Chelator-Accelerated One-Pot ‘Click’ Labeling of Small Molecule Tracers with 2-[18F]Fluoroethyl Azide

Abstract
:1. Introduction
2. Results and Discussion
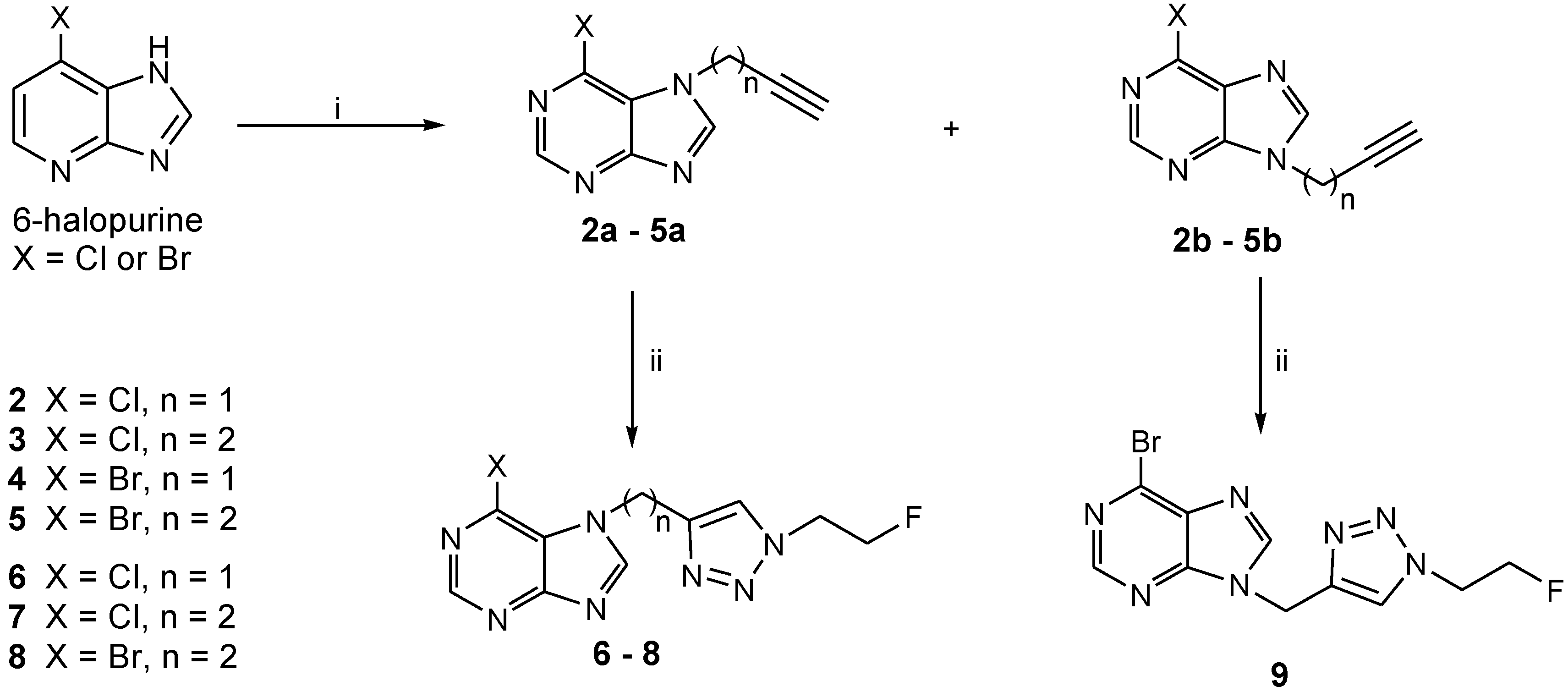
2.1. Chemistry

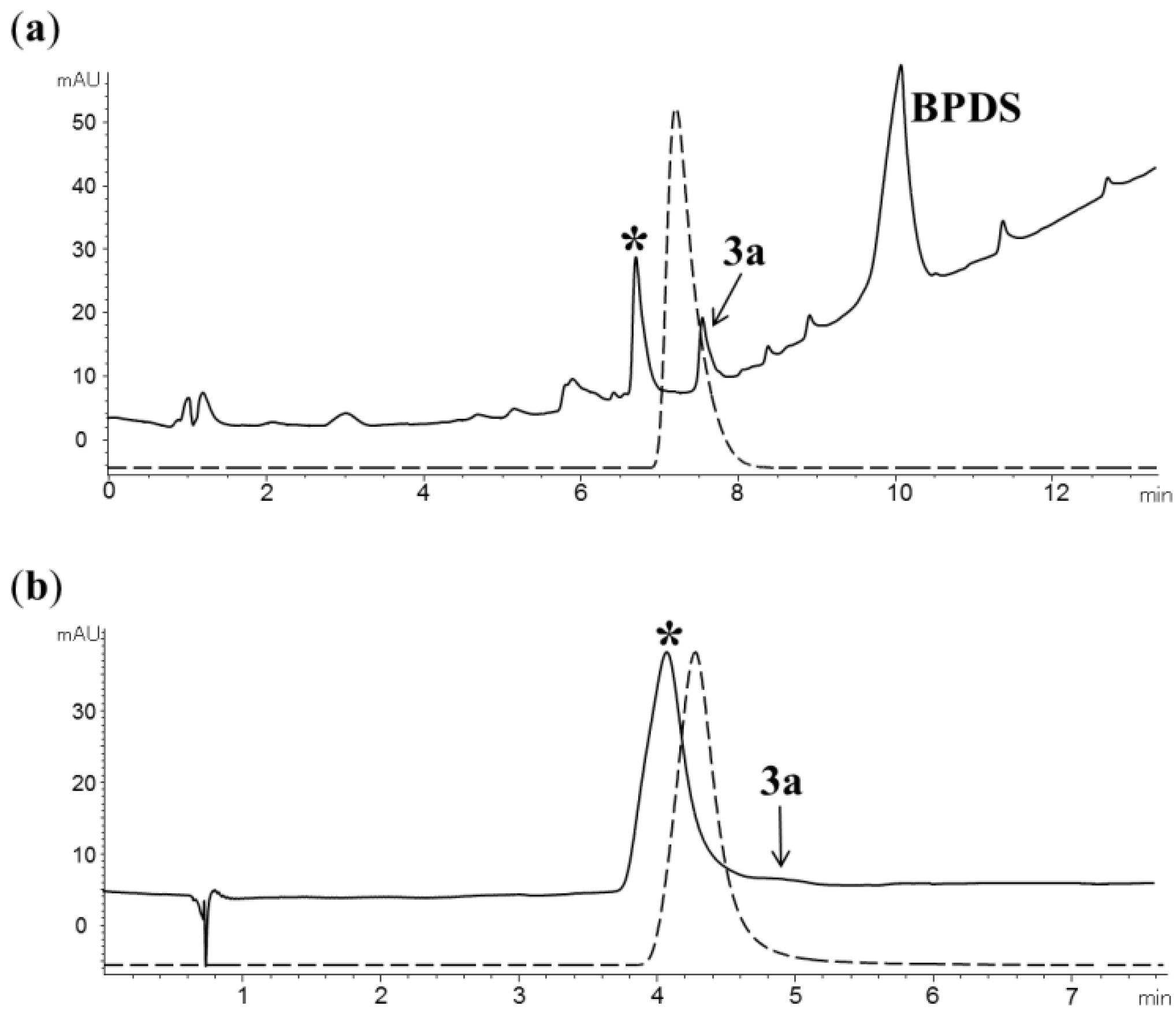
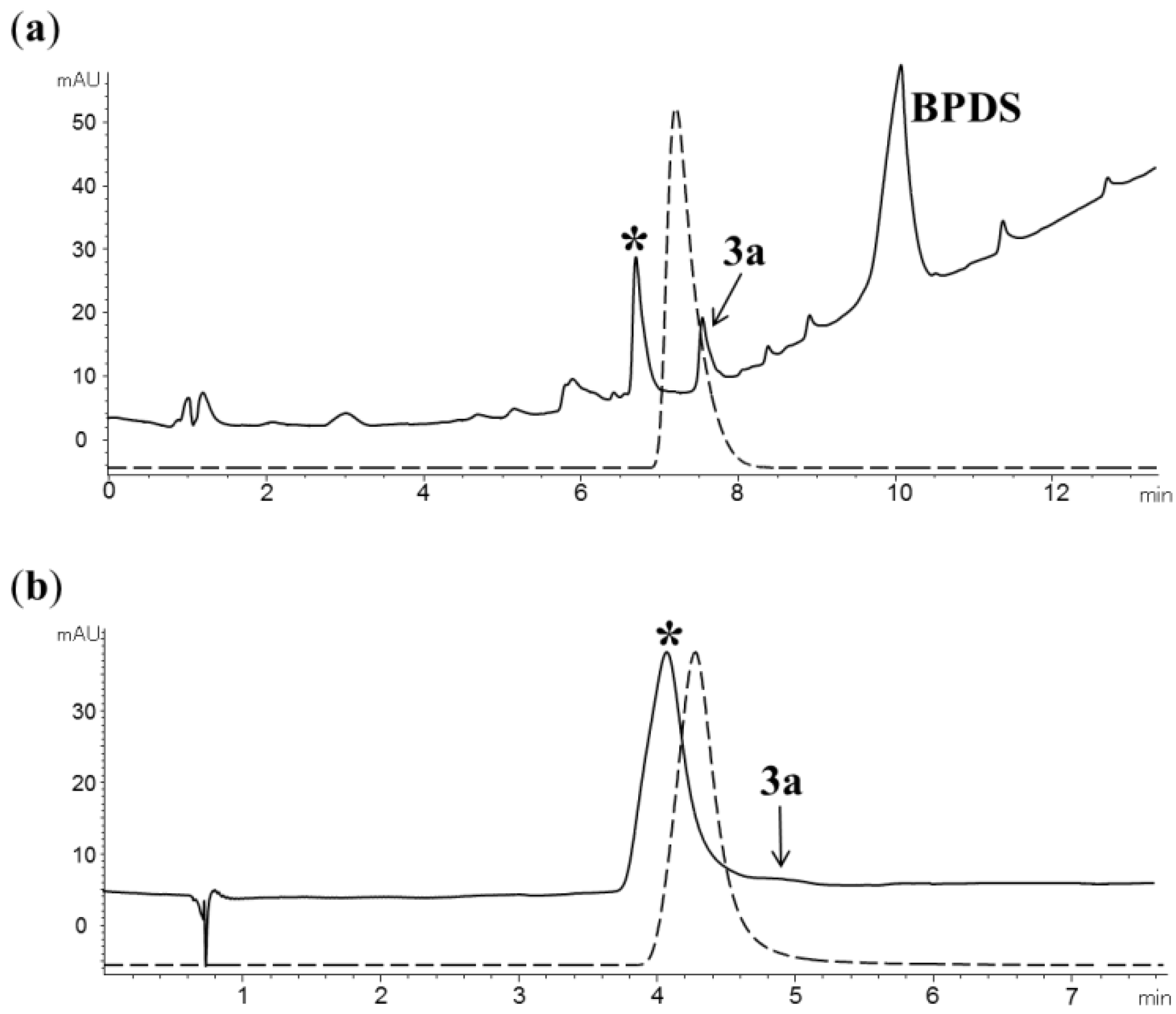
2.2. Radiochemistry
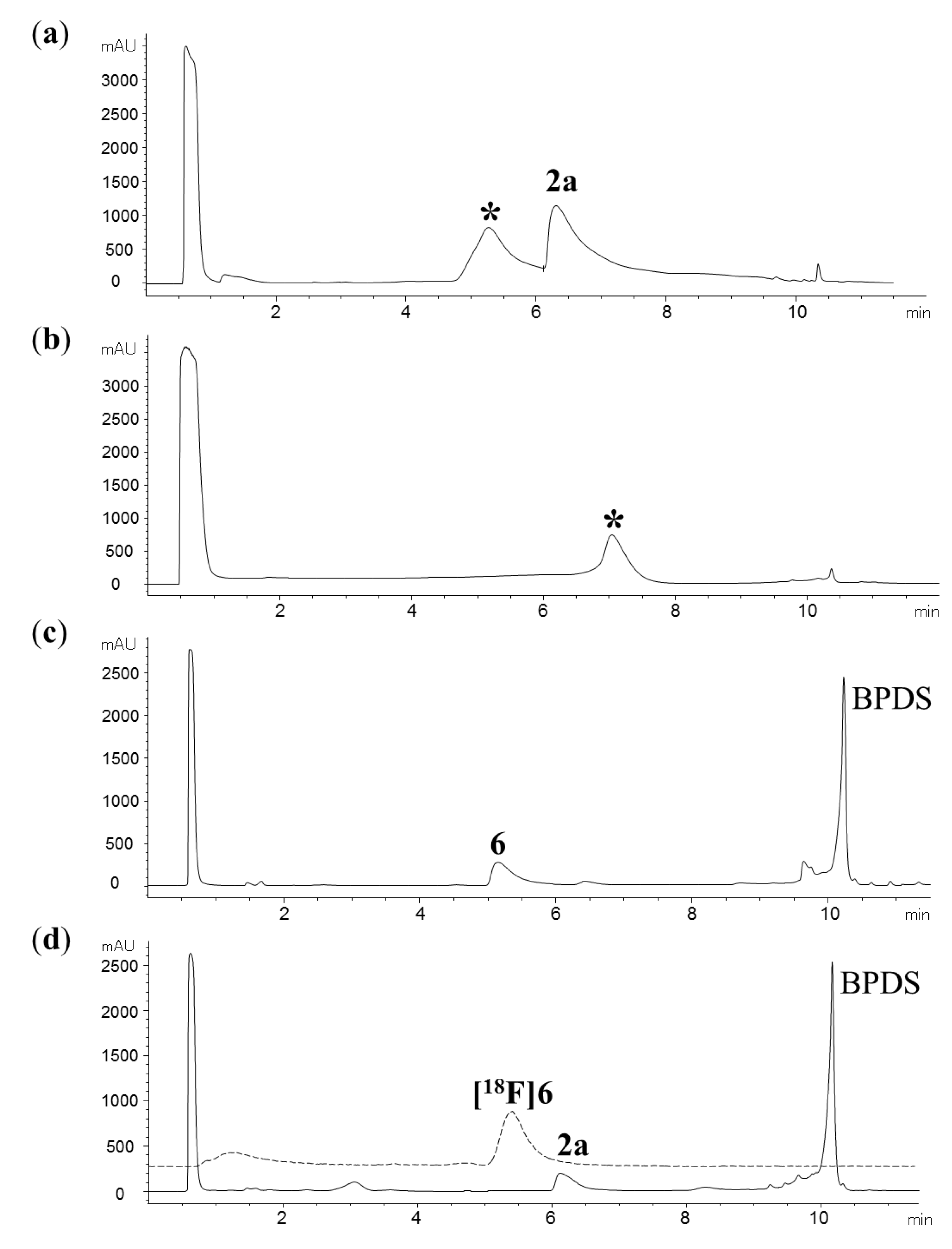

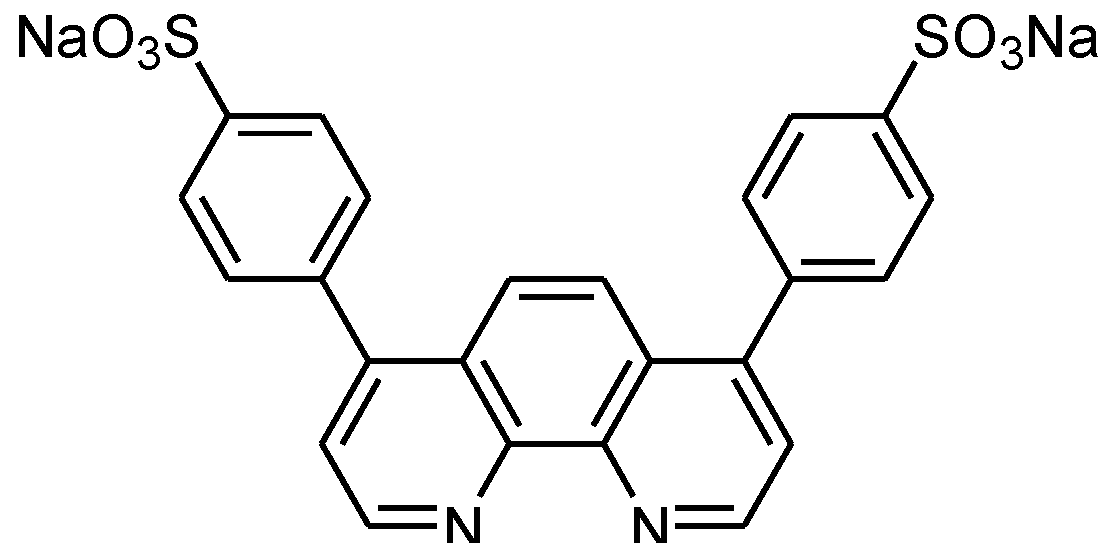
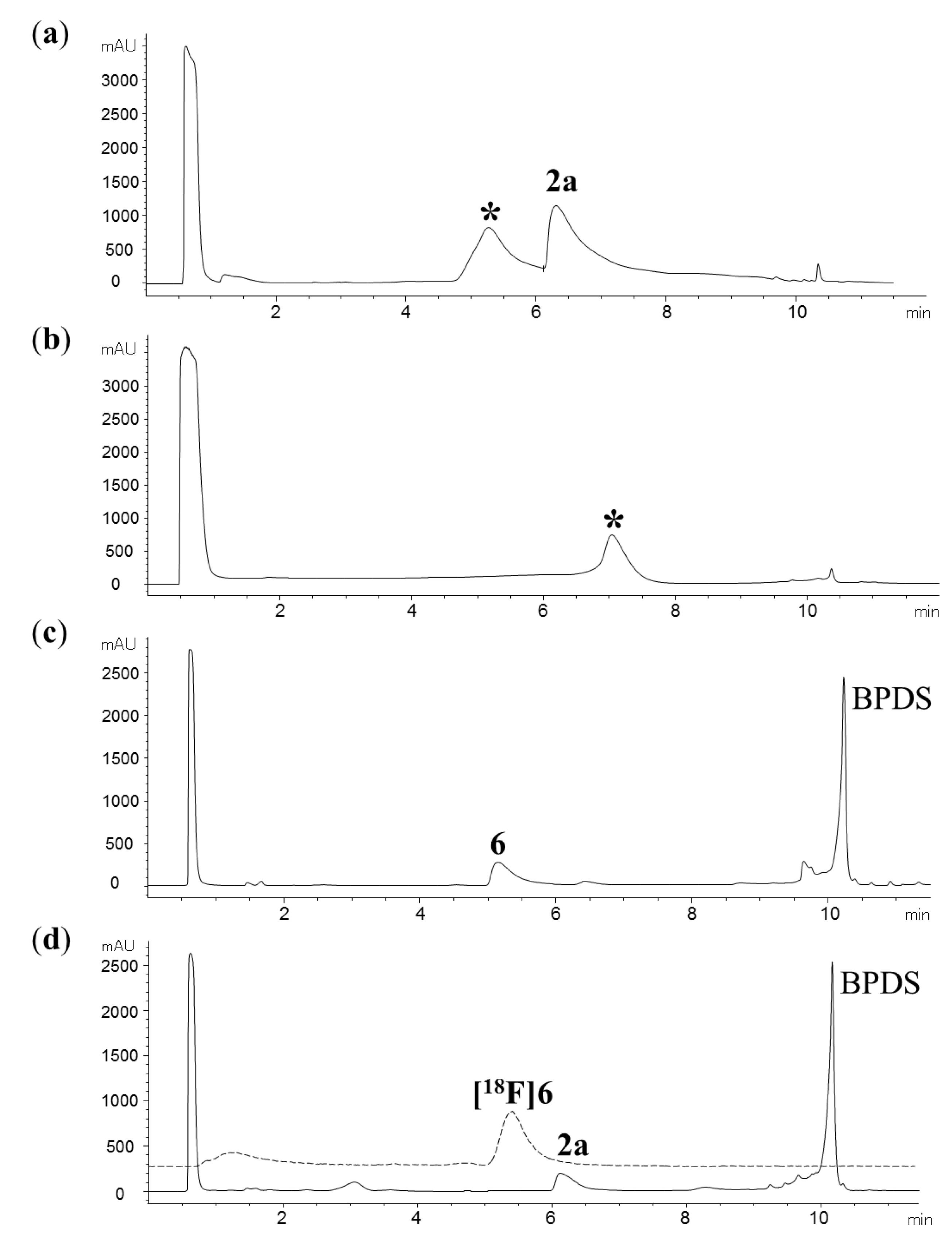
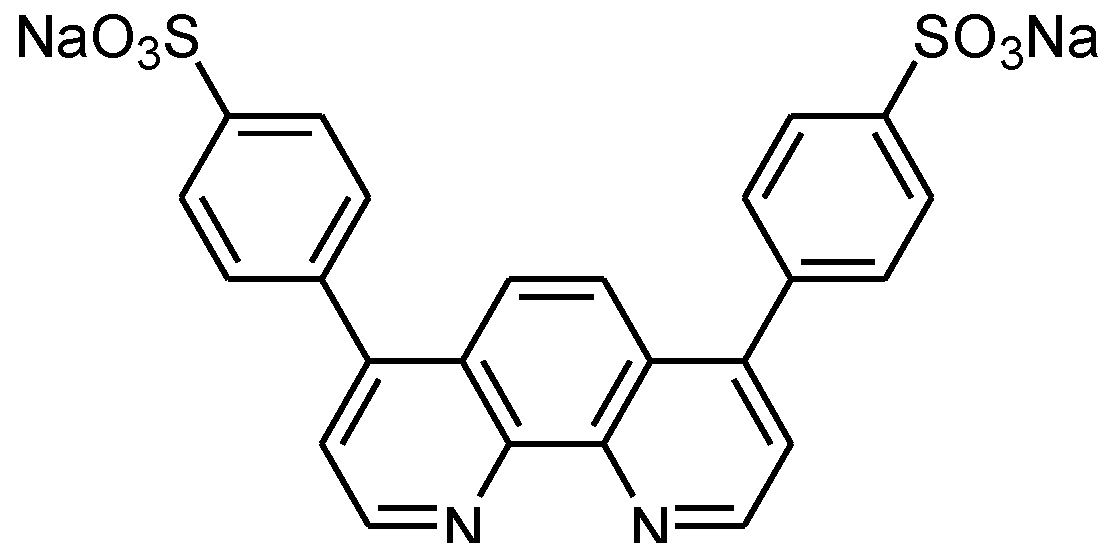{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Product | RCY a (%) | |
|---|---|---|---|
| 1 | [18F]6 | 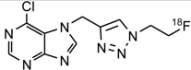 | 60 ± 1 (41) b |
| 2 | [18F]7 |  | 76 ± 1 (40) b,c |
| 3 | [18F]8 |  | 55 ± 2 (55) b |
| 4 | [18F]9 | 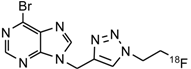 | 76 ± 1 (57) b |
