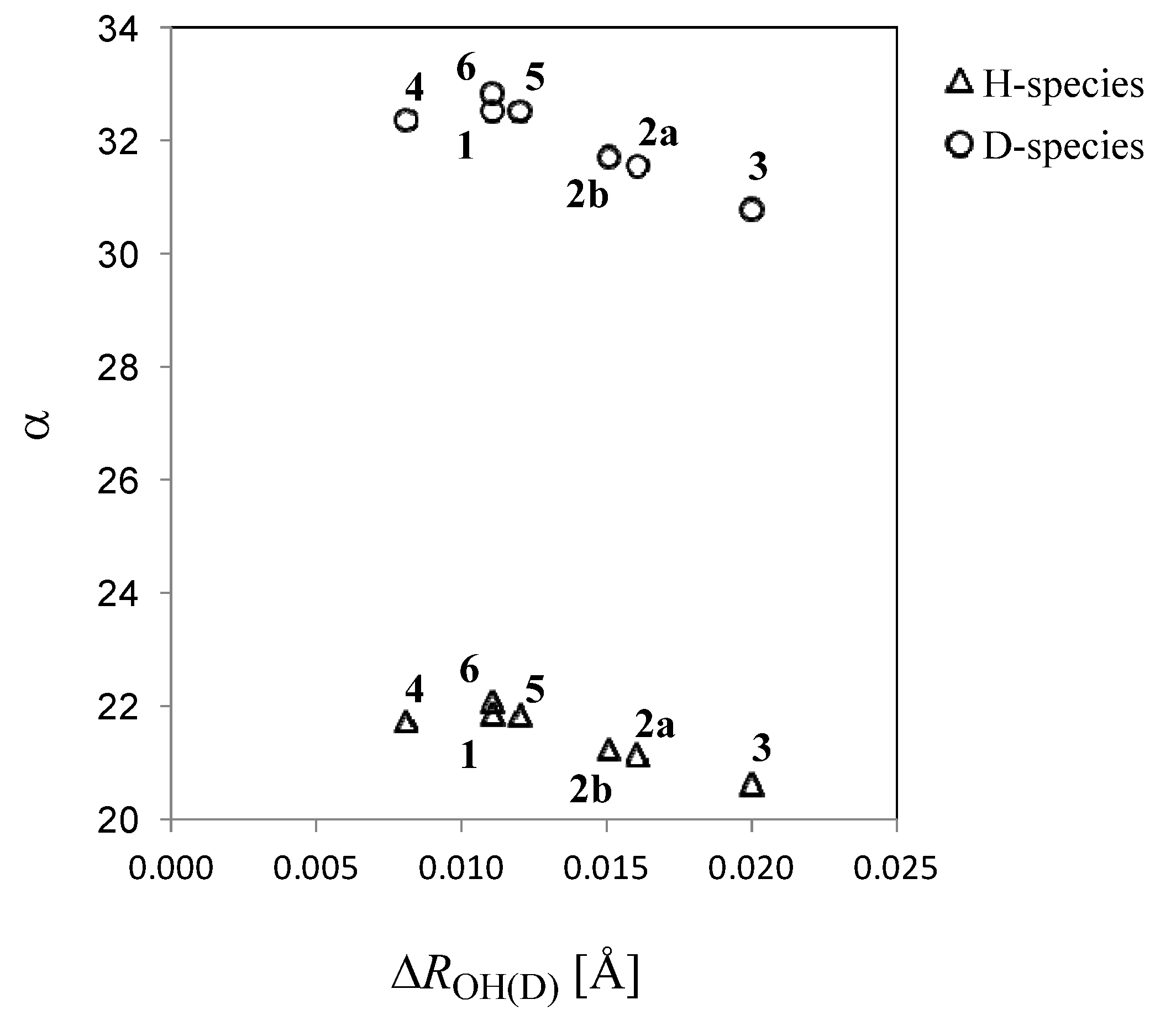
| 1 | 2a | 2b | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|
| H | 21.88 | 21.15 | 21.27 | 20.62 | 21.75 | 21.85 | 22.11 |
| D | 32.56 | 31.57 | 31.71 | 30.79 | 32.39 | 32.54 | 32.87 |
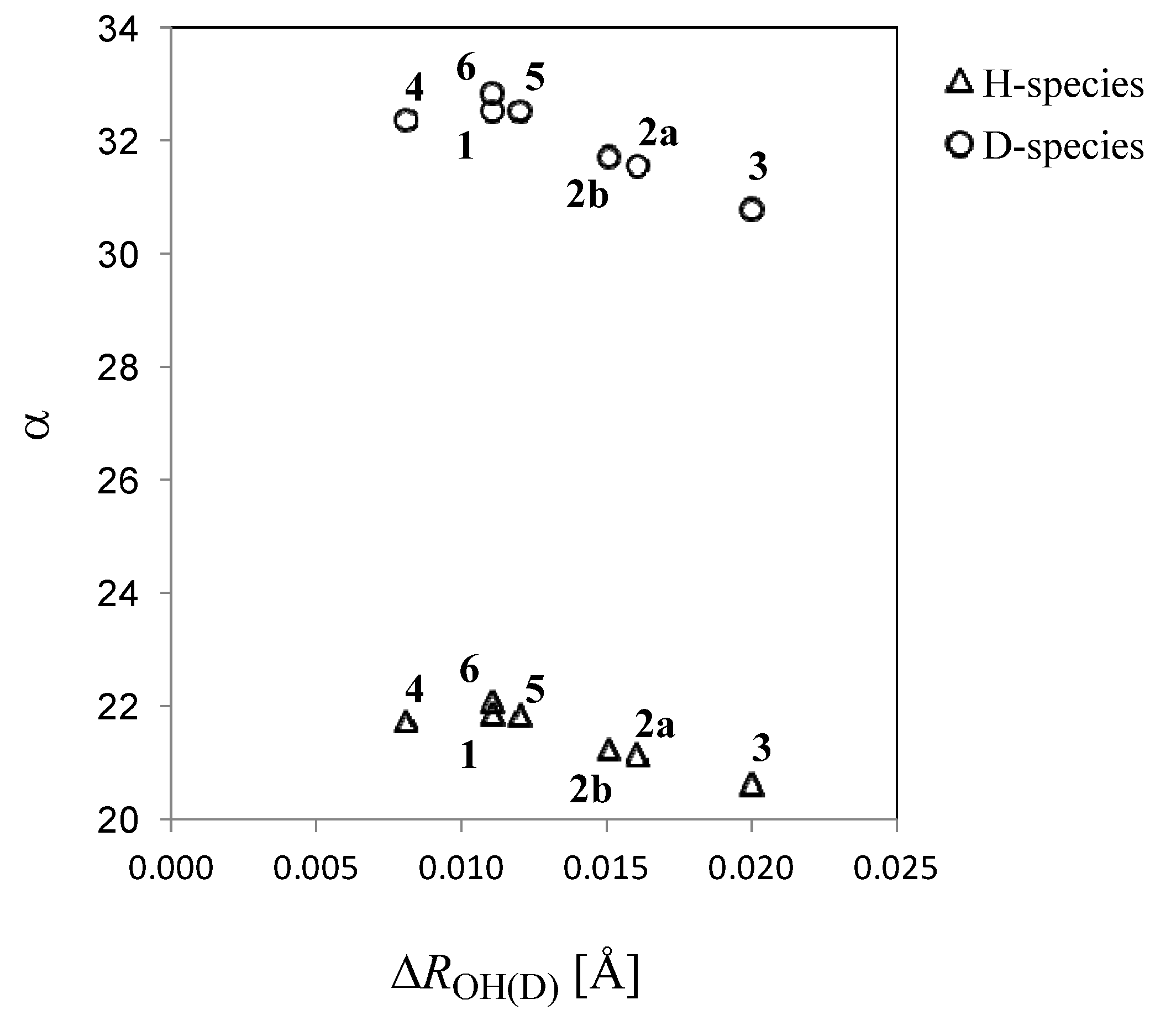
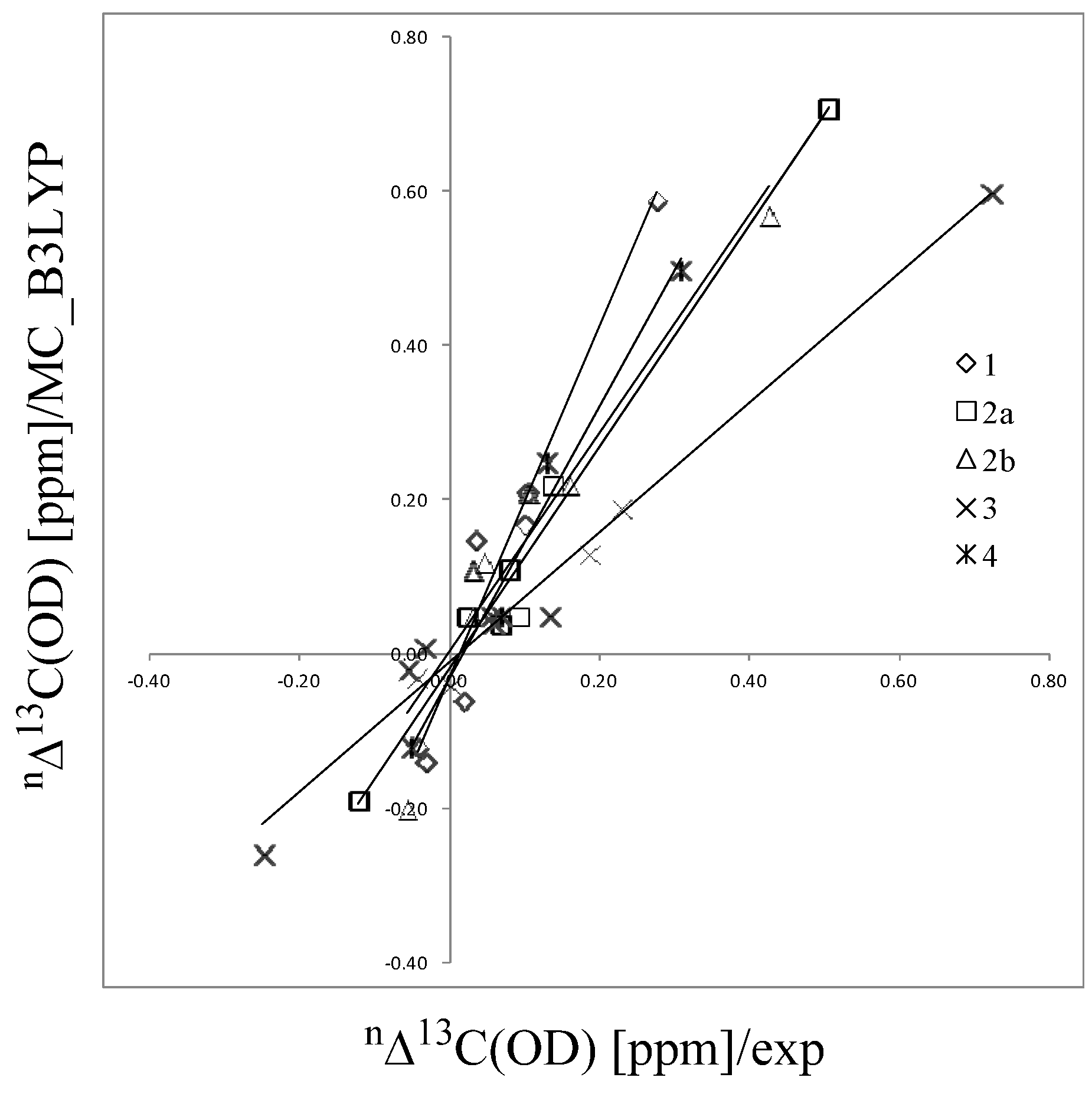
2.2. Isotope Effects on Nuclear Magnetic Shieldings
| MC_B3LYP | B3LYP//MC_B3LYP | B3LYP | |||
|---|---|---|---|---|---|
| H | D | H | D | Conv | |
| C1 | 77.02 | 76.96 | 76.63 | 76.49 | 74.24 |
| C2 | 66.76 | 66.72 | 66.30 | 66.25 | 66.19 |
| C3 | 80.70 | 80.56 | 80.21 | 80.08 | 79.89 |
| C4 | 60.95 | 61.10 | 60.62 | 60.73 | 61.06 |
| C5 | 77.58 | 77.75 | 77.53 | 77.58 | 77.73 |
| C6 | 30.61 | 31.20 | 30.43 | 30.93 | 32.10 |
| C7 | −5.18 | −4.97 | −5.41 | −5.21 | −4.64 |
| C8 | 166.61 | 166.49 | 166.22 | 166.10 | 165.84 |
| O1 | 196.05 | 199.37 | 194.29 | 197.87 | 205.55 |
| O2 | −151.26 | −158.69 | −156.44 | −162.30 | −174.63 |
| H1 | 14.86 | 15.92 | 16.31 | 16.96 | 18.47 |
| H2 | 24.18 | 24.18 | 24.18 | 24.17 | 24.17 |
| H3 | 25.04 | 25.03 | 25.03 | 25.02 | 25.00 |
| H4 | 24.34 | 24.34 | 24.33 | 24.33 | 24.34 |
| H5 | 24.83 | 24.83 | 24.83 | 24.82 | 24.82 |
| H6 | 29.80 | 29.80 | 29.81 | 29.81 | 29.82 |
| H7 | 29.06 | 29.07 | 29.07 | 29.07 | 29.08 |
| H8 | 29.06 | 29.07 | 29.07 | 29.07 | 29.08 |
| MC_B3LYP | B3LYP//MC_B3LYP | B3LYP | ||||
|---|---|---|---|---|---|---|
| H | D | H | D | Conv | ||
| 1 | 14.86 | 15.92 | 16.31 | 16.96 | 18.47 | |
| 2a | 11.90 | 13.03 | 13.30 | 14.03 | 15.84 | |
| 2b | 12.21 | 13.30 | 13.65 | 14.33 | 16.03 | |
| 3 | 9.68 | 10.72 | 11.07 | 11.72 | 13.55 | |
| 4 | 14.49 | 15.55 | 15.95 | 16.60 | 18.12 | |
