Diorganotin(IV) Derivatives of N-Methyl p-Fluorobenzo-Hydroxamic Acid: Preparation, Spectral Characterization, X-ray Diffraction Studies and Antitumor Activity

Abstract
:1. Introduction
2. Results and Discussion
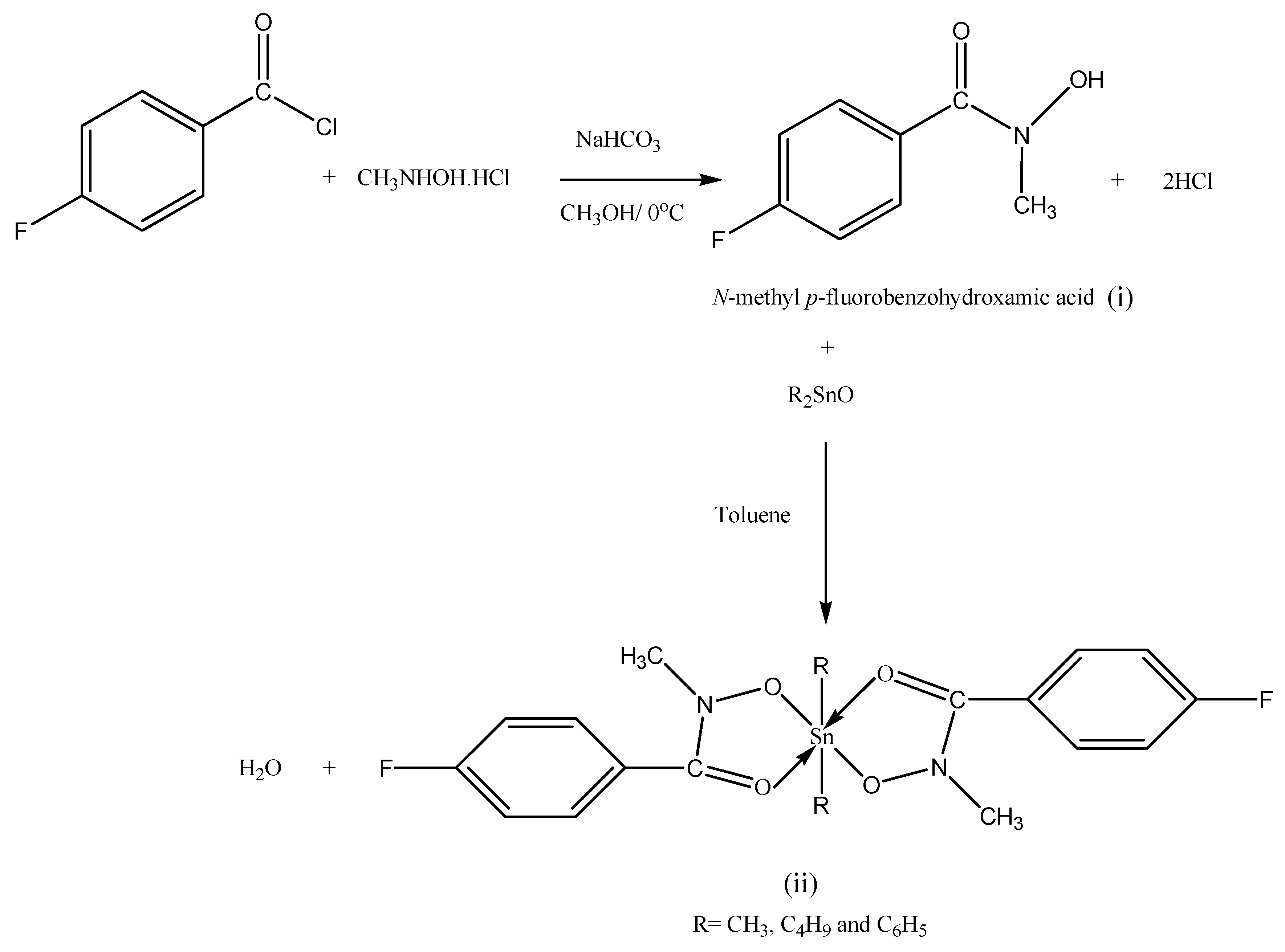
2.1. Synthesis

2.2. Infra-Red Spectroscopy
2.3. NMR Spectroscopy
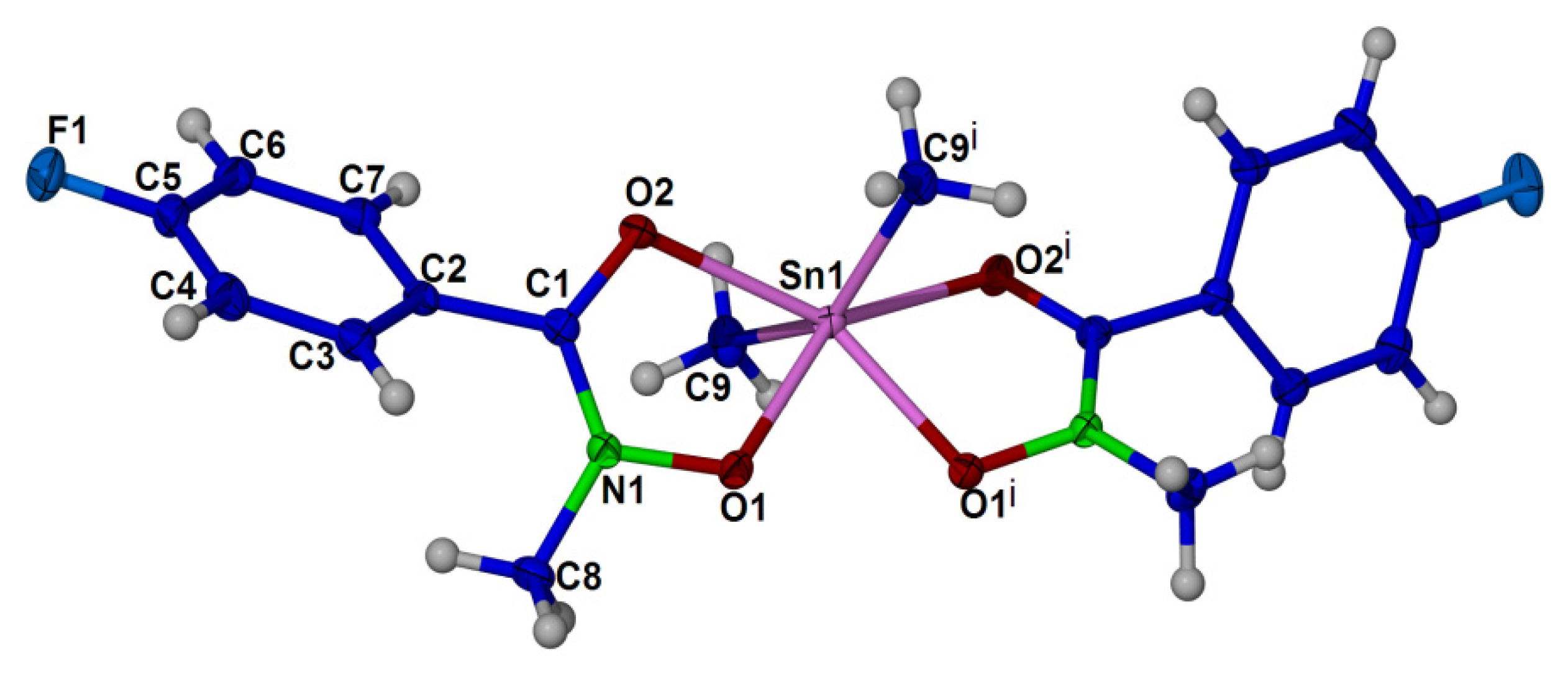
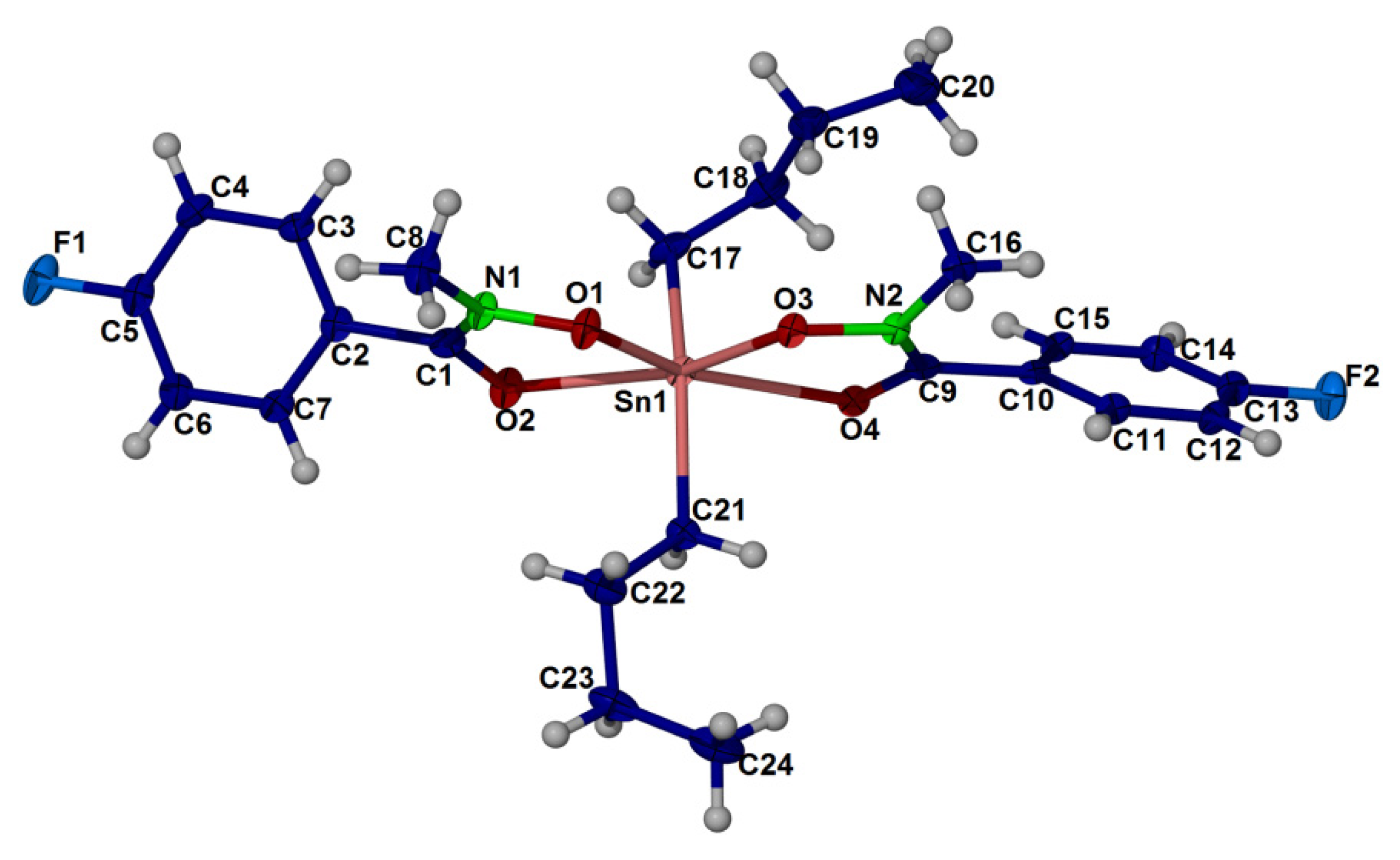
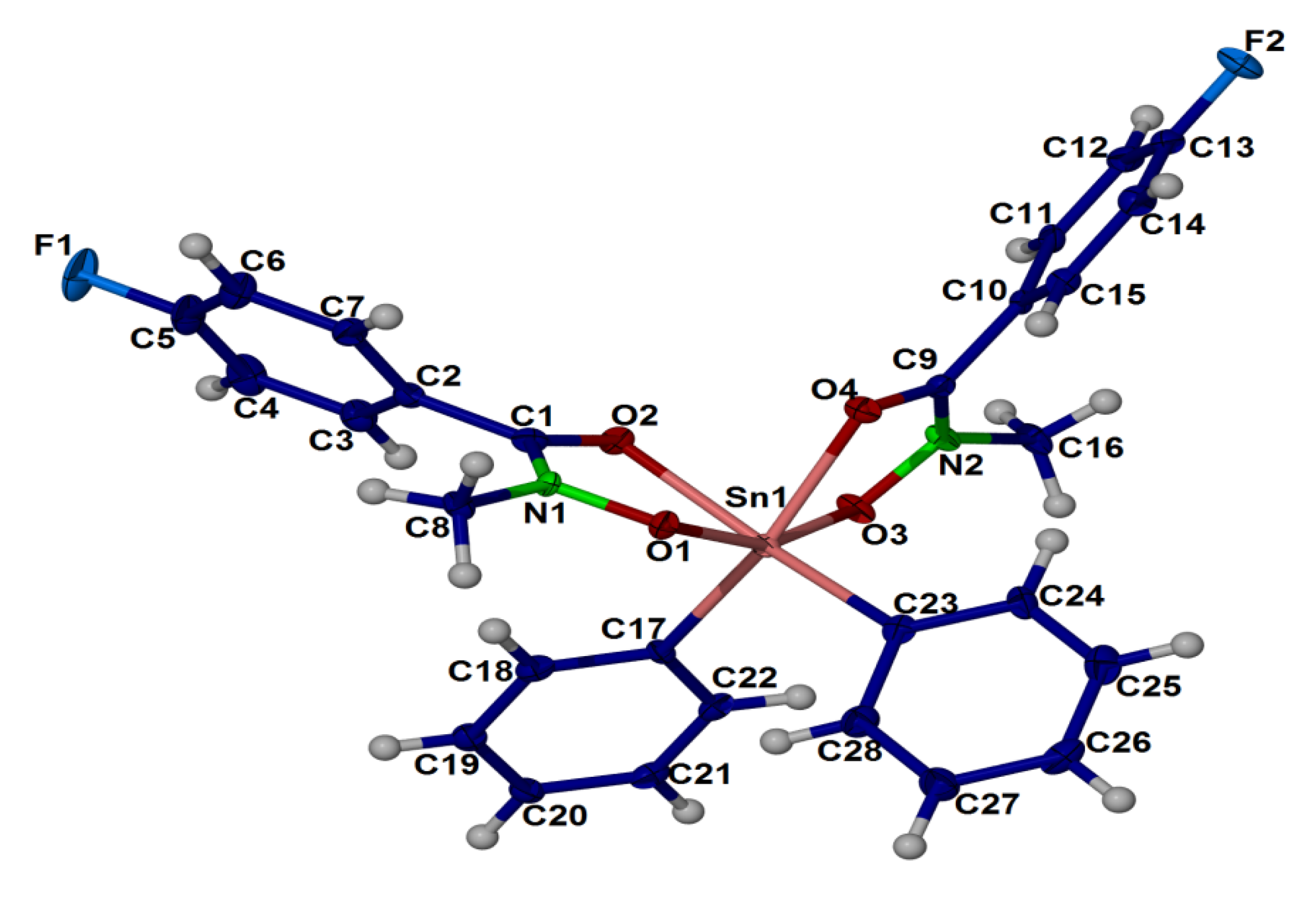
2.4. X-ray Crystallography




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1) | (2) | (3) | |||
|---|---|---|---|---|---|
| Sn1-O1 2.0921(9) | F1-C5 1.3583(15) | Sn1 O1 2.117(3) | N1 C1 1.312(5) | Sn1 O3 2.103(2) | N1 C1 1.326(4) |
| Sn1-O1 i 2.0921(9) | O1-N1 1.3807(13) | Sn1 C21 2.125(4) | N1 O1 1.385(4) | Sn1 O1 2.110(2) | N1 O1 1.379(3) |
| Sn1-C9 i 2.1184(13) | O2-C1 1.2696(15) | Sn1 O3 2.132(3) | N1 C8 1.453(5) | Sn1 C17 2.144(3) | N1 C8 1.452(4) |
| Sn1-C9 2.1184(13) | N1-C1 1.3192(16) | Sn1 C17 2.143(4) | N2 C9 1.321(5) | Sn1 C23 2.156(3) | N2 C9 1.322(4) |
| Sn1-O2 2.3778(9) | N1-C8 1.4558(16) | Sn1 O2 2.356(3) | N2 O3 1.375(4) | Sn1 O4 2.183(2) | N2 O3 1.380(3) |
| Sn1-O2 i 2.3778(9) | Sn1 O4 2.407(3) | O2 C1 1.262(5) | Sn1 O2 2.221(2) | O2 C1 1.277(4) | |
| F1 C5 1.354(5) | O4 C9 1.263(5) | F1 C5 1.353(4) | O4 C9 1.285(4) | ||
| (1) | (2) | (3) | ||||
|---|---|---|---|---|---|---|
| O1-Sn1-O1i 76.58(5) | C9i-Sn1-O2i 82.95(4) | O1 Sn1 C21 105.01(13) | C9 N2 O3 119.0(3) | O3 Sn1 O1 158.08(9) | C9 N2 O3 117.6(2) | |
| O1-Sn1-C9i 97.86(5) | C9-Sn1-O2i 85.64(4) | O1 Sn1 O3 76.51(10) | O3 N2 C16 111.9(3) | O3 Sn1 C17 89.96(10) | O1 N1 C8 112.8(2) | |
| O1i-Sn1-C9i 110.40(5) | O2-Sn1-O2i 142.49(4) | C21 Sn1 O3 103.19(13) | N1 O1 Sn1 114.8(2) | O1 Sn1 C17 103.49(10) | O3 N2 C16 112.7(2) | |
| O1-Sn1-C9 110.40(5) | N1-O1-Sn1 113.09(7) | O1 Sn1 C17 104.85(14) | C1 O2 Sn1 110.1(2) | O3 Sn1 C23 105.08(10) | C1 O2 Sn1 112.7(18) | |
| O1i-Sn1-C9 97.86(5) | C1-O2-Sn1 107.76(8) | C21 Sn1 C17 141.14(16) | C9 O4 Sn1 111.7(2) | O1 Sn1 C23 88.55(10) | N1 O1 Sn1 113.1(16) | |
| C9i-Sn1-C9 143.98(8) | C1-N1-O1 118.20(10) | O3 Sn1 C17 107.52(14) | N2 O3 Sn1 117.7(2) | C17 Sn1C23 104.16(11) | N2 O3 Sn1 113.1(16) | |
| O1-Sn1-O2 71.22(3) | C1-N1-C8 128.68(11) | O1 Sn1 O2 71.96(10) | C18 C17 Sn1 118.9(3) | O3 Sn1 O4 74.15(8) | C9 O4 Sn1 113.8(18) | |
| O1i-Sn1-O2 145.75(3) | O1-N1-C8 112.43(10) | C21 Sn1 O2 85.83(13) | Sn1 C17 H17A 107.6 | O1 Sn1 O4 88.51(8) | C22 C17 Sn1 120.5(2) | |
| C9i-Sn1-O2 85.64(4) | O2-C1-N1 120.10(12) | O3 Sn1 O2 148.46(10) | Sn1 C17 H17B 107.6 | C17 Sn1 O4 159.85(10) | C18 C17 Sn1 122.0(2) | |
| C9-Sn1-O2 82.95(4) | O2-C1-C2 119.21(11) | C17 Sn1 O2 80.36(14) | C22 C21Sn1 117.2(3) | C23 Sn1 O4 92.10(10) | C24 C23 Sn1 126.3(2) | |
