Antitumor, Antioxidant and Antimicrobial Studies of Substituted Pyridylguanidines

Abstract
:1. Introduction
2. Results and Discussions
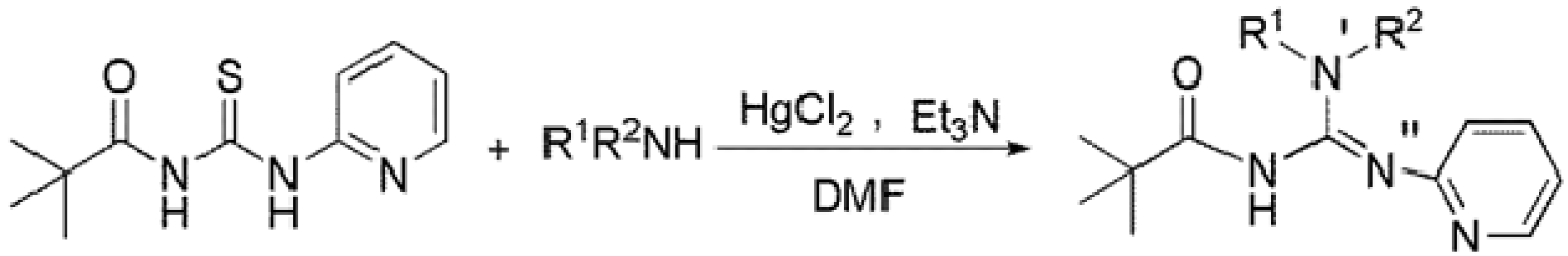
2.1. Synthesis and Characterization

2.1.1. IR Spectra

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | 1H | 13C | |||
|---|---|---|---|---|---|---|---|
| NH | NH | CN3 | C=O | ||||
| 1 | Phenyl | H | 11.39 | 14.52 | 161.6 | 180.6 | |
| 2 | 2-chlorophenyl | H | 11.91 | 14.49 | 161.2 | 180.2 | |
| 3 | 3-chlorophenyl | H | 11.32 | 14.46 | 161.3 | 180.9 | |
| 4 | 4-chlorophenyl | H | 11.26 | 14.48 | 161.3 | 180.7 | |
| 5 | 2-methoxyphenyl | H | 11.93 | 14.54 | 161.8 | 179.9 | |
| 6 | 4-tolyl | H | 11.36 | 14.53 | 161.8 | 180.6 | |
| 7 | 2-fluorophenyl | H | 11.79 | 14.47 | 161.3 | 180.5 | |
| 8 | n-propyl | H | 9.02 | 14.59 | 162.7 | 180.3 | |
| 9 | iso-propyl | H | 8.99 | 14.57 | 162.7 | 180.4 | |
| 10 | iso-butyl | H | 8.98 | 14.56 | 162.7 | 180.4 | |
| 11 | 2-pyridyl | H | 12.08 | 14.36 | 161.3 | 180.0 | |
| 12 | n-propyl | n-propyl | 12.27 | ---- | 161.8 | 176.7 | |
2.1.2. NMR Spectra
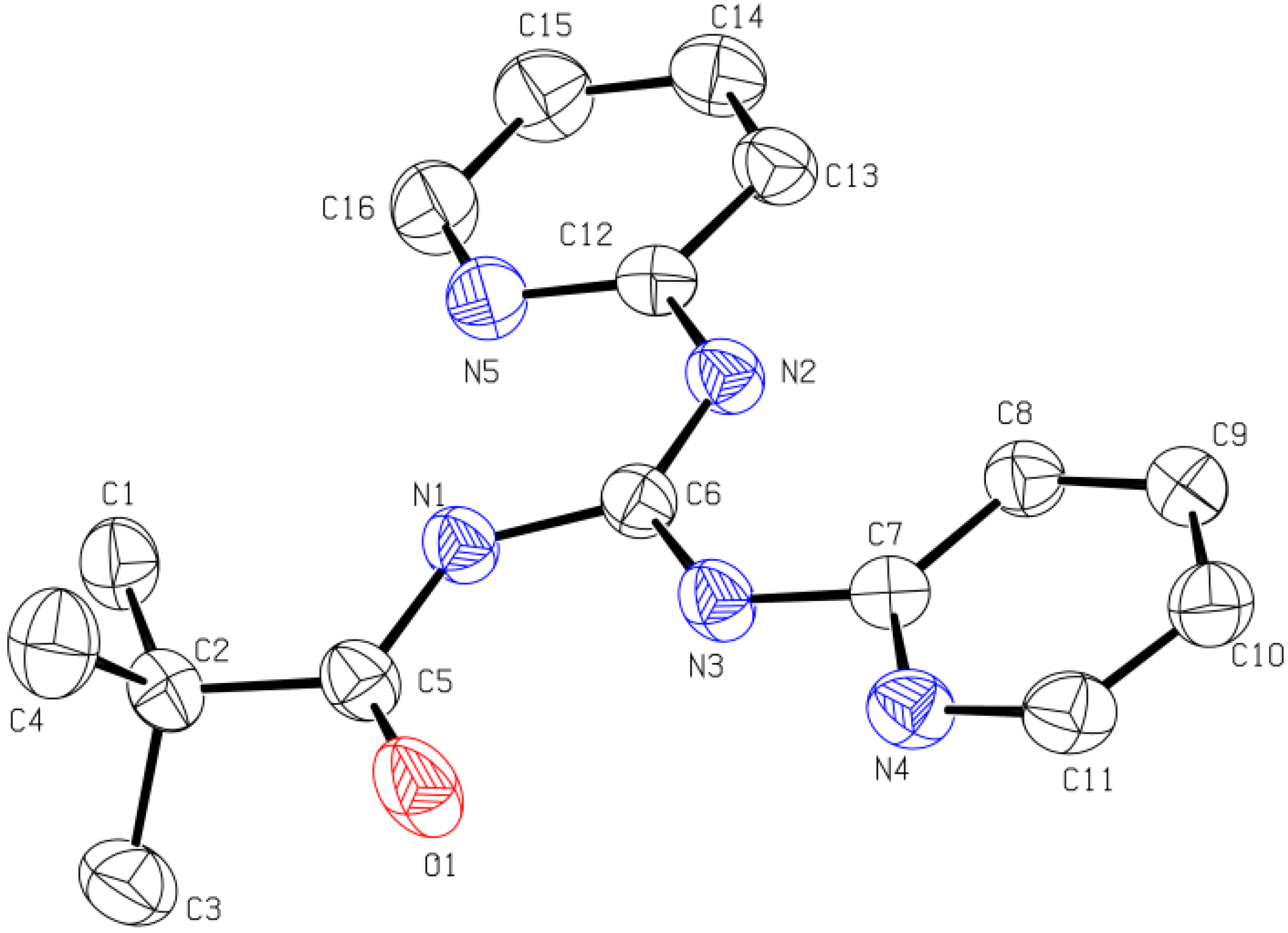
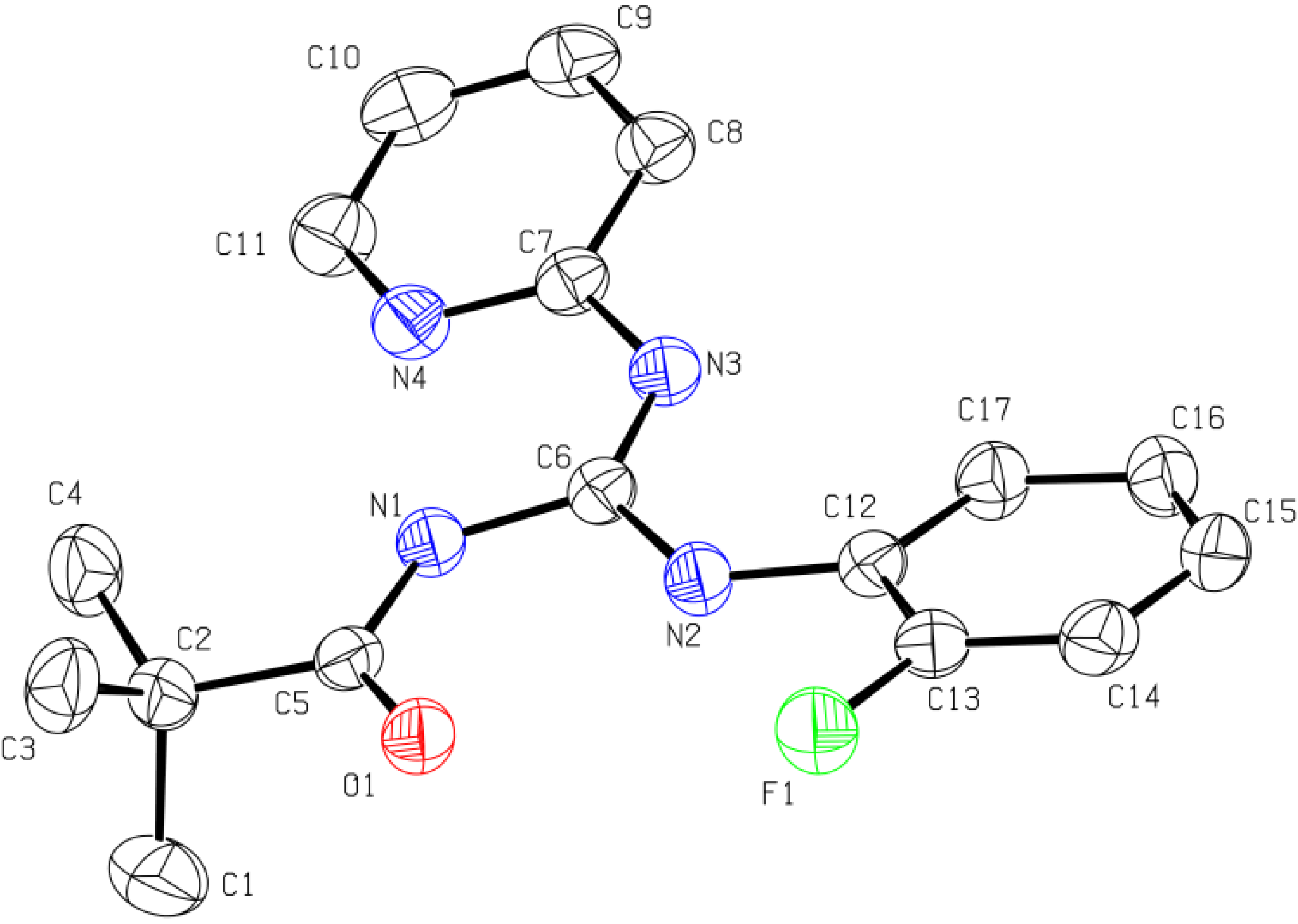
2.1.3. X-ray Diffraction Analysis
| Crystal parameters | 7 | 11 | |
|---|---|---|---|
| Empirical formula | C17H19N4OF | C16H19N5O | |