| 5 | 14.57 | 15.68 | 16.03 | 16.71 | 18.31 | |
| 6 | 16.11 | 17.13 | 17.58 | 18.18 | 19.63 | |
| H | D | |
|---|---|---|
| 1 | −1.45 | −1.04 |
| 2a | −1.40 | −1.00 |
| 2b | −1.44 | −1.04 |
| 3 | −1.39 | −1.00 |
| 4 | −1.46 | −1.05 |
| 5 | −1.46 | −1.03 |
| 6 | −1.47 | −1.05 |
| MC_B3LYP a | B3LYP//MC_B3LYP a | |
|---|---|---|
| C1 | −0.06 | −0.14 |
| C2 | −0.04 | −0.05 |
| C3 | −0.14 | −0.13 |
| C4 | 0.15 | 0.11 |
| C5 | 0.17 | 0.05 |
| C6 | 0.59 | 0.50 |
| C7 | 0.21 | 0.20 |
| C8 | −0.21 | −0.12 |
| O1 | 3.32 | 3.58 |
| O2 | −7.43 | −5.86 |
| H1 | 1.06 | 0.65 |
| H2 | 0.00 | −0.01 |
| H3 | −0.01 | −0.01 |
| H4 | 0.00 | 0.00 |
| H5 | 0.00 | −0.01 |
| H6 | 0.00 | 0.00 |
| H7 | 0.01 | 0.00 |
| H8 | 0.01 | 0.00 |

3. Theory and Computational Details


 is given as:
is given as:

4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Melander, L.; Saunders, W.H. Reaction Rates of Isotopic Molecules; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Dalal, N.; Klymachyov, A.; Bussmann-Holder, A. Coexistence of order-disorder and displacive features at the phase transitions in hydrogen-bonded solids: Squaric cid and its analogs. Phys. Rev. Lett. 1998, 81, 5924–5927. [Google Scholar] [CrossRef]
- Abildgaard, J.; Bolvig, S.; Hansen, P.E. Unraveling the electronic and vibrational contributions to deuterium isotope effects on 13C chemical shifts using ab initio model calculations. Analysis of the observed isotope effects on sterically perturbed intramolecular hydrogen-bonded σ-hydroxy acyl aromatics. J. Am. Chem. Soc. 1998, 120, 9063–9069. [Google Scholar] [CrossRef]
- Born, M.; Oppenheimer, J.R. Zur quantentheorie der molekeln. Ann. Phys. 1927, 84, 457–484. [Google Scholar] [CrossRef]