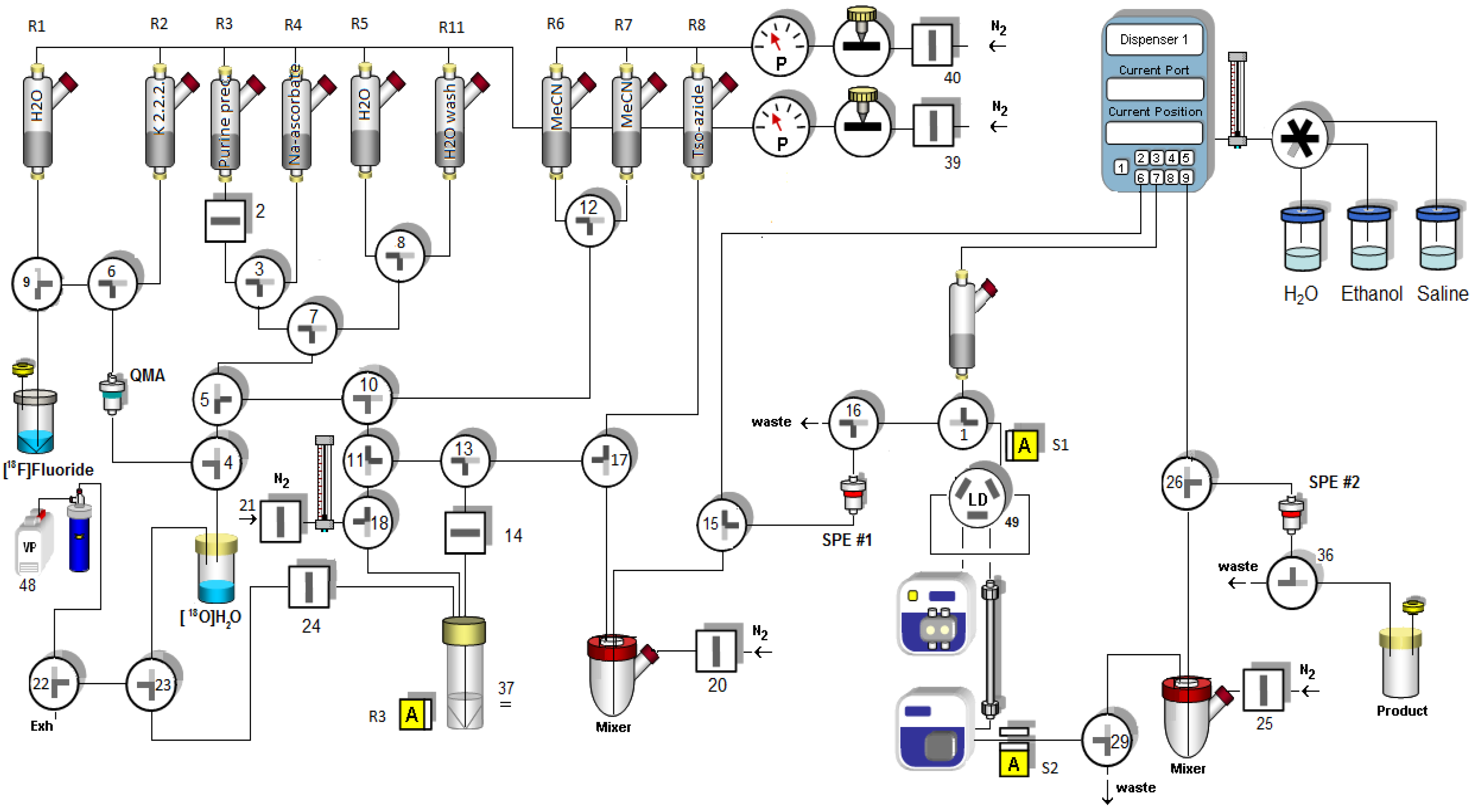
2.3. Automated radiosynthesis of purine [18F] 7

3. Experimental
3.1. General
3.2. Chemistry
3.2.1. General Procedure for Preparation of the Alkyne-functionalized 6-Halopurines
3.2.2. General Procedure for Cycloaddition of 2-fluoroethyl azide (FEA) with Alkyne-functionalized Purines
3.3. Radiochemistry
3.3.1. One-pot Method for Cycloaddition of [18F]FEA to the Alkyne-Functionalized Purine Precursors
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Marik, J.; Sutcliffe, J.L. Click for PET: rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Glaser, M; Årstad, E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjug. Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Glaser, M; Robins, E.G. ‘Click labeling’ in PET radiochemistry. J. Labelled Compd. Radiopharm. 2009, 52, 407–414. [Google Scholar] [CrossRef]
