Abstract
A series of 3,4-dihydroisoquinoline-3-carboxylic acid derivatives were synthesised and tested for their free-radical scavenging activity using 2,2-diphenyl-1-picrylhydrazyl radical (DPPH·), 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) radical (ABTS·+), superoxide anion radical (O2·−) and nitric oxide radical (·NO) assays. We also studied d-amino acid oxidase (DAAO), acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitory activity. Almost each of newly synthesised compounds exhibited radical scavenging capabilities. Moreover, several compounds showed moderate inhibitory activities against DAAO, AChE and BuChE. Compounds with significant free-radical scavenging activity may be potential candidates for therapeutics used in oxidative-stress-related diseases.
1. Introduction
3,4-Dihydroisoquinoline derivatives have several important biological properties and are in the area of interest of the pharmaceutical industry. Published activities include antimetastatic [1], anti-inflammatory and analgesic [2], antiarrhythmic, and antiaggregatory [3] antihypertensive [4], and antibacterial [5]. They also inhibit catechol O-methyltransferase [6] and kinase JNK3 [7]. Most of investigated 3,4-dihydroisoquinoline derivatives have substituents at C1.
We have recently described the isolation, synthesis and antimicrobial activity of 6,7-dihydroxy-3,4-dihydroisoquinoline-3-carboxylic acid (5e), a C1-unsubstituted secondary metabolite of the Streptomyces sp. 8812 fermentation process [8,9]. This prompted us to prepare a series of hydroxy- and halogeno-substituted 3,4-dihydroisoquinoline-3-carboxylic acids and investigate their biological activity. We also included in our investigation 2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]isoquinoline analogue, as well as a 4,5-dihydro-3H-2-benzazepine analogue.
Isoquinoline alkaloid derivatives (benzyltetrahydroisoquinolines (±)-coclaurine and (±)-norarmepavine) also present antioxidative properties [10]. Berberine, with widely demonstrated antioxidative activities, exhibited acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitory activities, as well [11].
Our main interests are concerned with the determination of antioxidative properties due to the fact that phenolic compounds often present potent free radical scavenging activities. Several known, potent free-radical scavengers such as flavonols (e.g., quercetin, kaempferol), flavones (e.g., luteolin, apigenin), flavanols (e.g., catechin) and isoflavones (e.g., genistein) contain phenols and polyphenolic moiety [12]. Previous studies have shown that polyphenolic structures are more potent free-radical scavengers than those with only one hydroxyl group [13]. Also, the ortho location of two hydroxyl groups increases the antioxidative properties [13,14]. The free-radical scavenging potential of polyphenolic compounds is associated with their reducing (hydrogen- or electron-donating) activities [12,15].
Oxidative stress is a process that arises when free radicals and oxidants are produced in excess and cells cannot destroy them properly. In other words, an imbalance between the formation and neutralisation of free radicals can occur, owing to a depletion of antioxidants or the accumulation of reactive oxygen species (ROS), and can result in the appearance of oxidative stress [16,17,18,19]. The latter may lead to the change of cell components such as proteins, lipids, lipoproteins, nucleic acids and so forth [18,19,20].
Oxidative stress plays a crucial role in the development of various diseases such as cardiovascular disorders; atherosclerosis; hypertension or heart failure; diabetes mellitus; cancer; rheumatoid arthritis; ocular diseases; neurodegenerative diseases such as Parkinson’s and Alzheimer’s disease (AD), as well as psychic impairments such as schizophrenia and is linked to aging [15,16,17,18].
The synthesised compounds were investigated for their free-radical scavenging activity against 2,2-diphenyl-1-picrylhydrazyl radical (DPPH·), 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) radical (ABTS·+), superoxide anion radical (O2·−) and nitric oxide radical (·NO). Additionally, we evaluated compounds for some activities associated with oxidative stress–inhibitory activity against d-amino acid oxidase (DAAO, a potential target in schizophrenia treatment) and for the inhibition of two enzymes important in pathophysiology of Alzheimer’s disease: acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE).