- Shibl, M.F.; Pietrzak, M.; Limbach, H.-H.; Kühn, O. Geometric isotope effects and cooperativity of the hydrogen bonds in porphycene. ChemPhysChem 2007, 8, 315–321. [Google Scholar] [CrossRef]
- Del Bene, J.E. Anharmonicities, Isotopes, and IR and NMR Properties of Hydrogen-Bonded Complexes. In Isotope Effects in Chemistry and Biology; Kohen, A., Limbach, H.-H., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 153–174. [Google Scholar]
- Tachikawa, M.; Mori, K.; Nakai, H.; Iguchi, K. An extension of ab initio molecular orbital theory to nuclear motion. Chem. Phys. Lett. 1998, 290, 437–442. [Google Scholar] [CrossRef]
- Tachikawa, M.; Mori, K.; Suzuki, K.; Iguchi, K. Full variational molecular orbital method: application to the positron-molecule complexes. Int. J. Quantum Chem. 1998, 70, 491–501. [Google Scholar] [CrossRef]
- Tachikawa, M. Simultaneous optimization of Gaussian type function exponents for electron and positron with full-CI wavefunction—Application to ground and excited states of positronic compounds with multi-component molecular orbital approach. Chem. Phys. Lett. 2001, 350, 269–276. [Google Scholar] [CrossRef]
- Tachikawa, M. Multi-component molecular orbital theory for electrons and nuclei including many-body effect with full configuration interaction treatment: Isotope effects on hydrogen molecules. Chem. Phys. Lett. 2002, 360, 494–500. [Google Scholar] [CrossRef]
- Udagawa, T.; Tachikawa, M. H/D isotope effect on porphine and porphycene molecules with multicomponent hybrid density functional theory. J. Chem. Phys. 2006, 125, 244105:1–244105:9. [Google Scholar]