| O1-Sn1-O2i 145.75(3) | N1-C1-C2 120.64(11) | O1 Sn1 O4 146.74(9) | Sn1 C21 H21A 108.0 | O3 Sn1 O2 90.35(8) | C28 C23 Sn1 116.7(2) | |
| O1i-Sn1-O2i 71.23(3) | F1-C5-C6 118.22(13) | C21 Sn1 O4 80.23(12) | Sn1 C21 H21B 108.0 | O1 Sn1 O2 73.26(8) | O2 C1 N1 118.9(3) | |
| O3 Sn1 O4 70.36(9) | O2 C1 N1 121.1(4) | C17 Sn1 O2 87.69(10) | F1 C5 C6 118.8(3) | |||
| C17 Sn1 O4 88.16(13) | F1 C5 C6 118.9(4) | C23 Sn1 O2 160.33(10) | F1 C5 C4 118.1(3) | |||
| O2 Sn1 O4 141.15(9) | F1 C5 C4 117.7(4) | O4 Sn1 O2 80.26(8) | O4 C9 N2 118.8(3) | |||
| C1 N1 O1 118.8(3) | O4 C9 N2 120.1(3) | C1 N1 O1 117.6(2) | ||||
| O1 N1 C8 111.7(3) | O4 C9 C10 119.2(3) | |||||
2.5. Antitumor Activity in vitro
| Compounds | IC50 values (µM) |
|---|---|
| dimethyltin(IV)bis[N-methyl p-fluorobenzohydroxamate] | >40 |
| diphenyltin(IV)bis[N-methyl p-fluorobenzohydroxamate] | 2.45 |
| dibutyltin(IV)bis[N-methyl p-fluorobenzohydroxamate] | NA |
| triphenyltin(IV)N-methyl p-fluorobenzohydroxamate | 0.41 |
3. Experimental
3.1. General
3.2. Synthesis of Ligand
3.3. Synthesis of Complexes
3.4. X-ray Crystallography
| Compound | (1) | (2) | (3) |
|---|---|---|---|
| Gross formula | C18 H20 F2 N2 O4 Sn | C24 H32F2 N2 O4 Sn | C28 H24 F2 N2 O4 Sn |
| M | 485.05 | 569.21 | 609.15 |
| Crystal system, space group | Monoclinic, C2/c | Triclinic, P-1 | Triclinic, P-1 |
| Crystal shape | Block | Block | Block |
| Colour | Colourless | Colourless | White |
| a, Ǻ | 21.7581(2) | 11.0271(7) | 8.8999(2) |
| b, Ǻ | 11.2694(1) | 11.1155(7) | 12.3601(3) |
| c, Ǻ | 7.8964(1) | 11.1404(7) | 12.4362(3) |
| α, deg | 90 | 75.948(3) | 109.600(1) |
| β, deg | 94.357 | 82.636(3) | 99.770(1) |
| γ, deg | 90 | 77.919(3) | 98.015(1) |
| V, Ǻ 3 | 1930.58(3) | 1290.99(14) | 1241.34(5) |
| Z | 4 | 2 | 2 |
| dc, g/cm-3 | 1.669 | 1.464 | 1.37 |
| F(000) | 968 | 580 | 612 |
| Μ, mm-1 | 1.368 | 1.035 | 1.083 |
| T , K | 100(2) | 100(2) | 100(2) |
| Crystal size, mm | 0.24× 0.29× 0.35 | 0.40× 0.15× 0.05 | 0.10×0.05× 0.05 |
| T min | 0.6415 | 0.6824 | 0.8995 |
| T max | 0.7457 | 0.9501 | 0.9479 |
| measured reflections | 8979 | 10517 | 10184 |
| independent reflections | 2221 | 5056 | 4581 |
| reflections with I > 2s(I) | 2191 | 4593 | 4231 |
| R int | 0.0114 | 0.1236 | 0.0219 |
| θ max | 27.5 | 26 | 25.5 |
| θ min | 1.88 | 1.89 | 1.79 |
| Completeness to theta | 0.998 | 0.998 | 0.993 |
| h | −28 28 | −12 13 | −10 10 |
| k | −14 14 | −13 13 | −14 14 |
| l | −10 10 | −13 13 | −15 15 |
| R[F2 > 2s(F2)] | 0.0136 | 0.0539 | 0.0239 |
| wR(F2) | 0.0367 | 0.1444 | 0.0654 |
| S | 1.146 | 1.096 | 1.19 |
| reflections | 2221 | 5056 | 4581 |
| parameters | 125 | 302 | 336 |
| restraints | 0 | 0 | 0 |
| ρmax e Ǻ−3 | 0.261 | 1.538 | 0.532 |
| ∆ρmin e Ǻ−3 | −0.450 | −3.311 | −0.571 |