2. Results and Discussion
2.1. Chemistry
Derivatives of the isoquinoline 5 have been obtained in a multistep synthesis using the Bischler-Napieralski reaction [21] as a cyclization step (Scheme 1, Table 1). Benzyl alcohols 1 can be easily obtained by known methods from commercially available hydroxy- or methoxy-substituted benzaldehydes. We have used benzyl protection for hydroxyl groups. Some other protective groups like allyl, carbonate or acetal groups were not stable in the cyclization step. Reduction of the carbonyl group and chlorination with thionyl chloride gave benzyl chlorides 2. Alkylation of dibenzyl formamidomalonate with 2 gave Bischler-Napieralski reaction substrates 3. Cyclization was conducted under standard conditions using POCl3 in acetonitrile at 70–95 °C. Higher reaction temperatures resulted in extensive formation of polymers and lowered the yield. Two isomers can form in the case of 2,6-unsubstituted derivatives 3b, d, g and h (R1 = H). Usually only one product was obtained. For example cyclization of compound 3b, gives predominantly 8-benzyloxy-7-bromo-3,4-dihydroisoquinoline instead of the corresponding 6-benzyloxy-7-bromo derivative. Apparently the C2 carbon atom in formamidomalonate 3b is more nucleophilic than C6. The stereochemistry of 4d, g and h has been determined on the basis of HMBC correlation spectra, and derivative 4b showed a vicinal coupling constant between C5-H and C6-H. In the last step of the synthesis, all benzyl groups were removed with BBr3 [22] and decarboxylation took place gently to give acid 5 (Table 2). We have also used hydrogenation on Pd/C with 1,4-cyclohexadiene as hydrogen donor. This is very mild method which selectively removes benzyl groups in the presence of C=N bond. However formation of isoquinoline derivatives due to aromatization of the ring was noticed. More difficult was removal of methyl groups with BBr3. In the case of trimethoxy-substituted dihydroisoquinoline we have obtained mixture of isomers, where 5i was the major one.

Scheme 1.
Synthesis of 3,4-dihydroisoquinoline-3-carboxylic acid derivatives.

Table 1.
Formamidomalonates 3 and 3,4-dihydroisoquinoline derivatives 4.
| 3 (Yield,%) | 4 (Yield,%) | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|
| 3a (63) | 4a (31) | Br | H | H | OBn |
| 3b (95) | 4b (26) | H | H | Br | OBn |
| 3c (77) | 4c (94) | H | OBn | H | OBn |
| 3d (65) | 4d (94) | H | OMe | OBn | I |
| 3f (66) | 4f (58) | OMe | OMe | H | Br |
| 3g (65) | 4g (94) | H | OMe | OMe | Br |
| 3h (57) | 4h (95) | H | OMe | OMe | Cl |
| 3i (70) | 4i (95) | H | OMe | OMe | OMe |
| 3j (75) | 4j (85) | H | OBn | Br | OBn |
| 3k (88) | 4k (94) | Cl | OBn | H | OBn |
| 3l (56) | 4l (86) | Cl | OMe | OMe | H |
Table 2.
3,4-Dihydroisoquinoline-3-carboxylic acid derivatives.
| 5 (Yield,%) | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| 5a (22) | Br | H | H | OH |
| 5b (87) | H | H | Br | OH |
| 5c (62) | H | OH | H | OH |
| 5d (22) | H | OH | OH | I |
| 5e a | H | OH | OH | H |
| 5f (21) | OH | OH | H | Br |
| 5g (34) | H | OH | OH | Br |
| 5h (15) | H | OH | OH | Cl |
| 5i (60) | H | OH | OMe | OH |
| 5j (53) | H | OH | Br | OH |
| 5k (17) | Cl | OH | H | OH |
| 5l (8) | Cl | OH | OH | H |
a Reference [9].