- Udagawa, T.; Tachikawa, M. Review of Multi-Component Molecular Orbital Theory. In Progress in Quantum Chemistry Research; Hoffman, E.O., Ed.; NOVA Science Publishers: New York, NY, USA, 2007; pp. 123–162. [Google Scholar]
- Udagawa, T.; Ishimoto, T.; Tokiwa, H.; Tachikawa, M.; Nagashima, U. The geometrical isotope effect of C–H…O type hydrogen bonds revealed by multi-component molecular orbital calculation. Chem. Phys. Lett. 2004, 389, 236–240. [Google Scholar] [CrossRef]
- Udagawa, T.; Ishimoto, T.; Tokiwa, H.; Tachikawa, M.; Nagashima, U. Geometric isotope effect of various intermolecular and intramolecular C–H…O hydrogen bonds, using the multicomponent molecular orbital method. J. Phys. Chem. A 2006, 110, 7279–7285. [Google Scholar] [CrossRef]
- Itou, Y.; Mori, S.; Udagawa, T.; Tachikawa, M.; Ishimoto, T.; Nagashima, U. Quantum treatment of hydrogen nuclei in primary kinetic isotope effects in a thermal [1,5]-sigmatropic hydrogen (or deuterium) shift from (Z)-1,3-pentadiene. J. Phys. Chem. A 2007, 111, 261–267. [Google Scholar]
- Ditchfield, R. Molecular orbital theory of magnetic shielding and magnetic susceptibility. J. Chem. Phys. 1971, 56, 5688:1–5688:4. [Google Scholar]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Kita, Y.; Udagawa, T.; Tachikawa, M. Nuclear quantum effect on molecular magnetic properties for low barrier hydrogen-bonded systems based on multicomponent density functional theory. Chem. Lett. 2009, 38, 1156–1157. [Google Scholar] [CrossRef]
- Kita, Y.; Tachikawa, M. Nuclear quantum effects on molecular magnetic properties. J. Mol. Struct. THEOCHEM 2009, 912, 2–4. [Google Scholar] [CrossRef]
