Revealing the Properties of Plant Defensins through Dynamics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
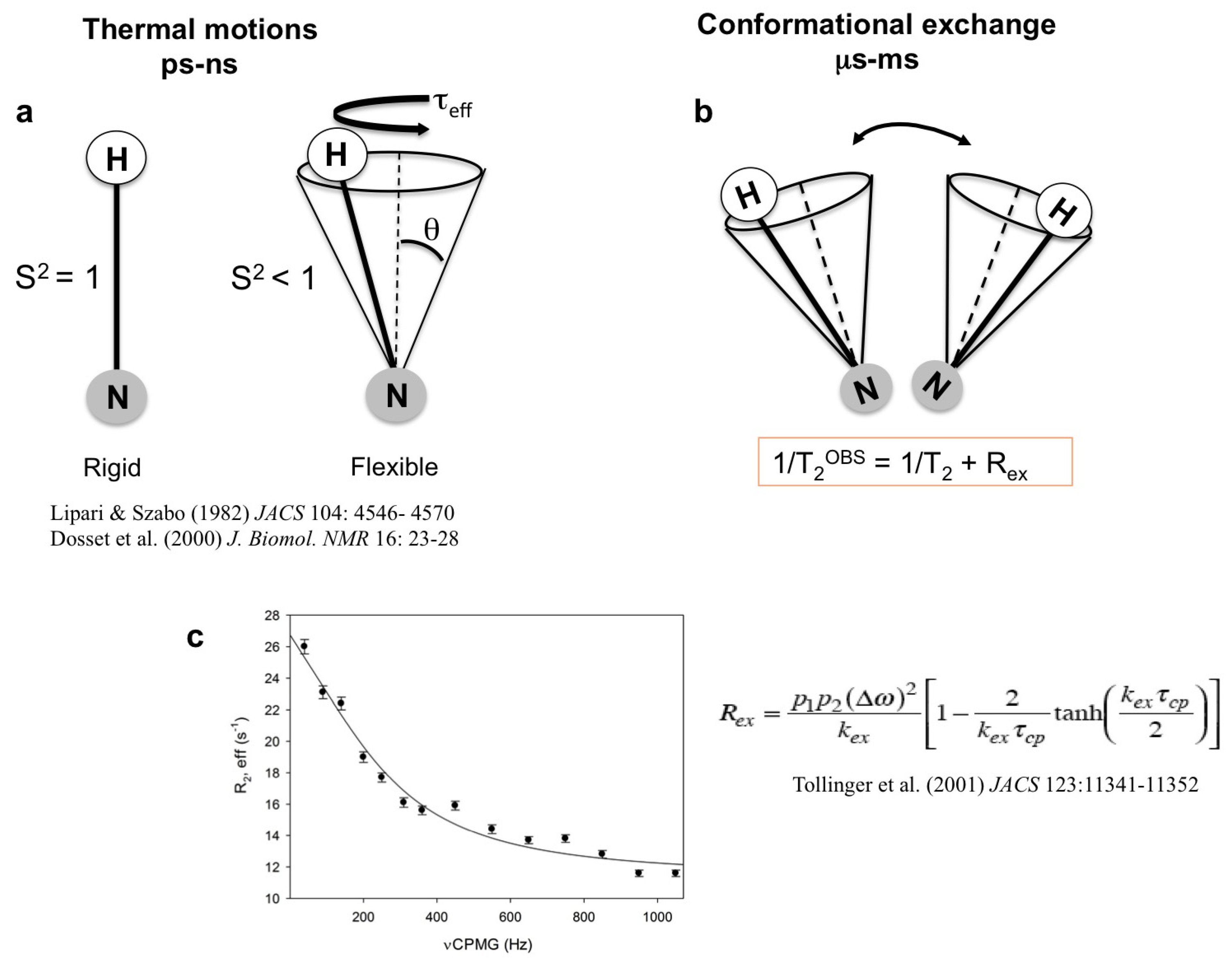
1.1. High-Energy States in Solution
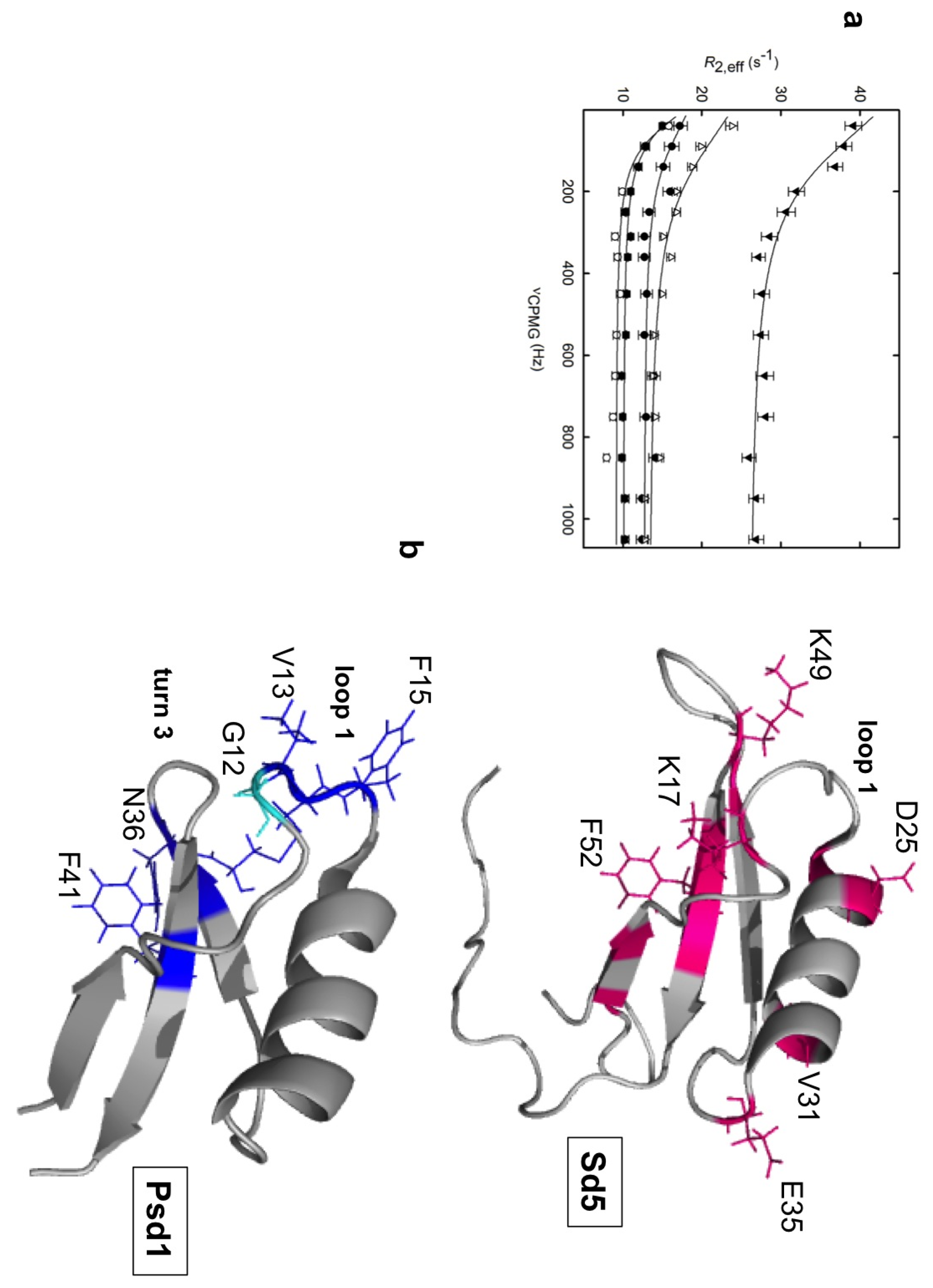
1.2. Plant Defensins
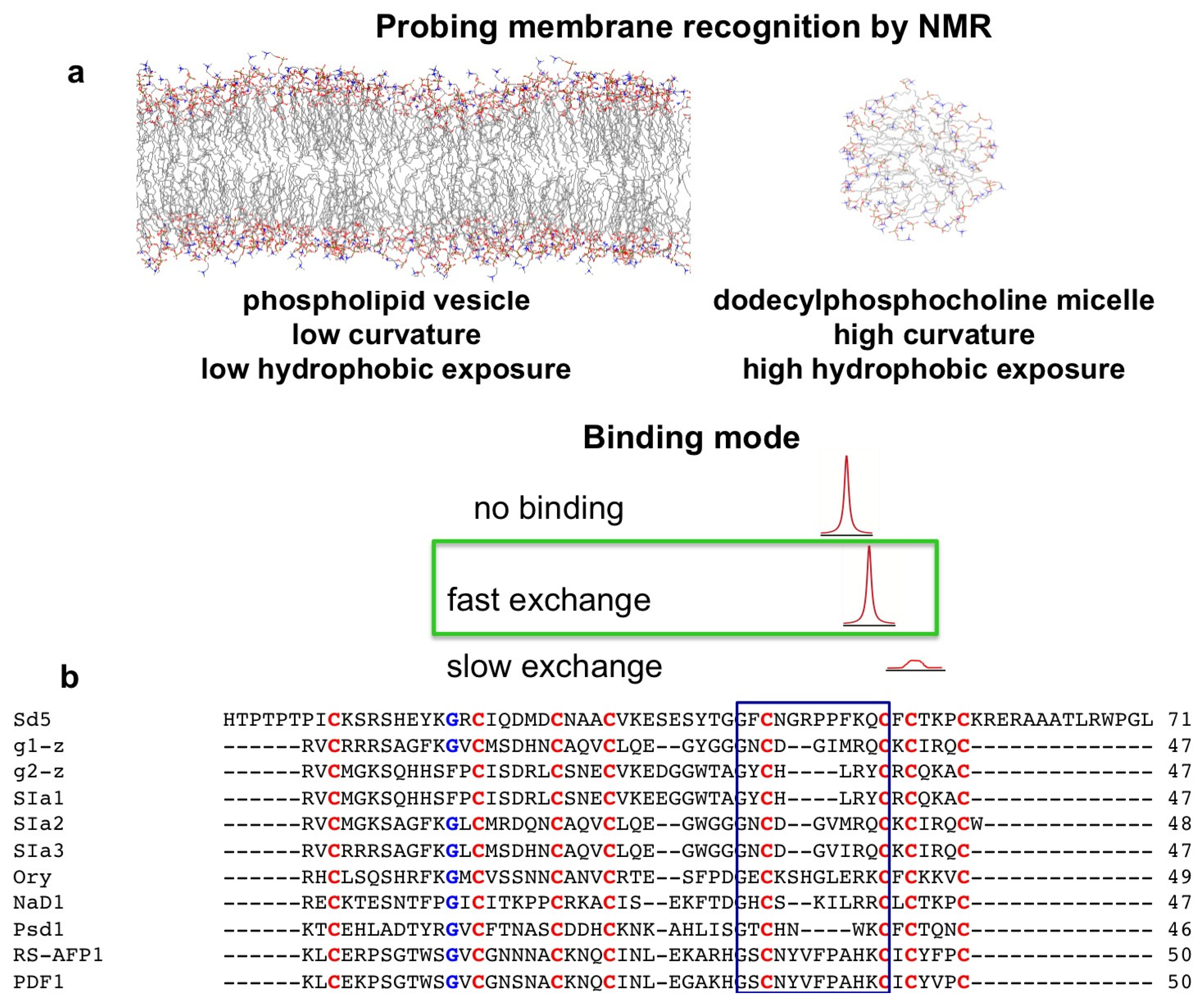
2. Probing Membrane Interaction
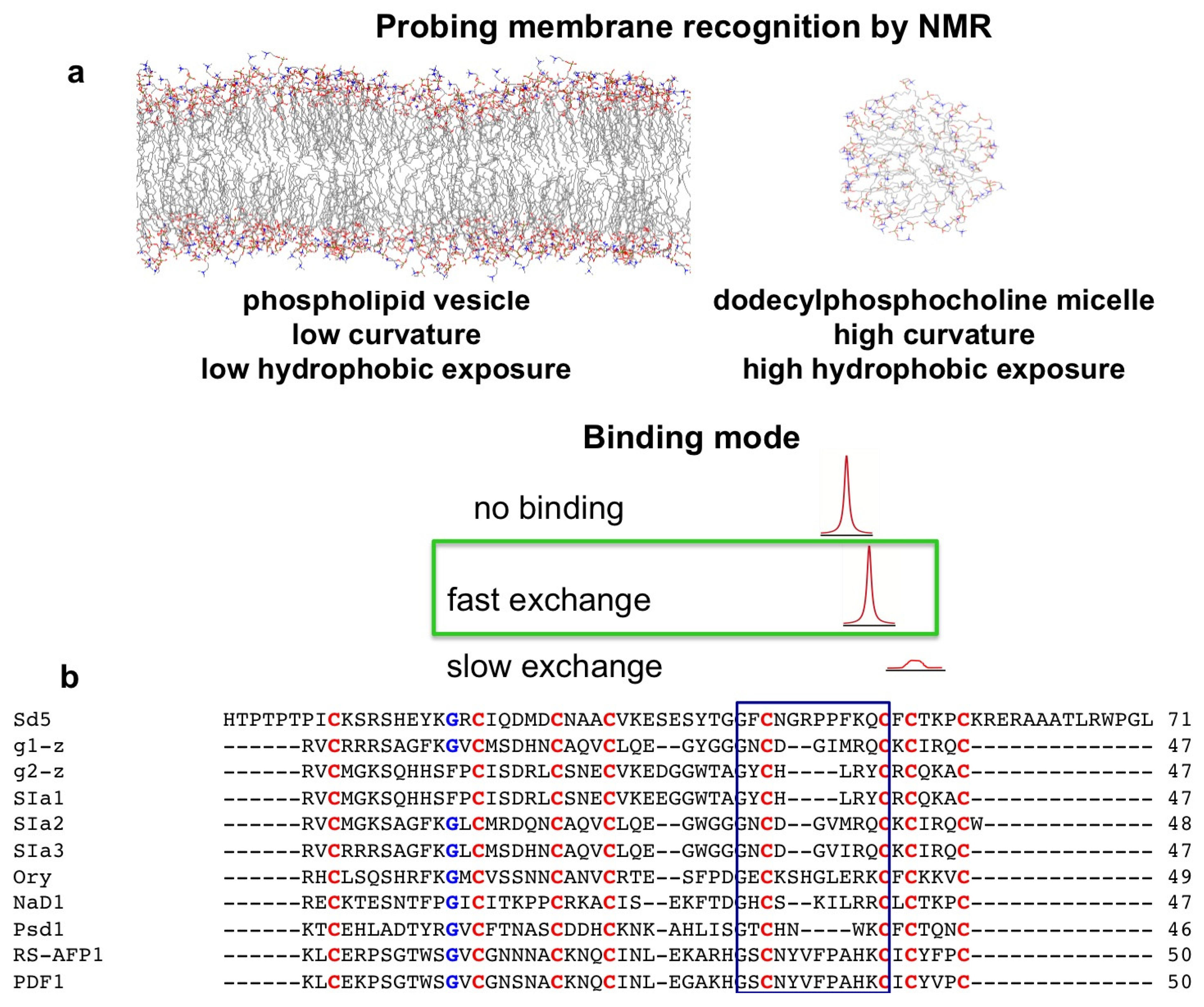
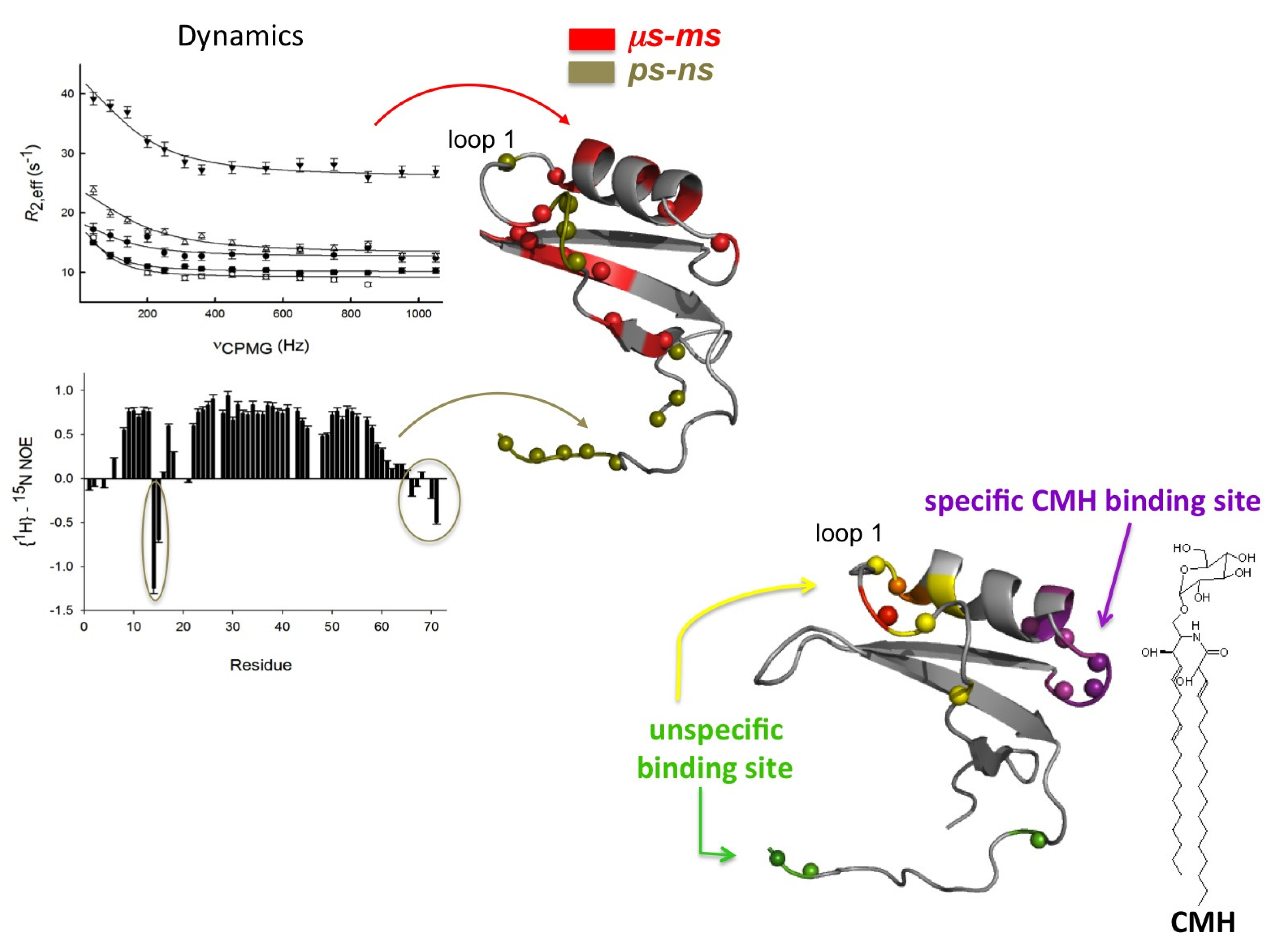
2.1. Sd5
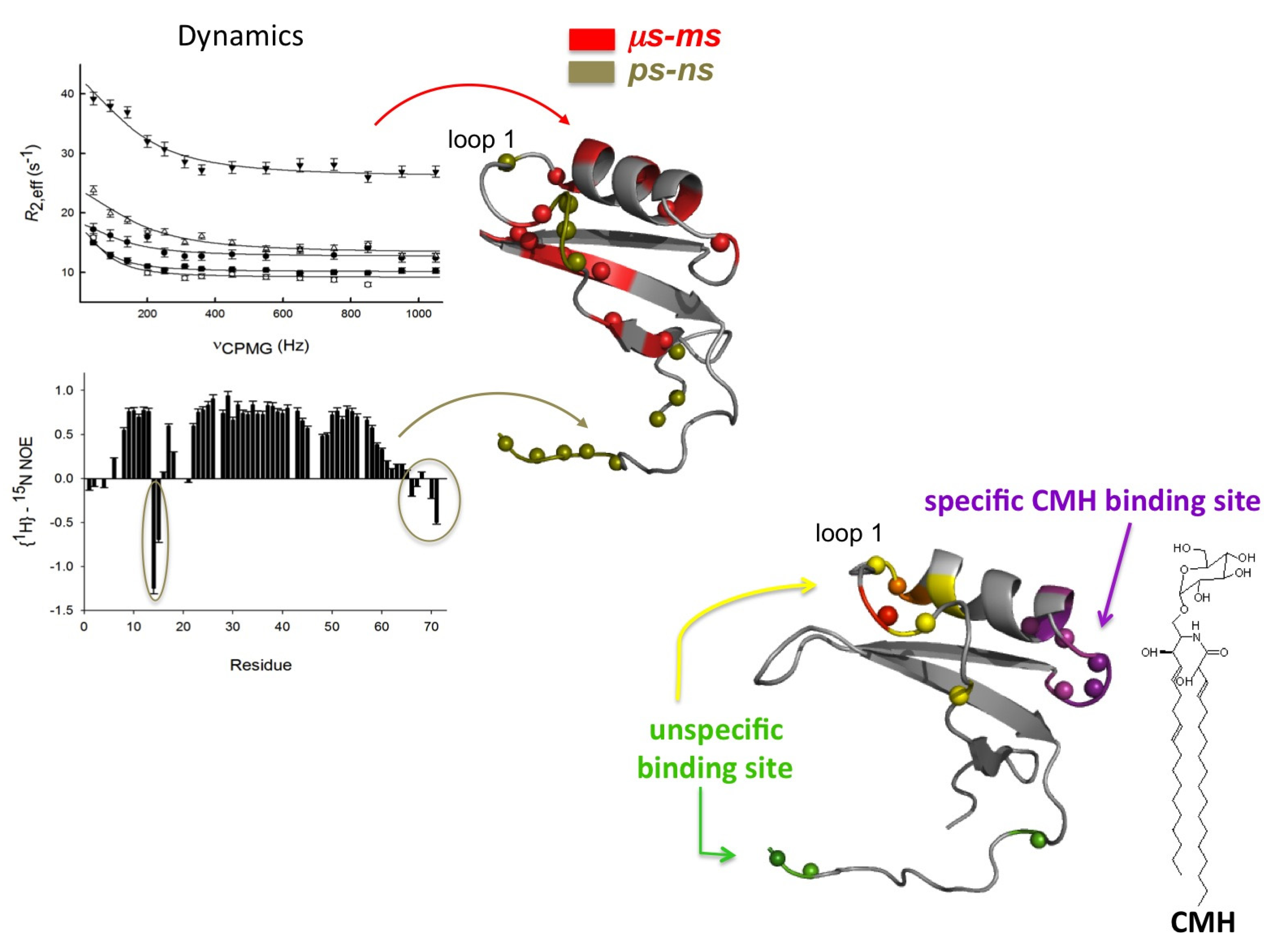
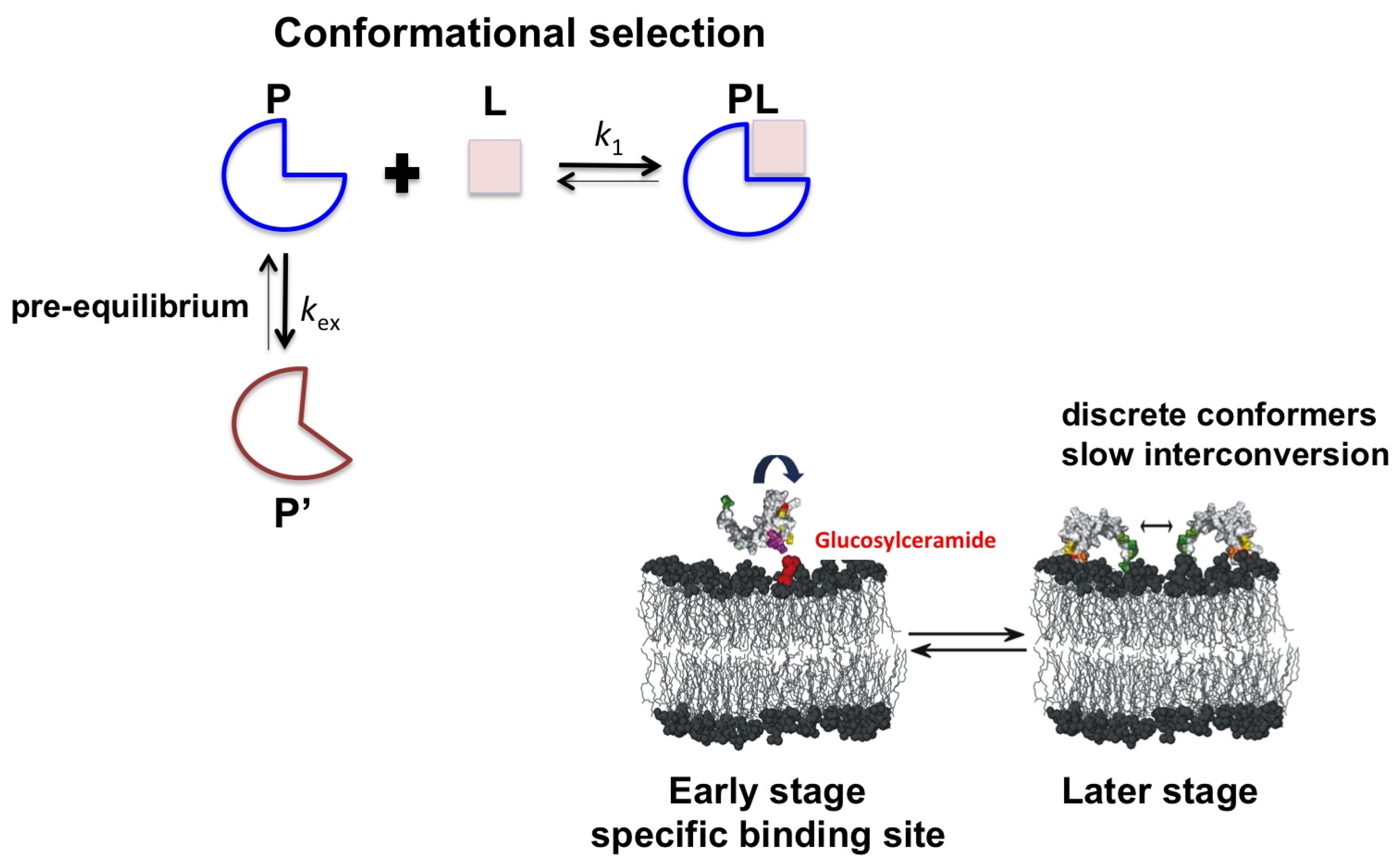
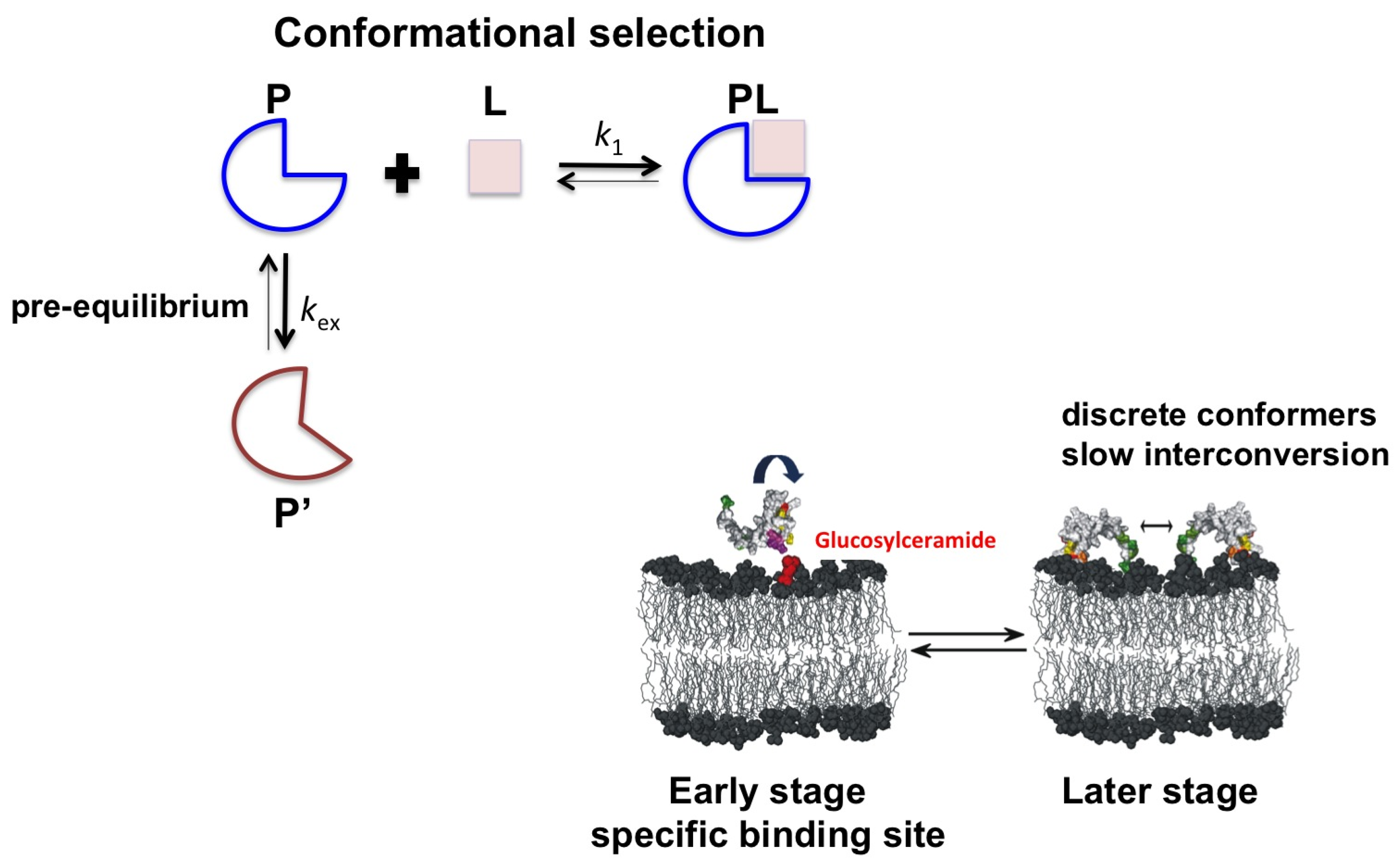
2.2. Psd1

3. Conclusions
Acknowledgments
Conflicts of Interest
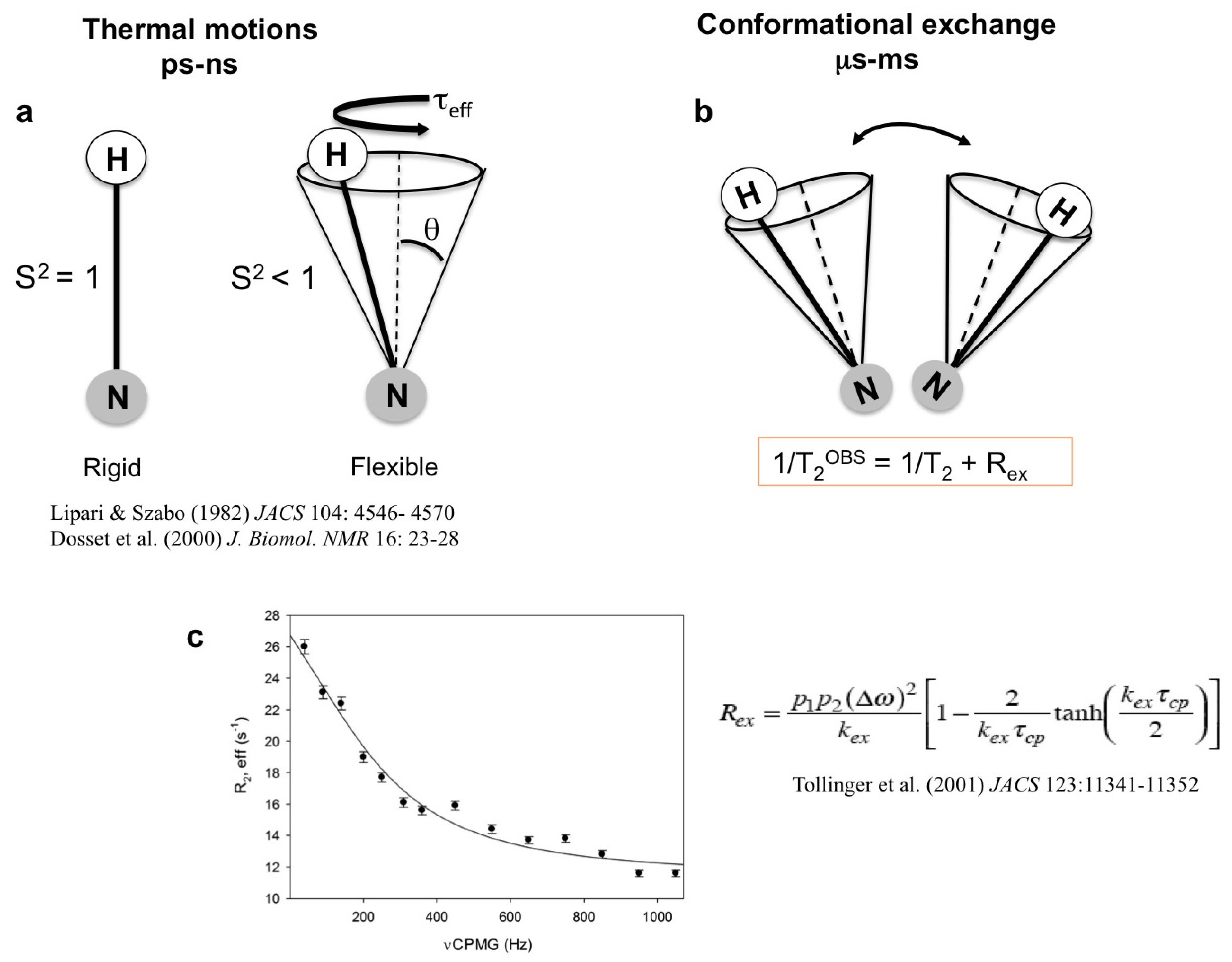
Appendix 1: The application of NMR to the study of interactions and dynamics
References
- Fisher, E. Eifluss der configuration auf die wirkung der enzyme. Ber. Dtsch. Chem. Ges. 1894, 27, 2984–2993. [Google Scholar]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Koshland, D.E.; Nemethy, G.; Filmer, D. Comparison of experiemtal binding data and theoretical models in proteins containing subunits. Biochemistry 1966, 5, 365–385. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions—A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef]
- Valente, A.P.; Miyamoto, C.; Almeida, F.C.L. Implications of protein conformational diversity for binding and development of new biological active compounds. Curr. Med. Chem. 2006, 13, 3697–3703. [Google Scholar] [CrossRef]
- Wu, Z.; Elgart, V.; Qian, H.; Xing, J.H. Amplification and detection of single-molecule conformational fluctuation through a protein interaction network with bomodal distributions. J. Phys. Chem. B 2009, 113, 12375–12381. [Google Scholar] [CrossRef]
- Grunberg, R.; Leckner, J.; Nilges, M. Complementarity of structure ensembles in protein-protein binding. Structure 2004, 12, 2125–2136. [Google Scholar] [CrossRef]