| Formula weight | 314.36 | 297.35 | |
| Temperature (K) | 296 | 200 | |
| Wavelength (Å) | 1.54178 | 1.54178 | |
| Crystal system | Monoclinic | Monoclinic | |
| Space group | P2(1)/n | P2(1)/n | |
| Unit cell dimensions | a (Å) | 10.7110(2) | 6.00430(10) |
| b(Å) | 9.9954(2) | 16.9550(2) | |
| c(Å) | 15.2679(4) | 15.2900(2) | |
| α(°) | 90 | 90 | |
| β(°) | 91.0430(10) | 92.4800(10) | |
| γ(°) | 90 | 90 | |
| V (Å3), Z | 1634.32(6),4 | 1555.11(4),4 | |
| Density (calcd) (g/cm3) | 1.278 | 1.270 | |
| Crystal size(mm3) | 0.10 × 0.08 × 0.08 | 0.14 × 0.12 × 0.10 | |
| Index ranges | −13 <= h <= 12 −12 <= k <= 12 −18 <= l <= 17 | −7 <= h <= 7 −20 <= k <= 20 −18 <= l <= 18 | |
| F(000) | 664 | 632 | |
| Total reflections | 21339 | 20265 | |
| Independent reflections | 3214 [Rint= 0.045] | 3032[Rint= 0.036] | |
| R indices (all data) | R1 = 0.0516 wR2 = 0.1220 | R1 = 0.0464, wR2 = 0.1154 | |
| Final R indices [I > 2σ(I)] | R1 = 0.0445, wR2 = 0.1141 | R1 = 0.0402, wR2 = 0.1093 | |
| Goodness-of-fit | 1.048 | 1.042 | |
| Theta range for data collection (°) | 5.00 to 72.64 | 3.89 to 72.53 | |
| Compound | Bond lengths (Å) | Bond angles (°) | Torsion angles (°) | |
|---|---|---|---|---|
| 7 | C5-O1 1.2306(17) | C6-N1 1.4034(18) | N1-C6-N2 114.25(12) | O1-C5-N1-C6 1.0(2) |