- Ullah, S.; Ishimoto, T.; Williamson, M.P.; Hansen, P.E. Ab initio calculations of deuterium isotope effects on chemical shifts of salt-bridged lysines. J. Phys. Chem. B 2011, 115, 3208–3215. [Google Scholar]
- Ubbelohde, A.R.; Gallagher, K.H. Acid-base effects in hydrogen bonds in crystals. Acta Crystallogr. 1955, 8, 71–83. [Google Scholar]
- Bolving, S.; Hansen, P.E.; Wemmer, D.; Williams, P. Deuterium isotope effects on 17O chemical shifts of intramolecularly hydrogen bonded systems. J. Mol. Struct. 1999, 509, 171–181. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648:1–5648:5. [Google Scholar]
- Tachikawa, M.; Sainowo, H.; Iguchi, K.; Suzuki, K. Ab initio calculation of [OH−;e+] system with consideration of electron correlation effect. J. Chem. Phys. 1994, 101, 5925:1–5925:4. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision B.03; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Udagawa, T.; Ishimoto, T.; Tachikawa, M. Theoretical Study of H/D Isotope Effects on Nuclear Magnetic Shieldings Using an ab initio Multi-Component Molecular Orbital Method. Molecules 2013, 18, 5209-5220. https://doi.org/10.3390/molecules18055209
Udagawa T, Ishimoto T, Tachikawa M. Theoretical Study of H/D Isotope Effects on Nuclear Magnetic Shieldings Using an ab initio Multi-Component Molecular Orbital Method. Molecules. 2013; 18(5):5209-5220. https://doi.org/10.3390/molecules18055209
Chicago/Turabian StyleUdagawa, Taro, Takayoshi Ishimoto, and Masanori Tachikawa. 2013. "Theoretical Study of H/D Isotope Effects on Nuclear Magnetic Shieldings Using an ab initio Multi-Component Molecular Orbital Method" Molecules 18, no. 5: 5209-5220. https://doi.org/10.3390/molecules18055209