The most problematic was purification of polyhydroxylated derivatives of 3,4-dihydroisoquinoline-3-carboxylic acid 5. The products are sensitive to air and bases giving aromatic derivatives of isoquinolines. Due to the very low solubility in organic solvents and high polarity of the products, we had to use chromatography on ion exchange and polyamide columns. In a similar way, we obtained 4,5-dihydro-3H-2-benzazepin-3-carboxylic acid 9 from 4-(2-iodoethyl)-1,2-dimethoxybenzene (Scheme 2). Optically active 3-methyl derivative of 3,4-dihydroisoquinoline-3-carboxylic acid 13 was obtained using l-methyl-DOPA by formylation with formyloxyacetonitrile and Bischler-Napieralski cyclization. We also synthesised a β-lactam derivative of dihydroisoquinoline 15 by the Bose method using [2+2] cycloaddition with chloroketene generated in situ from chloroacetyl chloride [23]. This type of reaction is usually highly stereoselective. Examination of the coupling constant of C1-H (1.7 Hz) suggests that we have obtained trans stereoisomer. The benzyl groups in 14 were removed by hydrogenolysis to give β-lactam 15 (Scheme 3).

Scheme 2.
Synthesis of 4,5-dihydro-3H-2-benzazepin-3-carboxylic acid 9.

Scheme 3.
Synthesis of the 3-methyl derivative of 3,4-dihydroisoquinoline-3-carboxylic acid 13 and β-lactam derivative 15.
2.2. Biological Activities
2.2.1. Free-Radical Scavenging Activity
All the synthesised derivatives were assayed in vitro for their antioxidant and free-radical scavenging activity. In these assays, the compounds were tested for their ability to scavenge DPPH·, ABTS·+, O2·− and ·NO. The EC50 values are summarized in Table 3. In general, almost all of the described compounds were potent scavengers on at least one free radical used in our study. Lower EC50 value indicates higher radical-scavenging activity. Free-radical scavenging activities of synthesised derivatives were compared to the properties shown by naturally obtained compound 5e.
Compounds 5e, 5g and 9 showed the best EC50 value, better than ascorbic acid on DPPH· scavenging. Compounds 5l, 5f and 5d also presented high DPPH· scavenging activity, comparable to the standard (Table 3).
Table 3.
Biological activities of synthesised compounds.
| Tested Compound | Scavenging Activity [EC50] a | DAAO Inhibition [IC50] c/[%] d | AChE Inhibition [IC50] c | BuChE Inhibition IC50] c | |||
|---|---|---|---|---|---|---|---|
| DPPH· | ABTS·+ | O2·− | ·NO | ||||
| 5a | 351.60 | 29.62 | 402.62 | 2196.69 | 8.46 c | - | - |
| 5b | 1103.08 | 280.47 | N.D. b | 901.25 | 13.88 d | N.A. e | N.A. |
| 5c | 520.32 | 16.22 | N.D. | 2459.99 | 1.66 c | N.A. | N.A. |
| 5d | 39.45 | 22.58 | 4.53 | 330.61 | 3.35 c | - | - |
| 5e | 23.26 | 7.19 | 35.96 | 386.38 | 7.52 d | 0.56 | 0.37 |
| 5f | 36.07 | 13.49 | 9.54 | 337.07 | 2.06 c | 0.54 | N.A. |
| 5g | 26.22 | 11.74 | 14.23 | 402.82 | 2.00 c | N.A. | N.A. |
| 5h | 52.69 | 21.48 | 116.09 | 3219.46 | 1.74 c | 0.82 | N.A. |
| 5i | 63.15 | 23.73 | 35.54 | 2236.12 | 3.28 c | N.A. | N.A. |
| 5j | 269.19 | 113.71 | 869.76 | 540.23 | 11.50 d | 0.47 | 0.64 |
| 5k | 494.52 | 28.85 | 49.21 | 979.18 | 7.27 d | 0.59 | 1.42 |
| 5l | 32.98 | 9.97 | 5.42 | 1644.66 | 1.29 c | N.A. | 0.17 |
| 9 | 27.03 | 12.02 | 5.92 | N.D. | 0.55 c | 2.08 | N.A. |
| 13 | 56.01 | 18.76 | 203.61 | 65.05 | 7.89 d | 1.78 | 0.62 |
| 15 | 84.62 | 10.62 | 4.40 | 1054.32 | 2.02 c | N.A. | 1.75 |
| Ascorbic acid | 42.11 | 28.25 | 98.77 | - | - | - | - |
| Trolox | - | - | - | 161.37 | - | - | - |
| Benzoic acid | - | - | - | - | 2.28 c | - | - |
| Galanthamine hydrobromide | - | - | - | - | - | 0.001 | 0.009 |
a The values were expressed as μM concentration; b N.D.—not determined; c The values were expressed as mM concentration; d % of DAAO inhibition at 3.57 mM concentration; e N.A.—not active.