- Wlodarski, T.; Zagrovic, B. Conformational selection and induced fit mechanism underlie specificity in non covalente interactions with ubiquitin. Proc. Natl. Acad. Sci. USA 2009, 106, 19346–19351. [Google Scholar] [CrossRef]
- Mittermaier, A.K.; Kay, L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009, 34, 601–611. [Google Scholar] [CrossRef]
- Kleckner, I.R.; Foster, M.P. An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 2011, 1814, 942–968. [Google Scholar] [CrossRef]
- Korzhnev, D.M.; Kay, L.E. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc. Chem. Res. 2008, 41, 442–451. [Google Scholar] [CrossRef]
- Cavanagh, J.; Akke, M. May the driving force be with you—Whatever it is. Nat. Struct. Biol. 2000, 7, 11–13. [Google Scholar] [CrossRef]
- Tsai, C.J.; Kumar, S.; Ma, B.; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999, 8, 1181–1190. [Google Scholar] [CrossRef]
- Tsai, C.J.; Ma, B.; Nussinov, R. Folding and binding cascades: Shifts in energy landscapes. Proc. Natl. Acad. Sci. USA 1999, 96, 9970–9972. [Google Scholar] [CrossRef]
- Kumar, S.; Ma, B.; Tsai, C.J.; Sinha, N.; Nussinov, R. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 2000, 9, 10–19. [Google Scholar]
- Peng, J.W. Exposing the moving parts of proteins with NMR spectroscopy. J. Phys. Chem. Lett. 2012, 3, 1039–1051. [Google Scholar] [CrossRef]
- Salmon, L.; Bouvignies, G.; Markwick, P.; Blackledge, M. Nuclear magnetic resonance provides a quantitative description of protein conformational flexibility on physiologically important time scales. Biochemistry 2011, 50, 2735–2747. [Google Scholar] [CrossRef]
- Lundström, P.; Vallurupalli, P.; Hansen, D.F.; Kay, L.E. Isotope labeling methods for studies of excited protein states by relaxation dispersion NMR spectroscopy. Nat. Protoc. 2009, 4, 1641–1648. [Google Scholar] [CrossRef]