3.5. MTT Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Nandurkar, N.S.; Petersen, R.; Qvortrup, K.; Komnatnyy, V.V.; Taveras, K.M.; Le Quement, S.T.; Frauenlob, R.; Givskov, M.; Nielsena, T.E. A convenient procedure for the solid-phase synthesis of hydroxamic acids on PEGA resins. Tetrahedron Lett. 2011, 52, 7121–7124. [Google Scholar] [CrossRef]
- Farkas, E.; Csoka, H.; Gama, S.; Santos, M.A. Dihydroxamate based siderophore model, piperazine-1,4-bis-(N-methyl-acetohydroxamic acid (PIPDMAHA), as a chelating agent of molybdenum(VI). Talanta 2002, 57, 935–943. [Google Scholar] [CrossRef]
- Jain, R.; Sundram, A.; Lopez, S.; Neckermann, G.; Wu, C.; Hackbarth, C.; Chen, D.; Wang, W.; Ryder, NS.; Weidmann, B.; et al. α-Substituted hydroxamic acids as novel bacterial deformylase inhibitor-based antibacterial agents. Bioorg. Med. Chem. Lett. 2003, 13, 4223–4228. [Google Scholar] [CrossRef]
- Farkas, E.; Enyedy, E.A.; Micera, G.; Garribba, E. Coordination modes of hydroxamicacids in copper(II), nickel(II) and zinc(II) mixed-ligand complexes in aqueous solution. Polyhedron 2000, 19, 1727–1736. [Google Scholar] [CrossRef]
- Sibi, M.P.; Hasegawa, H.; Ghorpade, S.R. A Convenient Method for the Conversion of N-Acyloxazolidinones to Hydroxamic Acids. Org. Lett. 2002, 4, 3343–3346. [Google Scholar] [CrossRef]
- Giacomelli, G.; Porcheddu, A.; Salaris, M. Simple One-Flask Method for the Preparation of Hydroxamic Acids. Org. Lett. 2003, 5, 2715–2717. [Google Scholar] [CrossRef]
- Porcheddu, A.; Giacomelli, G. Angeli—Rimini’s Reaction on solid support: A new approach to hydroxamic acids. J. Org. Chem. 2006, 71, 7057–7059. [Google Scholar] [CrossRef]
- Reddy, A.S.; Kumar, M.S.; Reddy, G.R. A convenient method for the preparation of hydroxamic acids. Tetrahedron Lett. 2000, 41, 6285–6288. [Google Scholar] [CrossRef]
- Volonterio, A.; Bellosta, A.S.; Bravo, P.; Canavesi, M.; Corradi, E.; Meille, S.V.; Monetti, M.; Moussier, N.; Zanda, M. Solution/Solid-Phase synthesis of partially modified Retro- and Retro-Inverso-ψ[NHCH (CF3)]-Peptidyl hydroxamates and their evaluation as MMP-9 inhibitors. Eur. J. Org. Chem. 2002, 2002, 428–438. [Google Scholar] [CrossRef]
- Price, S.; Osbourn, S.E. Solid-Phase Synthesis of N-Formylhydroxylamines (Reverse/RetroHydroxamates). Org. Lett. 2005, 7, 3761–3763. [Google Scholar] [CrossRef]
- Codd, R. Traversing the coordination chemistry and chemical biology of hydroxamic acids. Coord. Chem. Rev. 2008, 252, 1387–1408. [Google Scholar] [CrossRef]