Compounds 5h and 13, also showed good activity (slightly lower than standard). The presence of two hydroxyl groups at 6- and 7-position (5e, 5g, 5l, 5d, 5h, 13) and respectively at 7- and 8-position for compound 9 resulted in significant DPPH radical scavenging activity. Meta location of hydroxyl groups (compounds 5i, 5j, 5k, 5c) did not result in better activities in comparison to 5e (ortho location). Among compounds with meta location of hydroxyl groups (compounds 5i, 5j, 5k, 5c), compound 5i with an electron-donating group at 7-position presented better activity than others. Introduction of a halogen atom at 5- or 8-position of a compound, with two hydroxyl groups at 6- and 7-positions (5g, 5l, 5d, 5h), did not potentiate scavenging activity on DPPH· in comparison to 5e. DPPH radical was scavenged by compounds 5e, 5g, 9, 5l and ascorbic acid in a concentration-dependent manner (Figure 1).
Compound 5e, was the most active scavenger on ABTS·+. The EC50 value of the above-mentioned compound was approximately one quarter of that of ascorbic acid. Also, 5l, 15, 5g, 9, 5f, 5c, 13, 5h, 5d, 5i and 5k showed very potent activities. The presence of two hydroxyl groups at the 6- and 7-positions (5e, 5l, 5g, 13, 5h, 5d) and respectively at 7- and 8-positions in 15 and 9, resulted in significant ABTS·+ scavenging activity of the compounds. Halogen atoms (chlorine or bromine) at the 5- and 8-positions did not significantly influence the activity in comparison to 5e. ABTS·+ was scavenged by compounds 5e, 5l, 15, 5g, 9, 5f, 5c, 13 and ascorbic acid in a concentration-dependent manner (Figure 2).

Figure 1.
DPPH· scavenging of the most active compounds.

Figure 2.
ABTS·+ scavenging of the most active compounds.
We obtained nine compounds (15, 5d, 5l, 9, 5f, 5g, 5i, 5e, 5k) with superoxide anion radical (O2·−) scavenging activities better than the standard (ascorbic acid). Compounds 15, 5d, 5l, 9, and 5f presented very high superoxide anion radical scavenging activities. Their EC50 values were 22-times lower (15 and 5d) and more than 10-times lower (5l, 9, 5f) than the EC50 value for ascorbic acid. The chloroazetidinone moiety at [2,1a]-position in compound 15 and methyl group at the 4-position improved the activity in comparison to compound 5e. Also, the presence of two hydroxyl groups at the 6- and 7-positions and iodine at the 8-position (5d) or chlorine at the 5-position (5l) caused significant O2·− scavenging activity. Ortho position of two hydroxyl groups in the tested compounds contributed to significant superoxide anion radical scavenging activity. A bromine atom at the 8-position (compound 5g) enhanced O2·− scavenging activity of the compound, in comparison to a chlorine atom at the same position (compound 5h). The EC50 value for compound 5g was about sevenfold lower than the EC50 value for ascorbic acid. This data suggests that substitution of phenolic compounds with a halogen atom (I > Br > Cl) stabilizes the superoxide anion radical. Compound 9, with a dihydrobenzoazepine ring, showed 6-times greater O2·− scavenging activity in comparison to the similar compound, 5e, with a dihydropyridine ring. Tested compounds were able to scavenge superoxide anion radicals in a concentration-dependent manner (Figure 3).

Figure 3.
O2·− scavenging of the most active compounds.
Compound 13 was the most potent ·NO scavenger. Its EC50 value was lower than the standard (Trolox). All of the other tested compounds showed rather moderate scavenging activities on the nitric oxide radical. Comparing the results of all above assays, in general, compounds with two hydroxyl groups (5c–l, 9, 13, 15) were more potent free-radical scavengers than phenolic compounds (5a, 5b). Also, an ortho position of hydroxyl groups determined more significant free radical scavenging activity than the meta position.