| C6-N2 1.3613(17) | C6-N3 1.2929(18) | N1-C6-N3 123.83(12) | C5-N1-C6-N2 4.8(2) | |
| C5-N1 1.3644(18) | C7-N3 1.3985(17) | N2-C6-N3 121.92(13) | C5-N1-C6-N3 -175.45(14) | |
| C12-N2 1.4027(17) | C2-C5 1.526(2) | C5-N1-C6 128.79(12) | C6-N3-C7-N4 -5.5(2) | |
| 11 | C5-O1 1.2263(15) | C5-N1 1.3653(15) | N1-C6-N2 123.91(11) | O1-C5-N1-C6 0.2(2) |
| C6-N1 1.4017(15) | C6-N2 1.2918(15) | N1-C6-N3 114.67(10) | C5-N1-C6-N2 175.27(12) | |
| C6-N3 1.3623(15) | C7-N3 1.4003(15) | N2-C6-N3 121.42(11) | C5-N1-C6-N3 -5.09(18) | |
| C12-N2 1.3925(15) | C2-C5 1.5309(16) | C5-N1-C6 129.02(10) | C6-N3-C7-N4 177.02(12) | |
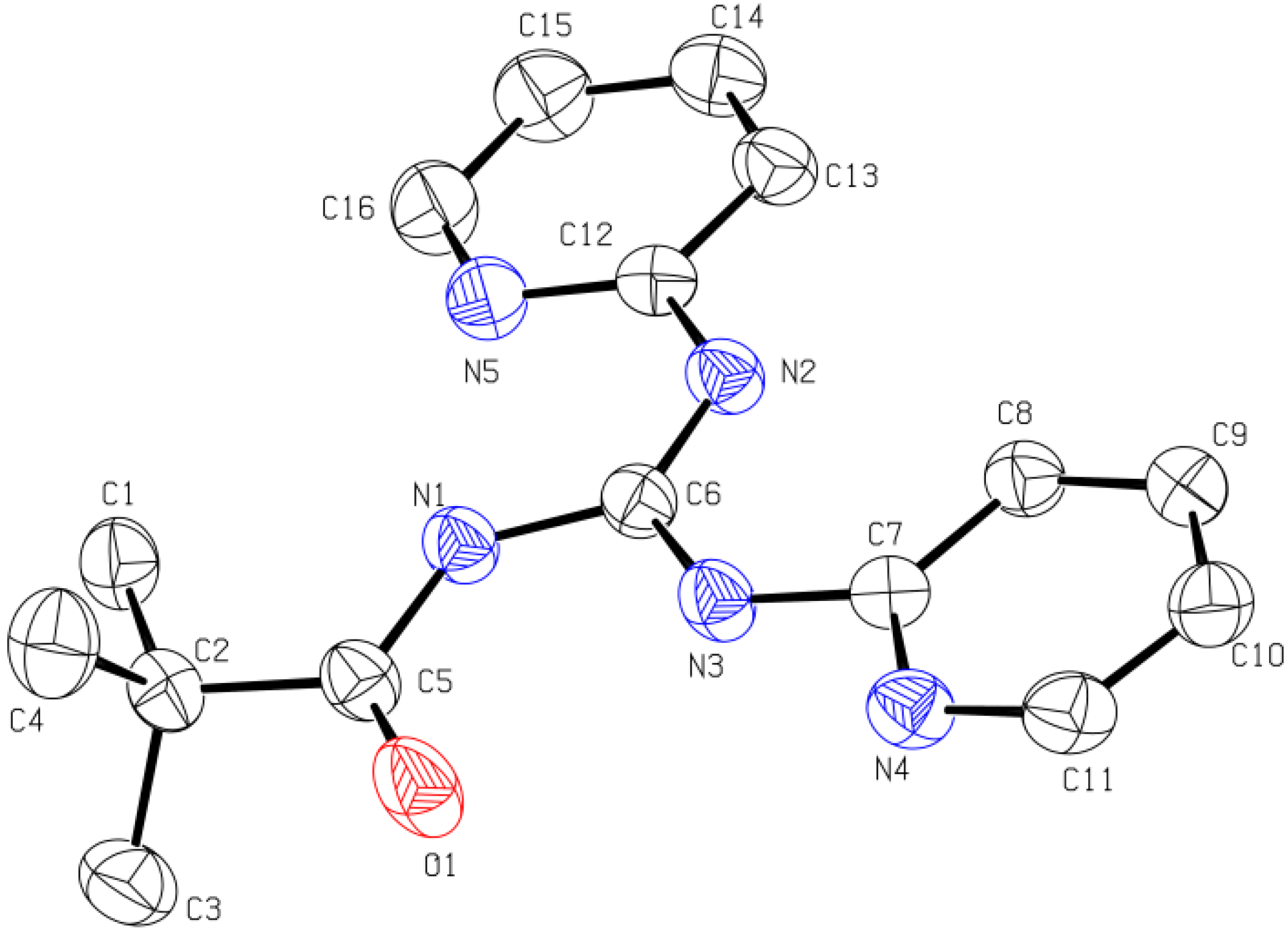
2.2. Biological Studies
2.2.1. Potato Disc Anti-Tumor Assay
| Compound | Average number of tumors per disc | % Inhibition of tumors |
|---|---|---|
| 1 | 2.0 | 73 |
| 2 | 1.5 | 80 |
| 3 | 2.0 | 73 |
| 4 | 3.0 | 60 |
| 5 | 2.0 | 73 |
| 6 | 1.5 | 80 |
| 7 | 2.0 | 73 |
| 8 | 3.0 | 60 |
| 9 | 2.0 | 73 |
| 10 | 2.0 | 73 |
| 11 | 3.0 | 60 |
| 12 | 1.5 | 73 |
| AT10 | 7.5 | --- |
| Blank | 00 | 100 |
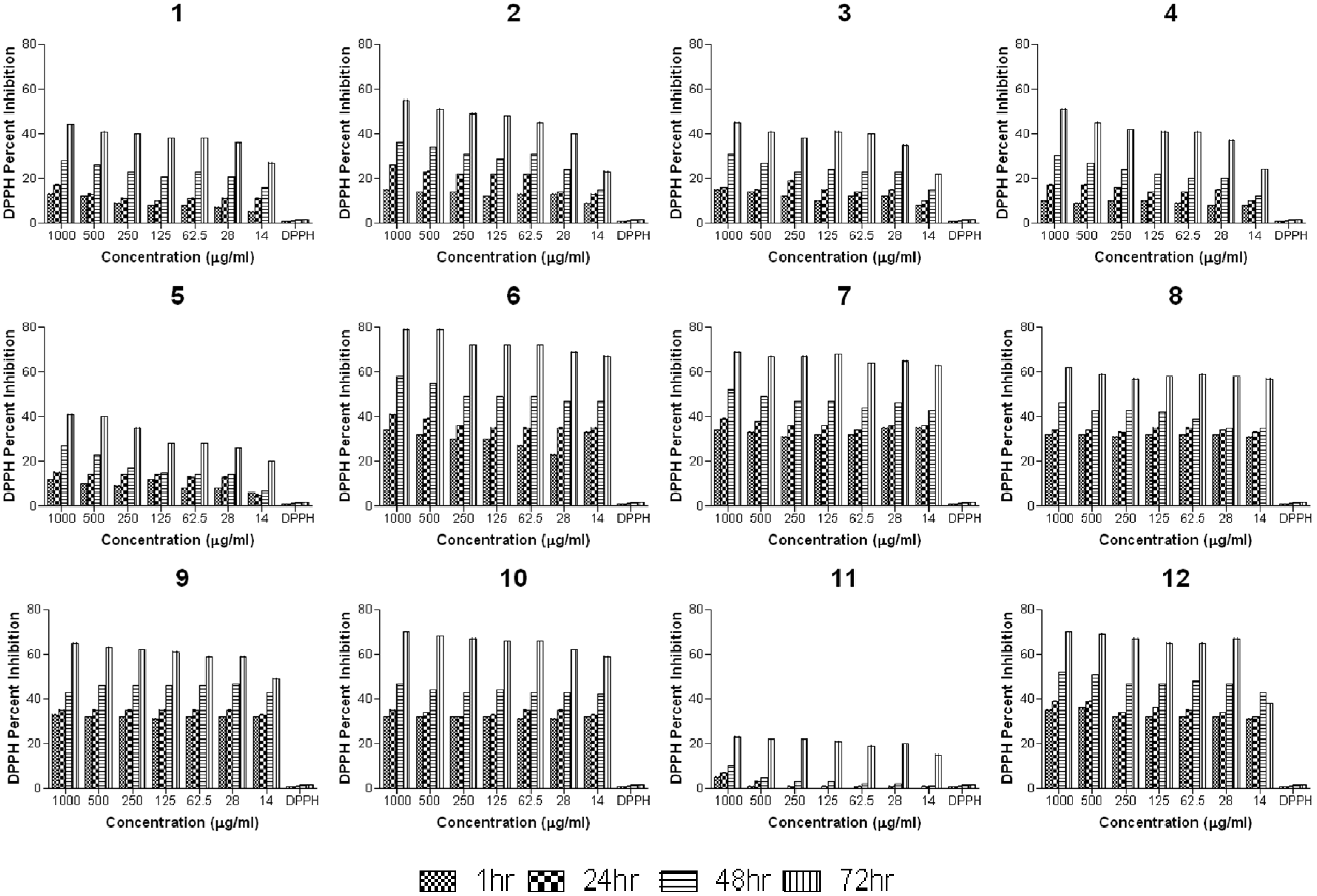
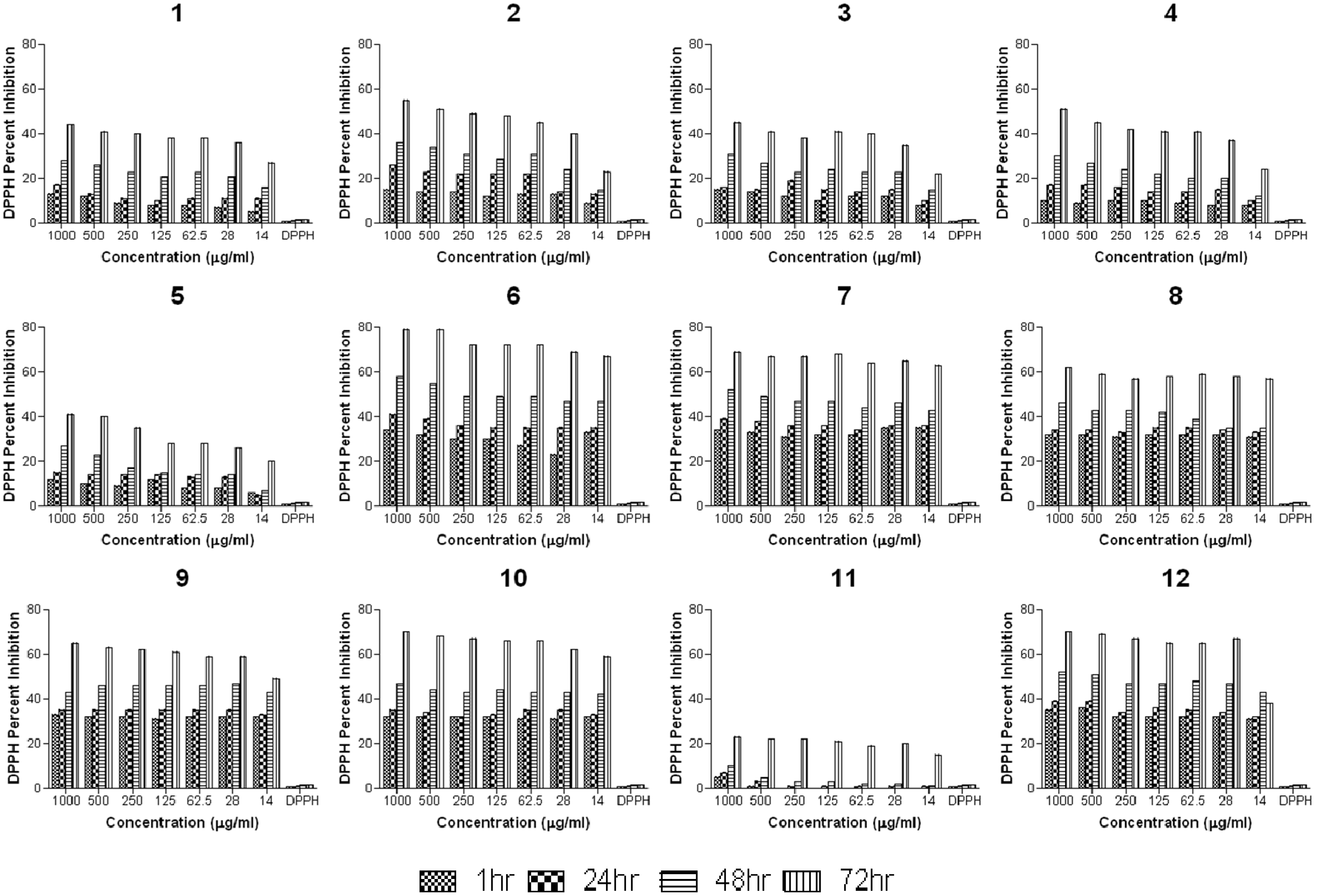
2.2.2. Anti-Oxidant Study
2.2.3. Antifungal Activity
| Compound | Mean Values of Percent Growth Inhibition | ||||
|---|---|---|---|---|---|