- Mamat, C; Ramenda, T; Wuest, F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Smith, G.; Glaser, M.; Perumal, M.; Nguyen, Q.-D.; Shan, B.; Årstad, E.; Aboagye, E.O. Design, Synthesis, and biological characterization of a Caspase 3/7 selective isatin labeled with 2-[18F]fluoroethylazide. J. Med. Chem. 2008, 51, 8057–8067. [Google Scholar] [CrossRef]
- Zhou, D.; Chu, W.; Dence, C.S.; Mach, R.H.; Welch, M.J. Highly efficient click labeling using 2-[18F]fluoroethyl azide and synthesis of an 18FN-hydroxysuccinimide ester as conjugation agent. Nucl. Med. Biol. 2012, 39, 1175–1181. [Google Scholar] [CrossRef]
- Laurens, E.; Dee Yeoh, S.; Rigopoulos, A.; Cao, D.; Cartwright, G.A.; O’Keefe, G.J.; Tochon-Danguy, H.J.; White, J.M.; Scott, A.M.; Ackermann, U. Radiolabelling and evaluation of novel haloethylsulfoxides as PET imaging agents for tumor hypoxia. Nucl. Med. Biol. 2012, 39, 871–882. [Google Scholar] [CrossRef]
- Smith, G.; Sala, R.; Carroll, L.; Behan, K.; Glaser, M.; Robins, E.; Nguyen, Q-D.; Aboagye, E.O. Synthesis and evaluation of nucleoside radiotracers for imaging proliferation. Nucl. Med. Biol. 2012, 39, 652–665. [Google Scholar] [CrossRef]
- Yan, R.; Sander, K.; Galante, E.; Rajkumar, V.; Badar, A.; Robson, M.; El-Emir, E.; Lythgoe, M.F.; Pedley, R.B.; Arstad, E. A one-pot three-component radiochemical reaction for rapid assembly of 125I-labeled molecular probes. J. Am. Chem. Soc. 2013, 135, 703–709. [Google Scholar]