2.2.2. DAAO Inhibitory Activity
Similarly, all newly synthesised compounds were screened for DAAO inhibitory activity. Compound 9 was the most active inhibitor of DAAO (Table 3). It showed a fourfold lower IC50 value than the standard benzoic acid. Other compounds, including 5l, 5c, 5h, 5g, 15 and 5f, showed comparable or better DAAO inhibitory activities than the standard. The presence of two hydroxyl groups at the meta position in the benzene ring (5c) as well as the ortho location of hydroxyl groups with additional chlorine or bromine atom at 5- or 8-positions (5g, 5h, 5l, 5f) exhibited good activity. Comparison of the compound with two hydroxyl groups at 6- and 7-positions (5e) and compounds with an additional halogen atom at the 8-position (5g, 5h, 5d) showed that halogen atoms enhanced DAAO inhibition. Moreover, the more electronegative the substituent (Cl > Br > I) at the 8-position, the stronger the resulting DAAO inhibitor. Compounds 5a–5l and 9 are obtained as racemic mixtures. Biological activities of separated enantiomers may be higher (even two times). However, the activity would remain in milimolar concentration range. Synthesised derivatives inhibited DAAO in a concentration-dependent manner (Figure 4).

Figure 4.
DAAO inhibition of the most active compounds.
2.2.3. AChE Inhibitory Activity
The effect of the synthesised compounds on AChE activity was evaluated, using a selective AChE inhibitor, galanthamine hydrobromide as a standard. Only a few of the synthesised compounds (5j, 5f, 5e, 5k) showed rather weak activity against AChE. None of the tested compounds were more potent than the standard. The results are presented in Table 3.
2.2.4. BuChE Inhibitory Activity
The reported compounds were tested for their BuChE inhibitory activity (IC50 values are summarised in Table 3). They showed moderate activities (5l, 5e, 13, 5j) against BuChE.
3. Experimental Section
3.1. General Information
d-Amino acid oxidase from porcine kidney (DAAO), d-alanine, nitroblue tetrazolium chloride (NBT), 2,2-diphenyl-1-picrylhydrazyl (DPPH·), 2,2'-azino bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS·+), potassium persulphate, sodium nitroprusside, Griess reagent, acetylthiocholine iodide, acetylcholinesterase from Electrophorus electricus (AChE), 5,5-dithiobis-2-nitrobenzoic acid (DTNB), galanthamine hydrobromide, butyrylcholinesterase from equine serum (BuChE) and butyrylthiocholine iodide were obtained from Sigma-Aldrich (Poznań, Poland) and 2,4-dinitro phenylhydrazine was purchased from POCH S.A. (Gliwice, Poland). Dibenzyl formamidomalonate has been prepared analogously to the procedure described for diethyl derivative [24]. All others reagents were of standard analytical grade.
NMR spectra were recorded using Varian 400 MR (400 MHz) spectrometer. Chemical shifts of aqueous solutions were referenced against solvent signal (4.75 ppm). Most of the 13C-NMR chemical shifts of deprotected phenols 5 could not be determined due to low solubility and deuteriation in the ring. IR spectra were recorded on Shimadzu IRAffinity-1S spectrophotometer using single reflection ATR (ZnSe). Mass spectra were obtained using LTQ Orbitrap Velos (Thermo Scientific) and QToF Premier (Waters), HR and LR respectively. The radical scavenging activity and enzymes inhibitory activity were assessed by UV-VIS spectrophotometer (FLUOstar Omega BMG LABTECH microplate reader). Copies of 1H and 13C-NMR spectra are available in the Supplementary Information file.
3.2. Synthesis and Characterisation
3.2.1. General Procedure for the Synthesis of Benzyl Chlorides 2
Pyridine (0.36 mL) and thionyl chloride (0.37 mL) were added dropwise to a solution of protected benzyl alcohol 1 (4.78 mmol) in CH2Cl2 (15 mL). The resultant mixture was then stirred at rt for 2.5 h. The reaction was quenched with 10 mL of water and stirred for 0.5 h to destroy excess of thionyl chloride. The organic layer was removed, and the aqueous layer was extracted twice with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over MgSO4 and evaporated. The products were used in the next step without purification.