- Mishra, H.; Parrill, A.L.; Williamsom, J.S. Three-dimensional quantitative structure-activity relationship and comparative molecular field analysis of dipeptide hydroxamic acid. Antimicrob. Agents Chemother. 2002, 46, 2613–2618. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Houtman, J.C.; Witiak, D.T.; Seltzer, J.; Bertics, P.J.; Lauhon, C.T. Design, combinatorial chemical synthesis and in vitro characterization of novel urea basedgelatinase inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 2823–2826. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Itakura, H.; Sato, K.; Fukuyama, K.; Miura, S.; Takahashi, S.; Ikezawa, H.; Hosoya, T. Binding of salicylhydroxamic acid and several aromatic donor molecules toArthromyces ramosus peroxidase, investigated by X-ray crystallography, Optical differencespectroscopy, NMR relaxation, molecular dynamics, and kinetics. Biochemistry 1999, 38, 12558–12568. [Google Scholar] [CrossRef]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: current status and future prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef]
- Hidalgo, M.; Eckhardt, S.G. Development of matrix metalloproteinase inhibitors incancer Therapy. J. Natl. Cancer Inst. 2001, 93, 178–193. [Google Scholar] [CrossRef]
- Albrecht-Gary, A.M.; Libman, J.; Shanzer, A. Biomimetic iron(l11) trishydroxamate complexes and triple stranded diferric helices. Pure Appl. Chem. 1996, 68, 1243–1247. [Google Scholar] [CrossRef]
- Hara, Y.; Shen, L.; Tsubouchi, A.; Akiyama, M.; Umemoto, K. Tripodal peptide hydroxamates as siderophore models. iron(III) binding with ligands containing H-(Alanyl)n-β-(N-hydroxy)alanyl strands (n = 1−3) anchored by nitrilotriacetic acid. Inorg. Chem. 2000, 39, 5074–5082. [Google Scholar]
- Munster, P.N.; Troso-Sandoval, T.; Rosen, N.; Rifkind, R.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001, 61, 8492–8497. [Google Scholar]
- Steward, W.P.; Thomas, A.L. Marimastat: the clinical development of a matrix metallopro-teinase inhibitor. Expert Opin. Invest. Drugs 2000, 9, 2913–2922. [Google Scholar] [CrossRef]
- Jeng, A.Y.; De Lombaert, S. Endothelin converting enzyme inhibitors. Curr. Pharm. Des. 1997, 3, 597–614. [Google Scholar]
- Torres, G. Hydroxyurea, a potential new anti-HIV agent. GMHC Treatment Issues 1995, 9, 7–9. [Google Scholar]
- Parvathy, S.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Alzheimer's Amyloid Precursor Protein α-Secretase Is Inhibited by Hydroxamic Acid-Based Zinc Metalloprotease Inhibitors: Similarities to the Angiotensin Converting Enzyme Secretase. Biochem. 1998, 37, 1680–1685. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Iron Chelators as antimalarials: the biochemical basis of selective cytotoxicity. Parasitol. Today 1995, 11, 74–78. [Google Scholar] [CrossRef]
- Tsafack, A.; Golenser, J.; Libman, J.; Shanzer, A.; Cabantchik, Z.I. Mode of action of iron(III) chelators as antimalarials. III. Over additive effects in the combined action of hydroxamate-based agents on in vitro growth of Plasmodium falciparum. Mol. Pharmacol. 1995, 47, 403–409. [Google Scholar]