- Kjellberg, J.; Johansson, N.G. Charaterization of N7 and N9 alkylated purine by 1H and 13C NMR. Tetrahedron 1986, 42, 6541–6544. [Google Scholar] [CrossRef]
- Manuprasad, B.K.; Murthy, V.S.; Shashikanth, S.; Rakshith, D.; Satish, S.; Raveesha, K.A. Design, Synthesis and antibacterial activity of novel 1,3-thiazolidine purine nucleosides. Der Pharm. Chem. 2011, 3, 45–55. [Google Scholar]
- Trávníček, Z.; Maloň, M.; Šindelář, Z.; Doležal, K.; Rolčík, J.; Kryštof, V.; Strnad, M.; Marek, J. Preparation, Physicochemical properties and biological activity of copper(II) complexes with 6-(2-chlorobenzylamino)purine (HL1) or 6-(3-chlorobenzylamino)purine (HL2). The single-crystal X-ray structure of. [Cu(H+L2)2Cl3]Cl∙2H2O. J. Inorg. Biochem. 2001, 84, 23–32. [Google Scholar] [CrossRef]
- Colacio, E.; Cuesta, R.; Moreno, J.M. Copper(I)-purine-phosphine complexes. Syntheses and molecular structures of [Cu2(µ-HL1)2(µ-dppm)(η1-dppm)2] and {[Cu3(µ3-Cl)2(µ-dppm)3][Cu(HL2)2]} (H2L1 = 8-Mercaptotheophylline and H2L2 = 8-Ethyl-3-methylxanthine). Inorg. Chem. 1997, 36, 1084–1087. [Google Scholar] [CrossRef]
- Lewis, W.G.; Magallon, F.G.; Fokin, V.V.; Finn, M.G. Discovery and characterization of catalysts for azide-alkyne cycloaddition by fluorescence quenching. J. Am. Chem. Soc. 2004, 126, 9152–9153. [Google Scholar]
- Hassane, F.S.; Frisch, B.; Schuber, F. Targeted liposomes: convenient coupling of ligands to preformed vesicles using “click chemistry”. Bioconjug. Chem. 2006, 17, 849–854. [Google Scholar] [CrossRef]