| A. flavus | A. niger | F. solani | M. species | A. fumagatus | |
| 1 | 10 ± 1.5 | 12.5 ± 2.0 | 20 ± 1.0 | --- | 47.5 ± 1.5 |
| 2 | --- | 72.5 ± 1.5 | 62.5 ± 2.5 | --- | 5 ± 0 |
| 3 | --- | 2.5 ± 1.5 | 20 ± 2.0 | --- | --- |
| 4 | --- | 79 ± 1.0 | 32.5 ± 1.5 | 25 ± 1.0 | --- |
| 5 | --- | 66.5 ± 1.5 | 65.3 ± 1.7 | --- | 42.5 ± 2.4 |
| 6 | --- | 72.5 ± 0.5 | 62.5 ± 0.5 | 5 ± 0.5 | 55 ± 1.0 |
| 7 | --- | 70 ± 2.0 | --- | 50 ± 1.0 | 5 ± 1.0 |
| 8 | 64.5 ± 1.5 | 42.5 ± 2.5 | 62.5 ± 1.5 | 40 ± 2 | 20 ± 0.5 |
| 9 | --- | 32.5 ± 0.5 | 40 ± 2 | 12.5 ± 0.5 | 12.5 ± 0.5 |
| 10 | --- | 27.5 ± 0.5 | 47.5 ± 1.5 | --- | 20 ± 1 |
| 11 | --- | --- | 25 ± 1.0 | --- | 30 ± 2 |
| 12 | --- | --- | 39.5 ± 1.5 | --- | 10 ± 0 |
| Terbinafine | 100 | 100 | 100 | 100 | 100 |
| Vehicle control | --- | --- | --- | --- | --- |
2.2.4. Antibacterial Activity
| Compound | Mean zone of inhibition (mm) | |||||
|---|---|---|---|---|---|---|
| M. luteus | S. aureus | K. pneumoniae | E. aerogenes | E. coli | B. bordetella | |
| 1 | --- | --- | --- | --- | --- | --- |
| 2 | 15 ± 0.5 | 12 ± 0.8 | --- | --- | 15 ± 1.0 | 16 ± 0.6 |
| 3 | --- | --- | --- | --- | --- | --- |
| 4 | --- | --- | --- | --- | --- | --- |
| 5 | --- | --- | --- | --- | --- | --- |
| 6 | --- | --- | --- | --- | 18 ± 1.2 | --- |
| 7 | --- | --- | --- | --- | --- | --- |