- Farrow, N.A.; Zhang, O.; Forman-Kay, J.D.; Kay, L.E. A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J. Biomol. NMR 1994, 4, 727–734. [Google Scholar] [CrossRef]
- Hansen, D.F.; Vallurupalli, P.; Kay, L.E. An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J. Phys. Chem. B 2008, 112, 5898–5904. [Google Scholar] [CrossRef]
- Loria, J.P.; Rance, M.; Palmer, A.G. A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR 1999, 15, 151–155. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Hansen, D.F.; Stollar, E.; Meirovitch, E.; Kay, L.E. Measurement of bond vector orientations in invisible excited states of proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 18473–18477. [Google Scholar] [CrossRef]
- Ishima, R.; Torchia, D. Estimating the time scale of chemical exchange of proteins from measurements of transverse relaxation rates in solution. J. Biomol. NMR 1999, 14, 369–372. [Google Scholar] [CrossRef]
- Orekhov, V.Y.; Korzhnev, D.M.; Kay, L.E. Double- and zero-quantum NMR relaxation dispersion experiments sampling millisecond time scale dynamics in proteins. J. Am. Chem. Soc. 2004, 126, 1886–1891. [Google Scholar] [CrossRef]
- Hansen, D.F.; Vallurupalli, P.; Lundström, P.; Neudecker, P.; Kay, L.E. Probing chemical shifts of invisible states of proteins with relaxation dispersion NMR spectroscopy: How well can we do? J. Am. Chem. Soc. 2008, 130, 2667–2675. [Google Scholar] [CrossRef]
- Lundström, P.; Hansen, D.F.; Kay, L.E. Measurement of carbonyl chemical shifts of excited protein states by relaxation dispersion NMR spectroscopy: Comparison between uniformly and selectively (13)C labeled samples. J. Biomol. NMR 2008, 42, 35–47. [Google Scholar] [CrossRef]