- Valapour, M.; Gou, J.; Schroeder, J.T.; Keen, J.; Cianferoni, A.; Casolaro, V.; Georas, S.N. Histone deacetylation inhibits IL-4 gene expression in T cells. J. Allergy Clin. Immunol. 2002, 109, 238–245. [Google Scholar] [CrossRef]
- Drovetskaia, T.V.; Yashina, N.S.; Leonova, T.V.; Petrosyan, V.S.; Lorberth, J.; Wocadlo, S.; Massa, W.; Pebler, J. Synthesis and structure of some Diethyl and Diphenyltin Bis-Hydroxamates. J. Organomet. Chem. 1996, 507, 201–205. [Google Scholar] [CrossRef]
- Smith, F.E.; Khoo, L.E.; Goh, N.K.; Hynes, R.C.; Eng, G. The synthesis, and molecular and crystal structure of diphenyl(2-oxidonaphthylmethyl-iminoacetato)tin(lV). Can. J. Chem. 1996, 74, 2041–2047. [Google Scholar] [CrossRef]
- Pellerito, L.; Nagy, L. Organotin(IV) complexes formed with biologically active ligands: Equilibrium and structural studies and some biological aspects. Chem. Rev. 2002, 224, 111–150. [Google Scholar]
- Gielen, M. Organotin compounds and their therapeutic potential: a report from the Organo-metallic Chemistry Department of the Free University of Brussels. Appl. Organomet. Chem. 2002, 16, 481–494. [Google Scholar] [CrossRef]
- Nath, M.; Pokharia, S.; Yadav, R. Organotin(IV) complexes of amino acids and peptides. Coord. Chem. Rev. 2001, 215, 99–149. [Google Scholar] [CrossRef]
- Petrosyan, V.S.; Yashina, N.S.; Ponomarev, S.V. Syntheses, Structures and biological activites of Organogermaniun and Organotin derivatives of hydroxamic acids. Met. Based Drugs 1998, 5, 237–244. [Google Scholar] [CrossRef]
- Shang, X.M.; Wu, J.Z.; Li, Q.S. New Coordination Modes of Substituted Benzohydroxa-mic Acid with Dialkyltin(IV): Structural diversity through Ligand Isomerization. Eur. J. Inorg. Chem. 2006, 2006, 4143–4150. [Google Scholar] [CrossRef]
- Harrison, P.G.; King, T.J.; Phillips, R.C. Structural studies in main-group chemistry. Part XVI. Crystal and molecular structures of bis(N-acetylhydroxylamino)dimethyltin(IV) and its monohydrate. J. Chem. Soc. Dalton Trans. 1976, 33, 2317–2321. [Google Scholar]
- Omae, I. Organotin antifouling paints and their alternatives. App. Organomet. Chem. 2003, 17, 81–105. [Google Scholar] [CrossRef]
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar] [CrossRef]
- Jensen, K.G.; Andersen, O.; Ronne, M. Organotin compounds induce aneuploidy in human peripheral lymphocytes in vitro. Mutat. Res. 1999, 246, 109–112. [Google Scholar]
- Choa, J.S.; Wei, L.Y.; Huang, M.C.; Liang, S.C.; Chen, H.H.C. Genotoxic effects oftriphenyltin acetate and triphenyltin hydroxide on mammalian cells in vitro and in vivo. Mutat. Res. 1999, 444, 167–174. [Google Scholar] [CrossRef]