| 8 | 15 ± 1.5 | 15 ± 0.5 | --- | --- | --- | 13 ± 1.5 |
| 9 | 14 ± 0.6 | 13 ± 1.2 | --- | 12 ± 0.2 | 12 ± 0.4 | --- |
| 10 | 15 ± 0.5 | 15 ± 0.7 | 13 ± 0.4 | 14 ± 0.8 | 17 ± 1.5 | 15 ± 0.7 |
| 11 | --- | --- | --- | --- | --- | --- |
| 12 | 12 ± 0.8 | 12 ± 0.6 | 12 ± 0.4 | --- | 16 ± 0.8 | --- |
| Cefixime | 25 ± 1.0 | 22 ± 0.6 | 22 ± 0.5 | 23 ± 1.0 | 25 ± 1.0 | 20 ± 0.5 |
| Roxythromycine | 30 ± 1.5 | 25 ± 1 | 30 ± 2.0 | 25 ± 1.0 | 26 ± 2.0 | 25 ± 1.0 |
| Compound | Minimum inhibitory concentration (MIC) mg/mL DMSO | |||||
|---|---|---|---|---|---|---|
| M. luteus | S. aureus | K. pneumoniae | E. aerogenes | E. coli | B. bordetella | |
| 1 | --- | --- | --- | --- | --- | --- |
| 2 | 0.0312 | 0.0078 | --- | --- | 0.0019 | 0.25 |
| 3 | --- | --- | --- | --- | --- | --- |
| 4 | --- | --- | --- | --- | --- | --- |
| 5 | --- | --- | --- | --- | --- | --- |
| 6 | --- | --- | --- | --- | 0.25 | --- |
| 7 | --- | --- | --- | --- | --- | --- |
| 8 | 1 | 1 | --- | --- | --- | 1 |
| 9 | 0.25 | 0.0156 | --- | 0.5 | 1 | --- |
| 10 | 0.5 | 0.25 | 1 | 1 | 0.5 | 0.5 |
| 11 | --- | --- | --- | --- | --- | --- |
| 12 | 0.5 | 0.125 | 0.0019 | --- | 0.0312 | --- |
3. Experimental
3.1. General
3.2. Synthesis
3.3. Biological Screening Protocols
3.3.1. Potato Disc Anti-Tumor Assay
- a.