- Korzhnev, D.M.; Religa, T.L.; Banachewicz, W.; Fersht, A.R.; Kay, L.E. A transient and low-populated protein-folding intermediate at atomic resolution. Science 2010, 329, 1312–1316. [Google Scholar] [CrossRef]
- Li, Y.; Palmer, A.G. Narrowing of protein NMR spectral lines broadened by chemical Exchange. J. Am. Chem. Soc. 2010, 132, 8856–8857. [Google Scholar] [CrossRef]
- Wolf-Watz, M.; Thai, V.; Henzler-Wildman, K.; Hadjipavlou, G.; Eisenmesser, E.Z.; Kern, D. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 2004, 11, 945–949. [Google Scholar] [CrossRef]
- Volkman, B.F.; Lipson, D.; Wemmer, D.E.; Kern, D. Two-state allosteric behavior in a single-domain signaling protein. Science 2001, 291, 2429–2433. [Google Scholar] [CrossRef]
- Villali, J.; Kern, D. Choreographing an enzyme’s dance. Curr. Opin. Chem. Biol. 2010, 14, 636–643. [Google Scholar] [CrossRef]
- Bosco, D.A.; Eisenmesser, E.Z.; Clarkson, M.W.; Wolf-Watz, M.; Labeikovsky, W.; Millet, O.; Kern, D. Dissecting the microscopic steps of the cyclophilin A enzymatic cycle on the biological HIV-1 capsid substrate by NMR. J. Mol. Biol. 2010, 403, 723–738. [Google Scholar] [CrossRef]
- Mulder, F.A.; Mittermaier, A.; Hon, B.; Dahlquist, F.W.; Kay, L.E. Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 2001, 8, 932–935. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982, 104, 4559–4570. [Google Scholar] [CrossRef]
- Dosset, P.; Hus, J.C.; Blackledge, M.; Marion, D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR 2000, 16, 23–28. [Google Scholar] [CrossRef]
- Tollinger, M.; Skrynnikov, N.R.; Mulder, F.A.; Forman-Kay, J.D.; Kay, L.E. Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 2001, 123, 11341–11352. [Google Scholar]
- The Palmer Lab. Available online: http://www.palmer.hs.columbia.edu/ (accessed on 10 September 2013).