- White, J.S.; Tobin, J.M.; Cooney, J.J. Organotin compounds and their interactions with microorganisms. Can. J. Microbiol. 1999, 45, 541–554. [Google Scholar] [CrossRef]
- Wong, P.T.S.; Chau, Y.K.; Kramar, O.; Bengert, G.A. Structure–toxicity relationship of tin compounds on Algae. Can. J. Fish. Aquat. Sci. 1982, 39, 483–488. [Google Scholar] [CrossRef]
- Berg, M.; Arnold, C.G.; Muller, S.R.; Muhlemann, J.; Schwarzenbach, R.P. Sorption and desorption behavior of organotin compounds in sediment−pore water systems. Environ. Sci. Technol. 2001, 35, 3151–3157. [Google Scholar] [CrossRef]
- Gielen, M. Tin-Based Antitumour Drugs. Coord. Chem. Rev. 1996, 151, 41–51. [Google Scholar]
- Gielen, M.; Biesemans, M.; Willem, R. Organotin compounds: from kinetics to stereo-chemistry and antitumour activities. Appl. Organomet. Chem. 2005, 19, 440–450. [Google Scholar] [CrossRef]
- Baul, T.; Masharing, C.; Ruisi, G.; Jirasko, R.; Holcapek, M.; De Vos, D.; Wolstenholme, D.; Linden, A. Self-assembly of extended Schiff base amino acetate keletons,2-{[(2Z)-(3-hydroxy-1-Methyl-2-butenylidene)]amino}Phenylpropio-nate and 2-{[(E)-1-(2-hydroxy-aryl) alkylidene] amino} Phenyl-propionate skeletons incorporating organotin(IV) moieties: Synthesis, spectroscopic characterization, crystal structures, and in vitro cytotoxic activity. J. Organomet. Chem. 2007, 692, 4849–4862. [Google Scholar] [CrossRef]
- Saad, E.; Farina, Y.; Baba, I.; Othman, H. Synthesis and characterization of some diorganotin(IV) bis(N-methyl o-nitrobenzohydroxamate). Sains Malays. 2003, 32, 79–86. [Google Scholar]
- Shahid, S.; Ali, S.; Hussain, M.; Mazhar, M.; Mahmood, S.; Rehman, S. Synthesis, characterization and thermal analysis of organotin(IV) derivatives of 4-(N-Maleoyl) butanoate. Turk. J. Chem. 2002, 26, 589–597. [Google Scholar]
- Li, Q.S.; Guedes da Silva, M.F.C.; Zhao, J.H.; Pombeiro, A.J.L. Diorganotin(IV) derivatives of arylhydroxamic acids: Synthesis, Properties and antitumor activity. J. Organomet. Chem. 2004, 689, 4584–4591. [Google Scholar] [CrossRef]
- Lockhart, T.P.; Manders, W.F. Structural determination by NMR spectroscopy. Dependence of [2J(119Sn,1H)] on the Me-Sn-Me angle in methyltin (IV) compounds. Inorg. Chem. 1986, 25, 892–895. [Google Scholar]
- Nadvornik, M.; Holeček, J.; Handlir, K.; Lycka, A. The [13]C and [119]Sn NMR spectra of Some four- and five-coordinate tri-n-butyltin (IV) compounds. J. Organomet. Chem. 1984, 275, 43–51. [Google Scholar] [CrossRef]
- Mohammad, M.; Khadija, S.; Sohail, M.; Saqib, A.; Moazzam, B. Synthesis, Spectral Characterization and Biological Applications of Tri- and Diorganotin(IV) Derivatives of 2[N-(2,6- Dichloro-3-methyl phenyl)amino]benzoic acid. Turk. J. Chem. 2004, 28, 17–26. [Google Scholar]