- Positive Control; prepared by taking 150 µL of DMSO in autoclaved Eppendorfs and then adding 1,350 µL of autoclaved distilled water;
- b.
- Negative control; prepared by taking 150 µL of DMSO in autoclaved Eppendorfs, then adding 750 µL of autoclaved distilled water and 600 µL of bacterial culture;
- c.
- Blank potato discs used as control.

3.3.2. Anti-Oxidant Study

3.3.3. Antifungal Activity

3.3.4. Antibacterial Activity
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Singh, E.K.; Ravula, S.; Pan, C.M.; Pan, P.S.; Vasko, R.C.; Lapera, S.A.; Weerasinghe, S.V.W.; Pflum, M.K.H.; McAlpine, S.R. Synthesis and biological evaluation of histone deacetylase inhibitors that are based on FR235222: A cyclic tetrapeptide scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 2549–2554. [Google Scholar] [CrossRef]
- Hranjec, M.; Pavlovic, G.; Karminski-Zamola, G. Synthesis, crystal structure determination and antiproliferative activity of novel 2-amino-4-aryl-4,10-dihydro[1,3,5]triazino[1,2-a]benzimidazoles. J. Mol. Struct. 2012, 1007, 242–251. [Google Scholar] [CrossRef]
- Singh, N.; Pandey, S.K.; Anand, N.; Dwivedi, R.; Singh, S.; Sinha, S.K.; Chaturvedi, V.; Jaiswal, N.; Srivastava, A.K.; Shah, P.; et al. Synthesis, molecular modeling and bio-evaluation of cycloalkyl fused 2-aminopyrimidines as antitubercular and antidiabetic agents. Bioorg. Med. Chem. Lett. 2011, 21, 4404–4408. [Google Scholar] [CrossRef]
- Saczewski, F.; Balewski, L. Biological activities of guanidine compounds, 2008–2012 update. Expert Opin. Ther. Patents 2013. [Google Scholar] [CrossRef]
- Kumar, N.; Chauhan, A.; Drabu, S. Synthesis of cyanopyridine and pyrimidine analogues as new anti-inflammatory and antimicrobial agents. Biomed. Pharmacother. 2011, 65, 375–380. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Jones, R.; Bryant, C.; Boddeker, N.; Mabery, E.; Bahador, G.; Watson, J.; Clough, J.; Arimilli, M.; et al. SAR studies on dihydropyrimidinone antibiotics. Bioorg. Med. Chem. Lett. 2011, 21, 1670–1674. [Google Scholar] [CrossRef]
- Coghlan, D.R.; Bremner, J.B.; Keller, P.A.; Pyne, S.G.; David, D.M.; Somphol, K.; Baylis, D.; Coates, J.; Deadman, J.; Rhodes, D.I.; et al. Synthesis and antibacterial activity of some binaphthyl-supported macrocycles containing a cationic amino acid. Bioorg. Med. Chem. 2011, 19, 3549–3557. [Google Scholar] [CrossRef]
- Kumar, V.V.; Singh, S.K.; Sharma, S.; Bhaduri, A.P.; Gupta, S.; Zaidi, A.; Tiwari, S.; Katiyat, J.C. Design, synthesis and biological evaluation of 1,3-diaminopropanes: a new class of polyamine analogs as leishmanicidal agents. Bioorg. Med. Chem. Lett. 1997, 7, 675–680. [Google Scholar] [CrossRef]
- Pandey, S.; Suryawanshi, S.N.; Gupta, S.; Srivastava, V.M.L. Synthesis and antileishmanial profile of some novel terpenyl pyrimidines. Eur. J. Med. Chem. 2004, 39, 969–973. [Google Scholar] [CrossRef]
- Suryawanshi, S.N.; Bhat, B.A.; Pandey, S.; Chandra, N.; Gupta, S. Chemotherapy of leishmaniasis. Part VII: Synthesis and bioevaluation of substituted terpenyl pyrimidines. Eur. J. Med. Chem. 2007, 42, 1211–1217. [Google Scholar] [CrossRef]
- Chandra, N.; Ramesh; Ashutosh; Goyal, N.; Suryawanshi, S.N.; Gupta, S. Antileishmanial agents part-IV: synthesis and antileishmanial activity of novel terpenyl pyrimidines. Eur. J. Med. Chem. 2005, 40, 552–556. [Google Scholar] [CrossRef]
- Chandra, N.; Pandey, S.; Ramesh; Suryawanshi, S.N.; Gupta, S. hemotherapy of leishmaniasis part III: Synthesis and bioevaluation of novel aryl substituted terpenyl pyrimidines as antileishmanial agents. Eur. J. Med. Chem. 2006, 41, 779–785. [Google Scholar] [CrossRef]
- Sunduru, N.; Agarwal, A.; Katiyar, S.B.; Nishi; Goyal, N.; Gupta, S.; Chauhan, P.M.S. Synthesis of 2,4,6-trisubstituted pyrimidine and triazine heterocycles as antileishmanial agents. Bioorg. Med. Chem. 2006, 14, 7706–7715. [Google Scholar] [CrossRef]
- Singh, S.; Jhingran, A.; Sharma, A.; Simonian, A.R.; Soininen, P.; Vepsalainen, J.; Khomutov, A.R.; Madhubala, R. Novel agmatine analogue, γ-guanidinooxypropylamine (GAPA) efficiently inhibits proliferation of Leishmania donovani by depletion of intracellular polyamine levels. Biochem. Biophys. Res. Commun. 2008, 375, 168–172. [Google Scholar] [CrossRef]
- Hua, H.M.; Peng, J.; Dunbar, D.C.; Schinazi, R.F.; Andrews, A.G.C.; Cuevas, C.; Garcia-Fernandez, L.F.; Kelly, M.; Hamann, M.T. Batzelladine alkaloids from the caribbean sponge Monanchora unguifera and the significant activities against HIV-1 and AIDS opportunistic infectious pathogens. Tetrahedron 2007, 63, 11179–11188. [Google Scholar] [CrossRef]