3.2.2. General Procedure for Alkylation of Dibenzyl Formamidomalonate 3a–3l
Dibenzyl formamidomalonate (315 mg, 0.96 mmol) followed by K2CO3 (1.76 g, 12.75 mmol) and KI (478 mg, 2.88 mmol) were added to a solution of respective benzyl chlorides (0.96 mmol) in acetone (20 mL). The reaction mixture was stirred under reflux (oil bath, 60 °C) for 20 h. When complete, the reaction mixture was cooled and filtered through Celite. The solvent was evaporated to give a solid.
Dibenzyl 2-(2-bromo-5-benzyloxybenzyl)-2-formamidomalonate (3a). The compound was purified by chromatography on silica gel (EtOAc-hexane, 1:3). White crystals. Yield 0.363 g (63%). 1H-NMR (CDCl3) δ: 3.84 (s, 2H, CH2); 4.98 (s, 2H, OCH2Ph); 5.12 and 5.16 (AB, J = 14, 4H, CH2Ph); 6.58 (bs, 1H, NH); 6.69 (d, J = 3.3 Hz, 1H, C6-H); 6.76 (dd, J = 3.3, 8.8 Hz, 1H, C4-H); 7.40 (d, J = 8.8 Hz; 1H, C3-H); 7.16–7.38 (m, 15H, Ar); 7.94 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.6; 65.5; 68.5; 70.0; 116.0; 116.1; 118.7; 127.2; 128.1; 128.3; 128.5; 128.55; 128.6; 133.7; 134.5; 135.4; 136.5; 157.6; 160.1; 166.8. HR MS ESI calculated for C32H29BrNO6 (M+H) 602.1173. Found 602.1170.
Dibenzyl 2-(3-benzyloxy-4-bromobenzyl)-2-formamidomalonate (3b). This compound was used in the next step without purification. Beige solid. Yield 95%. 1H-NMR (CDCl3) δ: 3.84 (s, 2H, CH2); 4.98 (s, 2H, OCH2Ph); 5.12 and 5.16 (AB, J = 12, 4H, CH2Ph); 6.76 (dd, J = 2.9 Hz, J = 8.8, 1H, C6-H); 6.89 (d, J = 2.9, 1H, C2-H); 6.60 (bs, 1H, NH); 7.14–7.38 (m, 15H, Ar); 7.40 (d, J = 8.8, 1H, C5-H); 7.93 (d, J = 0.8 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.6; 65.5; 68.4; 69.9; 116.04; 116.05 118.6; 127.2; 128.0; 128.3; 128.5; 128,6; 133.7; 134.4; 135.4; 136.5; 157.6; 160.1, 166.8. HR MS ESI calculated for C32H29BrNO6 (M+H) 602.1172. Found 602.1170.
Dibenzyl (3,5-dibenzyloxybenzyl)-2-formamidomalonate (3c). The product was purified on silica gel (EtOAc-hexane 1:2.5). White solid. Yield 77%. 1H-NMR (CDCl3) δ: 3.59 (s, 2H, CH2); 4.93 (s, 4H, OCH2Ph); 5.08 and 5.12 (AB, J = 12 Hz, 4H, CH2Ph); 6.17 (d, J = 2.1 Hz, 2H, C2-H, C6-H); 6.52 (t, J = 2.1 Hz, 1H, C4-H); 6.55 (bs, 1H, NH); 7.16–7.44 (m, 20H, Ar); 7.91 (d, J = 0.8 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 38.2; 66.7; 68.3; 69.9; 101.2; 109.2; 127.3; 127.9; 128.3; 128.5; 128.61; 128.62; 134.5; 136.7; 136.8; 159.7; 159.8; 166.6. HR MS ESI calculated for C39H36NO7 (M+H) 630.2486. Found 630.2466.