- Das, M.K.; Nath, M.; Zuckermann, J.J. Di- And Triorganotin (IV) Derivatives of N, N-Substituted hydroxylamines. Inorg. Chem. Acta 1983, 71, 49–59. [Google Scholar] [CrossRef]
- Ahmed, F.; Ali, S.; Parvez, M.; Munir, A.; Mazhar, M.; Shah, T.A. Synthesis, Characterization and biological studies of tri- and diorganotin (IV) complexes with 2,4-difluoro-4-hydroxy-[1,1]-biphenyle-3-carbolic acid: Crystal structure of [(CH3)3Sn(C13H7O3F2)]. Heteroatom. Chem. 2002, 13, 638–642. [Google Scholar] [CrossRef]
- Ali, S.; Ahmad, F.; Mazhar, M.; Munir, A.; Masood, M.T. Synthesis and spectral studies of di- and triorganotin (IV) complexes with 2-(6-methoxynaphthyl) propionic acid (Naproxen). Synth. React. Inorg. Met-Org. Chem. 2001, 32, 357–372. [Google Scholar]
- Yoder, C.H.; Margolis, L.A.; Horne, J.M. A Tin-119 NMR investigation of phosphine and phosphine oxide adducts of organotin chlorides. J. Organomet. Chem. 2001, 633, 33–38. [Google Scholar] [CrossRef]
- Holecek, J.; Nadvornik, M.; Handlír, K.; Lycka, A. 13C and 119Sn NMR spectra of di-n-butyltin(IV) compounds. J. Organomet. Chem. 1986, 315, 299–308. [Google Scholar] [CrossRef]
- Choudhary, M.A.; Mazhar, M.; Ali, S.; Song, X.; Eng, G. Synthesis, characterization and biological activity of dimethyltin dicarboxylates containing germanium. Met. Based Drugs 2002, 8, 275–281. [Google Scholar] [CrossRef]
- Harrison, P.G. Chemistry of Tin; Harrison, P.G, Ed.; Chapman and Hall: New York, NY, USA, 1989; pp. 76 and references therein. [Google Scholar]
- Nath, M.; Yadav, R.; Gielen, M.; Dalil, H.; de Vos, D.; Eng, G. Synthesis, Characteristic spectral studies and in vitro antimicrobial and antitumour activities of organotin (IV) complexes of Schiff bases derived from amino acids. App. Organomet. Chem. 1997, 11, 727–736. [Google Scholar] [CrossRef]
- Harrison, P.G.; King, T.J.; Richards, J.A. Structural studies in main group chemistry. Part VIII. The crystal and molecular structure of Bis(N-methyl acetylhydroxylamino) dimethyltin(IV). J. Chem. Soc. Dalton Trans. 1975, 9, 826–830. [Google Scholar]
- Shang, X.M.; Li, Q.S.; Wu, J.Z. Synthesis and crystal structure of a mixed-ligand compound di-n-butyl-(4-chlorobenzohydroxamato)tin(IV). J. Organomet. Chem. 2005, 17, 3997–4000. [Google Scholar] [CrossRef]
- Gomez-Ruiz, S.; Kaluderovic, G.N.; Prashar, S.; Hey-Hawkins, E.; Eric, A.; Zizak, Z.; Juranic, Z.D. Study of the cytotoxic activity of di and triphenyltin(IV) carboxylate complexes. J. Inorg. Biochem. 2008, 102, 2087–2096. [Google Scholar] [CrossRef]
- Gielen, M.; Handlir, K.; Hollein, M.; De Vos, D. Synthesis, characterization and anti-tumour activity of some butyltin (IV) cysteaminates and N,N-dimethyl-cysteaminates. Met. Based Drugs 2000, 7, 233–236. [Google Scholar] [CrossRef]