- Hua, H.M.; Peng, J.; Fronczek, F.R.; Kellye, M.; Hamann, M.T. Crystallographic and NMR studies of antiinfective tricyclic guanidine alkaloids from the sponge Monanchora unguifera. Bioorg. Med. Chem. 2004, 12, 6461–6464. [Google Scholar] [CrossRef]
- Saczewski, F.; Kornicka, A.; Rybczynska, A.; Hudson, A.L.; Miao, S.S.; Gdaniec, M.; Boblewski, K.; Lehmann, A. 1-[(Imidazolidin-2-yl)imino]indazole. highly α2/I1 selective agonist: Synthesis, X-ray structure, and biological activity. J. Med. Chem. 2008, 51, 3599–3608. [Google Scholar] [CrossRef]
- Wender, P.A.; Galliher, W.C.; Goun, E.A.; Jones, L.R.; Pillow, T.H. The design of guanidinium-rich transporters and their internalization mechanisms. Adv. Drug Deliv. Rev. 2008, 60, 452–472. [Google Scholar] [CrossRef]
- Muller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.H.; Koepsell, H.; Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860. [Google Scholar] [CrossRef]
- Spannhoff, A.; Sippl, W.; Jung, M. Cancer treatment of the future: Inhibitors of histone methyltransferases. Int. J. Biochem. Cell Biol. 2009, 41, 4–11. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Kang, T.S.; Jo, H.O.; Park, W.K.; Kim, J.P.; Konishi, Y.; Kong, J.Y.; Park, N.S.; Jung, Y.S. Synthesis and antioxidant activities of 3,5-dialkoxy-4-hydroxycinnamamides. Bioorg. Med. Chem. Lett. 2008, 18, 1663–1667. [Google Scholar] [CrossRef]
- Noguchi, N. Novel insights into the molecular mechanisms of the antiatherosclerotic properties of antioxidants: the alternatives to radical scavenging. Free Radical Biol. Med. 2002, 33, 1480–1489. [Google Scholar] [CrossRef]
- Makeeva, A.V.; Popova, T.N.; Sukhoveeva, O.V.; Panchenko, L.F. Effects of guanidine derivatives on the activities of superoxide dismutase and catalase during postischemic reperfusion in the rat brain. Neurochem. J. 2010, 4, 217–221. [Google Scholar] [CrossRef]
- Chern, J.W.; Leu, Y.L.; Wang, S.S.; Jou, R.; Lee, C.F.; Tsou, P.C.; Hsu, S.C.; Liaw, Y.C; Lin, H.M. Synthesis and cytotoxic evaluation of substituted sulfonyl-N-hydroxyguanidine derivatives as potential antitumor agents. J. Med. Chem. 1997, 40, 2276–2286. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Saczewski, F.; Gdaniec, M. Synthesis, molecular structure and anticancer activity of 1-allyl-3-amino-2-(4-chloro-2-mercaptobenzenesulphonyl)guanidine derivatives. Eur. J. Med. Chem. 2002, 37, 285–293. [Google Scholar] [CrossRef]
- Kamal, A.; Khan, M.N.A.; Srikanth, Y.V.V.; Reddy, K.S.; Juvekar, A.; Sen, S.; Kurian, N.; Zingde, S. Synthesis, DNA-binding ability and evaluation of antitumour activity of triazolo[1,2,4]benzothiadiazine linked pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg. Med. Chem. 2008, 16, 7804–7810. [Google Scholar] [CrossRef]
- Dolzhenko, A.V.; Tan, B.J.; Dolzhenko, A.V.; Chiu, G.N.C.; Chui, W.K. Synthesis and biological activity of fluorinated 7-aryl-2-pyridyl-6,7-dihydro[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-amines. J. Fluorine Chem. 2008, 129, 429–434. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Castaneda, M.; Sanz, M.A.; Bautista, D.; Molina, P. An iminophosphorane-based approach for the synthesis of spiropyrrolidine–imidazole derivatives. Tetrahedron 2007, 63, 1849–1856. [Google Scholar] [CrossRef]
- Shchekotikhin, A.E.; Glazunova, V.A.; Dezhenkova, L.G.; Luzikov, Y.N.; Sinkevich, Y.B.; Kovalenko, L.V.; Buyanov, V.N.; Balzarini, J.; Huang, F.C.; Lin, J.J.; et al. Synthesis and cytotoxic properties of 4,11-bis[(aminoethyl)amino]anthra[2,3-b]thiophene-5,10-diones, novel analogues of antitumor anthracene-9,10-diones. Bioorg. Med. Chem. 2009, 17, 1861–1869. [Google Scholar] [CrossRef]
- Zarraga, M.; Zarraga, A.M.; Rodriguez, B.; Perez, C.; Paz, C.; Paz, P.; Sanhueza, C. Synthesis of a new nitrogenated drimane derivative with antifungal activity. Tetrahedron Lett. 2008, 49, 4775–4776. [Google Scholar] [CrossRef]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Trindade-Silva, A.E.; Romminger, S.; Morais, R.P.; Bandeira, K.; Mizuno, C.M. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2010, 27, 1871–1907. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Rodriguez, A.D.; Berlinck, R.G.S.; Hamann, M.T. Marine pharmacology in 2003–4: Marine compounds with anthelmintic antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiplatelet, antiprotozoal, antituberculosis, and antiviral activities; affecting the cardiovascular, immune and nervous systems, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. Part C. Pharmacol. Toxicol. 2007, 145, 553–581. [Google Scholar] [CrossRef]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954. [Google Scholar] [CrossRef]