Dibenzyl 2-(3-iodo-4-benzyloxy-5-methoxybenzyl)-2-formamidomalonate (3d). This compound was used in the next step without purification. Beige solid. Yield 65%. 1H-NMR (CDCl3) δ: 3.63 (s, 2H, CH2); 3.67 (s, 3H, OCH3); 4.97 (s, 2H, OCH2Ph); 5.13 and 5.20 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.51 (d, J = 1.8 Hz, 1H, C6-H); 6.80 (bs, 1H, NH); 6.99 (d, J = 1.8 Hz, 1H, C2-H); 7.24–7.40 (m, 13H, Ar); 7.53–7.57 (m, 2H, Ar); 8.20 (d, J = 1.1 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.2; 55.91; 55.94; 66.8; 68.5; 74.4; 92.8; 114.8; 128.0; 128.3; 128.37; 128.4; 128.69; 128.74; 131.6; 132.7; 134.4; 137.0; 147.2; 152.4; 159.9; 166.5. HR MS ESI calculated for C33H30INO7 (M+H) 680.1139. Found 680.1131.
Dibenzyl 2-(5-bromo-2,3-dimethoxybenzyl)-2-formamidomalonate (3f). The product was purified on silica gel (EtOAc-hexane 1:2). White solid. Yield 66%. 1H-NMR (CDCl3) δ: 3.66 (s, 2H, CH2); 3.66 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 5.13 and 5.17 (AB, J = 12 Hz, 4H, OCH2Ph); 6.67 (bs, 1H, NH); 6.75 (d, J = 2.5 Hz, 1H, C6-H); 6.93 (d, J = 2.5 Hz, 1H, C4-H); 7.21–7.38 (m, 10H, Ar); 8.12 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 33.0; 55.9; 60.6; 65.8; 68.4; 115.2; 116.1; 126.3; 128,2; 128.5; 128.6; 130.1; 134.6; 147.3; 153.3; 160.2; 166.9. HR MS ESI calculated for C27H27BrNO7 (M+H) 556.0965. Found 556.0960.
Dibenzyl 2-(3-bromo-4,5-dimethoxybenzyl)-2-formamidomalonate (3g). The product was purified on silica gel (EtOAc-hexane 1:2). White solid. Yield 65%. 1H-NMR (CDCl3) δ: 3.62 (s, 2H, CH2); 3.66 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 5.12 and 5.20 (AB, J = 12 Hz, 4H, OCH2Ph); 6.44 (d, J = 2.1 Hz, 1H, C2-H); 6.71 (d, J = 2.1 Hz, 1H, C6-H); 6.76 (bs, 1H, NH); 7.24–7.38 (m, 10H, Ar); 8.20 (d, J = 0.8 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.4; 55.9; 60.5; 66.8; 68.5; 113.6; 117.5; 125.8; 128.4; 128.7; 128,8; 131.7; 134.4; 145.8; 153.4; 159.9; 166.5. HR MS ESI calculated for C27H27BrNO7 (M+H) 558.0965. Found 558.0935.
Dibenzyl 2-(3-chloro-4,5-dimethoxybenzyl)-2-formamidomalonate (3h). This compound was purified on silica gel (EtOAc-hexane 1:2.5). White solid. Yield 57%. 1H-NMR (CDCl3) δ: 3.62 (s, 2H, CH2); 3.67 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 5.12 and 5.20 (AB, J = 12.1 Hz, 2H, OCH2Ph); 6.40 (d, J = 1.7 Hz, 1H, C6-H); 6.52 (d, J = 1.7 Hz, 1H, C2-H); 6.76 (s, 1H, NH); 7.25–7.37 (m, 10H, Ar); 8.20 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.5; 56.0; 60.6; 66.7; 68.5; 112.9; 123.0; 128.1; 128.4; 128.7; 128.8; 131.0; 134.4; 144.8; 153.5; 159.9; 166.6. HR MS ESI calculated for C27H27ClNO7 (M+H) 512.1470. Found 512.1467.