- Baldwin, A.J.; Kay, L.E. NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol. 2009, 5, 808–814. [Google Scholar] [CrossRef]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef]
- Piazza, F.; Sanejouand, Y.H. Long-range energy transfer in proteins. Phys. Biol. 2009, 6, 046014. [Google Scholar] [CrossRef]
- Piazza, F.; Sanejouand, Y.H. Discrete breathers in protein structures. Phys. Biol. 2008, 5, 026001. [Google Scholar] [CrossRef]
- Leitner, D. Energy flow in proteins. Ann. Rev. Phys. Chem. 2008, 59, 233–259. [Google Scholar] [CrossRef]
- Bulet, P.; Stöcklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef]
- Thevissen, K.; Warnecke, D.C.; François, I.E.; Leipelt, M.; Heinz, E.; Ott, C.; Zähringer, U.; Thomma, B.P.; Ferket, K.K.; Cammue, B.P. Defensins from insects and plants interact with fungal glucosylceramides. J. Biol. Chem. 2004, 279, 3900–3905. [Google Scholar]
- De Coninck, B.; Cammue, B.P.A.; Thevissen, K. Modes of antifungal action and in planta functions of plant defensins and defensin-like peptides. Fungal Biol. Rev. 2013, 26, 109–120. [Google Scholar] [CrossRef]
- Sun, C.Q.; Arnold, R.; Fernandez-Golarz, C.; Parrish, A.B.; Almekinder, T.; He, J.; Ho, S.M.; Svoboda, P.; Pohl, J.; Marshall, F.F.; et al. Human beta-defensin-1, a potential chromosome 8p tumor suppressor: Control of transcription and induction of apoptosis in renal cell carcinoma. Cancer Res. 2006, 66, 8542–8549. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, W.; He, T.; Yu, J.; Caffrey, R.E.; Dalmasso, E.A.; Fu, S.; Pham, T.; Mei, J.; Ho, J.J.; et al. Contribution of human alpha-defensin 1, 2, and 3 to the anti-HIV-1 activity of CD8 antiviral factor. Science 2002, 298, 995–1000. [Google Scholar] [CrossRef]
- Droin, N.; Hendra, J.-B.; Ducoroy, P.; Solary, E. Human defensins as cancer biomarkers and antitumour molecules. J. Proteomics 2009, 72, 918–927. [Google Scholar] [CrossRef]