- Powell, D.A.; Ramsden, P.D.; Batey, R.A. Phase-Transfer-Catalyzed alkylation of guanidines by alkyl halides under biphasic conditions: A convenient protocol for the synthesis of highly functionalized guanidines. J. Org. Chem. 2003, 68, 2300–2309. [Google Scholar] [CrossRef]
- Kim, K.S.; Qian, L. Improved method for the preparation of guanidines. Tetrahedron Lett. 1993, 34, 7677–7680. [Google Scholar] [CrossRef]
- Levallet, C.; Lerpiniere, J.; Ko, S.Y. The HgCl2-Promoted guanylation reaction: The scope and limitations. Tetrahedron 1997, 53, 5291–5304. [Google Scholar] [CrossRef]
- Murtaza, G.; Rauf, M.K.; Badshah, A.; Ebihara, M.; Said, M.; Gielen, M.; de Vos, D.; Dilshad, E.; Mirza, B. Synthesis, structural characterization and in vitro biological screening of some homoleptic copper(II) complexes with substituted guanidines. Eur. J. Med. Chem. 2012, 48, 26–35. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar]
- Faraglia, G.; Fregona, D.; Sitran, S.; Giovagnini, L.; Marzano, C.; Baccichetti, F.; Casellato, U.; Graziani, R. Platinum(II) and palladium(II) complexes with dithiocarbamates and amines: synthesis, characterization and cell assay. J. Inorg. Biochem. 2001, 83, 31–40. [Google Scholar] [CrossRef]
- Cunha, S.; Rodrigues, M.T., Jr.; da Silva, C.C.; Napolitano, H.B.; Vencato, I.; Lariucci, C. The first synthesis of pyridinium N-benzoylguanidines by bismuth- and mercury-promoted guanylation of N-iminopyridinium ylide with thioureas. Tetrahedron 2005, 61, 10536–10540. [Google Scholar] [CrossRef]
- Turker, A.U.; Camper, N.D. Biological activity of common mullein, a medicinal plant. J. Ethnopharmacol. 2002, 82, 117–125. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Shah, N.A.; Khan, M.R.; Tahir, M.N. Synthesis, characterization, biological screenings and interaction with calf thymus DNA of a novel azomethine 3-((3,5-dimethylphenylimino)methyl)benzene-1,2-diol. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 94, 134–142. [Google Scholar] [CrossRef]
- Rehman, A.; Choudhary, M.I.; Thomesen, W.J. Bioassay Techniques for Drug Development; Harwood Academic Publishers: Amsterdam, The Netherland, 2001; p. 9. [Google Scholar]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth Heinemann: New York, NY, USA, 2003. [Google Scholar]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- SAINT, Release 6.06, Integration software for single crystal data, Bruker AXS Inc.: Madison, WI, USA, 1999.
- XPREP, Release 5.10, X-ray data preparation and reciprocal space exploration program, Bruker AXS Inc.: Madison, WI, USA, 1997.
- SHELXTL, Release 5.10, The complete software package for single crystal structure determination, Bruker AXS Inc.: Madison, WI, USA, 1997.
- Sheldrick, G.M. SHELXS97, Program for the solution of crystal structures; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL97, Program for the refinement of crystal structures; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Said, M.; Murtaza, G.; Freisinger, E.; Anwar, S.; Rauf, A. (Z)-2-Phenyl-3-pivaloyl-1,1-dipropylguanidine. Acta Cryst. 2009, E65, o2073–o2074. [Google Scholar]
- Saeed, A.; Zaman, S.; Jamil, M.; Mirza, B. Synthesis and antifungal activity of some novel N-(4-phenyl-3-aroylthiazol-2(3H)-ylidene) substituted benzamides. Turk. J. chem. 2008, 32, 585–592. [Google Scholar]
- Sample Availability: Samples of the compounds (1–12) are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Said, M.; Badshah, A.; Shah, N.A.; Khan, H.; Murtaza, G.; Vabre, B.; Zargarian, D.; Khan, M.R. Antitumor, Antioxidant and Antimicrobial Studies of Substituted Pyridylguanidines. Molecules 2013, 18, 10378-10396. https://doi.org/10.3390/molecules180910378
Said M, Badshah A, Shah NA, Khan H, Murtaza G, Vabre B, Zargarian D, Khan MR. Antitumor, Antioxidant and Antimicrobial Studies of Substituted Pyridylguanidines. Molecules. 2013; 18(9):10378-10396. https://doi.org/10.3390/molecules180910378
Chicago/Turabian StyleSaid, Muhammad, Amin Badshah, Naseer Ali Shah, Hizbullah Khan, Ghulam Murtaza, Boris Vabre, Davit Zargarian, and Muhammad Rashid Khan. 2013. "Antitumor, Antioxidant and Antimicrobial Studies of Substituted Pyridylguanidines" Molecules 18, no. 9: 10378-10396. https://doi.org/10.3390/molecules180910378
APA StyleSaid, M., Badshah, A., Shah, N. A., Khan, H., Murtaza, G., Vabre, B., Zargarian, D., & Khan, M. R. (2013). Antitumor, Antioxidant and Antimicrobial Studies of Substituted Pyridylguanidines. Molecules, 18(9), 10378-10396. https://doi.org/10.3390/molecules180910378