- Gill, H.S.; Marik, J. Preparation of 18F-labeled peptides using the copper(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition. Nat. Protoc. 2011, 6, 1718–1725. [Google Scholar] [CrossRef]
- Glaser, M.; Solbakken, M.; Turton, D.R.; Pettitt, R.; Barnett, J.; Arukwe, J.; Karlsen, H.; Cuthbertson, A.; Luthra, S.K.; Årstad, E. Methods for 18F-labeling of RGD peptides: Comparison of aminooxy [18F]fluorobenzaldehyde condensation with ‘click labeling’ using 2-[18F]fluoroethylazide, and S-alkylation with [18F]fluoropropanethiol. Amino Acids 2009, 37, 717–724. [Google Scholar]
- Sample Availability: Samples of the compounds 6–9 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Galante, E.; Schoultz, B.W.; Koepp, M.; Årstad, E. Chelator-Accelerated One-Pot ‘Click’ Labeling of Small Molecule Tracers with 2-[18F]Fluoroethyl Azide. Molecules 2013, 18, 5335-5347. https://doi.org/10.3390/molecules18055335
Galante E, Schoultz BW, Koepp M, Årstad E. Chelator-Accelerated One-Pot ‘Click’ Labeling of Small Molecule Tracers with 2-[18F]Fluoroethyl Azide. Molecules. 2013; 18(5):5335-5347. https://doi.org/10.3390/molecules18055335
Chicago/Turabian StyleGalante, Eva, Bent Wilhelm Schoultz, Matthias Koepp, and Erik Årstad. 2013. "Chelator-Accelerated One-Pot ‘Click’ Labeling of Small Molecule Tracers with 2-[18F]Fluoroethyl Azide" Molecules 18, no. 5: 5335-5347. https://doi.org/10.3390/molecules18055335
APA StyleGalante, E., Schoultz, B. W., Koepp, M., & Årstad, E. (2013). Chelator-Accelerated One-Pot ‘Click’ Labeling of Small Molecule Tracers with 2-[18F]Fluoroethyl Azide. Molecules, 18(5), 5335-5347. https://doi.org/10.3390/molecules18055335