- Gielen, M.; Biesemans, M.; De Vos, D.; Willem, R. Synthesis, characterization and in vitro antitumor activity of di- and triorganotin derivatives of polyoxa- and biologically relevant carboxylic acids. J. Inorg. Biochem. 2000, 79, 139–145. [Google Scholar] [CrossRef]
- Xanthopoulou, M.N.; Hadjikakou, S.K.; Hadjiliadis, N.; Schurmann, M.; Jurkschat, K.; Michaelides, A.; Skoulika, S.; Bakas, T.; Binolis, J.; Karkabounas, S.; et al. Synthesis, Structural characterization and in vitro cytotoxicity of organotin(IV) derivatives of heterocyclic thioamides, 2-mercaptobenzothiazole, 5-chloro-2-mercaptobenzothiazole, 3- methyl-2-mercaptobenzothiazole and 2-mercaptonicotinic acid. J. Inorg. Biochem. 2003, 96, 425–434. [Google Scholar] [CrossRef]
- Sharma, N.; Puneet, P.; Sharma, V.; Bhatt, S.S.; Chaudhary, S.C. Synthesis, characterization and thermal study of di- and triphenyltin(IV) complexes of p-chloro and p-nitro and benzohydroxamic acid. Synth. Reactiv. Inorg. Metal-Org. C 1997, 27, 1381–1397. [Google Scholar]
- APEX2 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2008.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
- Sheldrick, G.M. SHELXS-97, Program for Crystal Structure Determination; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Meth. 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the ligand and complexes are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Naqeebullah; Farina, Y.; Chan, K.M.; Mun, L.K.; Rajab, N.F.; Ooi, T.C. Diorganotin(IV) Derivatives of N-Methyl p-Fluorobenzo-Hydroxamic Acid: Preparation, Spectral Characterization, X-ray Diffraction Studies and Antitumor Activity. Molecules 2013, 18, 8696-8711. https://doi.org/10.3390/molecules18078696
Naqeebullah, Farina Y, Chan KM, Mun LK, Rajab NF, Ooi TC. Diorganotin(IV) Derivatives of N-Methyl p-Fluorobenzo-Hydroxamic Acid: Preparation, Spectral Characterization, X-ray Diffraction Studies and Antitumor Activity. Molecules. 2013; 18(7):8696-8711. https://doi.org/10.3390/molecules18078696
Chicago/Turabian StyleNaqeebullah, Yang Farina, Kok Meng Chan, Lo Kong Mun, Nor Fadilah Rajab, and Theng Choon Ooi. 2013. "Diorganotin(IV) Derivatives of N-Methyl p-Fluorobenzo-Hydroxamic Acid: Preparation, Spectral Characterization, X-ray Diffraction Studies and Antitumor Activity" Molecules 18, no. 7: 8696-8711. https://doi.org/10.3390/molecules18078696
APA StyleNaqeebullah, Farina, Y., Chan, K. M., Mun, L. K., Rajab, N. F., & Ooi, T. C. (2013). Diorganotin(IV) Derivatives of N-Methyl p-Fluorobenzo-Hydroxamic Acid: Preparation, Spectral Characterization, X-ray Diffraction Studies and Antitumor Activity. Molecules, 18(7), 8696-8711. https://doi.org/10.3390/molecules18078696