- Arnett, E.; Seveau, S. The multifaceted activities of mammalian defensins. Curr. Pharm. Des. 2011, 17, 4254–4569. [Google Scholar] [CrossRef]
- Silverstein, K.A.; Moskal, W.A., Jr.; Wu, H.C.; Underwood, B.A.; Graham, M.A.; Town, C.D.; VandenBosch, K.A. Small cysteine-rich peptides resembling antimicrobial peptides have been underestimated in plants. Plant J. 2007, 51, 262–280. [Google Scholar] [CrossRef]
- Conibear, A.C.; Rosengren, K.J.; Daly, N.L.; Henriques, S.T.; Craik, D.J. The cyclic cystine ladder in θ-defensins is important for structure and stability, but not antibacterial activity. J. Biol. Chem. 2013, 288, 10830–10840. [Google Scholar]
- Wu, Z.; Hoover, D.M.; Yang, D.; Boulègue, C.; Santamaria, F.; Oppenheim, J.J.; Lubkowski, J.; Lu, W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human beta-defensin 3. Proc. Natl. Acad. Sci. USA 2003, 100, 8880–8885. [Google Scholar] [CrossRef]
- Lobo, D.S.; Pereira, I.B.; Fragel-Madeira, L.; Medeiros, L.N.; Cabral, L.M.; Faria, J.; Bellio, M.; Campos, R.C.; Linden, R.; Kurtenbach, E. Antifungal Pisum sativum defensin 1 interacts with Neurospora crassa cyclin F related to the cell cycle. Biochemistry 2007, 46, 987–996. [Google Scholar] [CrossRef]
- Aerts, A.M.; Carmona-Gutierrez, D.; Lefevre, S.; Govaert, G.; François, I.E.; Madeo, F.; Santos, R.; Cammue, B.P.; Thevissen, K. The antifungal plant defensin RsAFP2 from radish induces apoptosis in a metacaspase independent way in Candida albicans. FEBS Lett. 2009, 6, 2513–2516. [Google Scholar]
- De Medeiros, L.N.; Angeli, R.; Sarzedas, C.G.; Barreto-Bergter, E.; Valente, A.P.; Kurtenbach, E.; Almeida, F.C.L. Backbone dynamics of the antifungal Psd1 pea defensin and its correlation with membrane interaction by NMR spectroscopy. Biochim. Biophys. Acta 2010, 1798, 105–113. [Google Scholar] [CrossRef]
- De Paula, V.S.; Razzera, G.; Barreto-Bergter, E.; Almeida, F.C.L.; Valente, A.P. Portrayal of complex dynamic properties of sugarcane defensin 5 by NMR: Multiple motions associated with membrane interaction. Structure 2011, 19, 26–36. [Google Scholar] [CrossRef]
- Yount, N.Y.; Yeaman, M.R. Multidimensional signatures in antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 7363–7368. [Google Scholar] [CrossRef]
- Yount, N.Y.; Yeaman, M.R. Structural congruence among membrane-active host defense polypeptides of diverse phylogeny. Biochim. Biophys. Acta 2006, 1758, 1373–1386. [Google Scholar] [CrossRef]
- Sagaram, U.S.; Pandurangi, R.; Kaur, J.; Smith, T.J.; Shah, D.M. Structure-activity determinants in antifungal plant defensins MsDef1 and MtDef4 with different modes of action against Fusarium graminearum. PLoS One 2011, 6, e18550. [Google Scholar]
- Ramamoorthy, V.; Cahoon, E.B.; Li, J.; Thokala, M.; Minto, R.E.; Shah, D.M. Glucosylceramide synthase is essential for alfalfa defensin-mediated growth inhibition but not for pathogenicity of Fusarium graminearum. Mol. Microbiol. 2007, 66, 771–786. [Google Scholar] [CrossRef]
- De-Paula, V.S.; Razzera, G.; Medeiros, L.; Miyamoto, C.A.; Almeida, M.S.; Kurtenbach, E.; Almeida, F.C.L.; Valente, A.P. Evolutionary relationship between defensins in the Poaceae family strengthened by the characterization of new sugarcane defensins. Plant Mol. Biol. 2008, 68, 321–335. [Google Scholar] [CrossRef]
- Cornilescu, G.; Delaglio, F.; Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 1999, 13, 289–302. [Google Scholar] [CrossRef]
- Almeida, M.S.; Cabral, K.M.S.; Kurtenbach, E.; Almeida, F.C.L.; Valente, A.P. Solution structure of Pisum sativum defensin 1 by high resolution NMR: Plant defensins, identical backbone with different mechanisms of action. J. Mol. Biol. 2002, 315, 749–757. [Google Scholar] [CrossRef]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef]
- Wishart, D.S. Interpreting protein chemical shift data. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87. [Google Scholar] [CrossRef]
- Gao, G.; Williams, J.G.; Campbell, S.L. Protein-protein interaction analysis by nuclear magnetic resonance spectroscopy. Methods Mol. Biol. 2004, 261, 79–92. [Google Scholar]
- Palmer, A.G., 3rd. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. [Google Scholar] [CrossRef]
- Clore, G.M.; Driscoll, P.C.; Wingfield, P.T.; Gronenborn, A.M. Analysis of the backbone dynamics of interleukin-1 beta using two-dimensional inverse detected heteronuclear 15N-1H NMR spectroscopy. Biochemistry 1990, 29, 7387–7401. [Google Scholar] [CrossRef]
- Cavalli, A.; Salvatella, X.; Dobson, C.M.; Vendruscolo, M. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2007, 104, 9615–9620. [Google Scholar] [CrossRef]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 2008, 105, 4685–4690. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Valente, A.P.; De Paula, V.S.; Almeida, F.C.L. Revealing the Properties of Plant Defensins through Dynamics. Molecules 2013, 18, 11311-11326. https://doi.org/10.3390/molecules180911311
Valente AP, De Paula VS, Almeida FCL. Revealing the Properties of Plant Defensins through Dynamics. Molecules. 2013; 18(9):11311-11326. https://doi.org/10.3390/molecules180911311
Chicago/Turabian StyleValente, Ana Paula, Viviane Silva De Paula, and Fabio C. L. Almeida. 2013. "Revealing the Properties of Plant Defensins through Dynamics" Molecules 18, no. 9: 11311-11326. https://doi.org/10.3390/molecules180911311