Dibenzyl 2-(3,4,5-trimethoxybenzyl)-2-formamidomalonate (3i). This compound was purified on silica gel (EtOAc-hexane 1:1.5). White solid. Yield 70%. 1H-NMR (CDCl3) δ: 3.63 (s, 6H, OCH3); 3.67 (s, 2H, CH2); 3.80 (s, 3H, OCH3); 5.10 and 5.20 (AB, J = 12.1 Hz, 4H, OCH2PH); 6.16 (s, 2H, C2-H, C6-H); 6.73 (bs, 1H, NH); 7.23–7.36 (m, 10H, Ar); 8.19 (d, J = 1.2 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 38.3; 55.9; 60.8; 66.9; 68.3; 106.9; 128.2; 128.67; 128.68; 130.1; 134.5; 137.3; 153.0; 159.8; 166.8. HR MS ESI calculated for C28H30NO8 (M+H) 508.1965. Found 508.1945.
Dibenzyl 2-(4-bromo-3,5-dibenzyloxybenzyl)-2-formamidomalonate (3j). This compound was purified on silica gel (EtOAc-hexane 1:4). Beige solid. Yield 75%. 1H-NMR (CDCl3) δ: 3.58 (s, 2H, CH2); 5.00 (s, 4H, OCH2Ph); 5.02 and 5.05 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.17 (s, 2H, C2-H, C6-H); 6.34 (bs, 1H, NH); 7.14–7.42 (m, 20H, Ar); 7.68 (d, J = 1.2 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 38.2; 66.6; 68.3; 70.6; 101.7; 108.5; 126.7; 127.8; 128.1; 128.5; 128.6; 128.7; 134.4; 134.9; 136.5; 155.9; 159.9; 166.4. HR MS ESI calculated for C39H35BrNO7 (M+H) 708.1591. Found 708.1593.
Dibenzyl (2-chloro-3,5-dibenzyloxybenzyl)-2-formamidomalonate (3k). This compound was used in the next step without purification. Beige solid. Yield 88%. 1H-NMR (CDCl3) δ: 3.87 (s, 2H, CH2); 4.94 (s, 2H, OCH2Ph); 5.07 (s, 2H, OCH2Ph); 5.11 and 5.16 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.30 (d, J = 2.9 Hz, 1H, C4-H); 6.56 (br, 1H, NH); 6.57 (d, J = 2.9 Hz, 1H, C6-H); 7.18–7.45 (m, 20H, Ar); 7.88 (d, J = 1.3, 1H, CHO). 13C-NMR (CDCl3) δ: 35.7; 65.5; 68.4; 70.1; 70.9; 101.7; 109.7; 116.3; 127.1; 127.2; 128.0; 128.1; 128.3; 128.5; 128.56; 128.59; 128.6; 134.47; 134.52; 136.2; 136.5; 155.0; 157.3; 160.1; 166.9. HR MS ESI calculated for C39H35ClNO7 (M+H) 664.2096. Found 664.2079.
Dibenzyl 2-(2-chloro-3,4-dimethoxybenzyl)-2-formamidomalonate (3l). This compound was purified on silica gel (EtOAc-hexane 1:2.5). White solid. Yield 56%. 1H-NMR (CDCl3) δ: 3.81 (s, 3H, OCH3); 3.82 (s, 2H, CH2); 3.83 (s, 3H, OCH3); 5.15 (s, 4H, CH2Ph); 6.63 and 6.70 (AB, J = 8.3 Hz, 2H, C6-H and C5-H); 6.66 (br, 1H, NH); 7.21–7.34 (m, 10H, Ar); 8.13 (s, 1H, CHO). 13C-NMR (CDCl3) δ: 35.1; 55.9; 60.5; 65.7; 68.4; 110.3; 125.4; 126.9; 128.4; 128.6; 129.6; 134.5; 145.4; 152.9; 154.4; 160.1; 166.9. HR MS ESI calculated for C27H27ClNO7 (M+H) 512.1470. Found 512.1448.
3.2.3. General Procedure for Bischler-Napieralski Cyclization. Preparation of Dibenzyl 3,4-Dihydro- isoquinolin-3,3-dicarboxylates 4a–4l
POCl3 (0.23 mL) was added to a stirred solution of the respective dibenzyl formamidomalonate 3a–3l (0.83 mmol) in acetonitrile (10 mL) and the mixture was heated under various conditions depending on the compound. The solvent was evaporated and residue was made alkaline with aqueous NaHCO3 (5 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried with MgSO4 and concentrated to give the product.
Dibenzyl 5-bromo-8-benzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4a). The reaction mixture was heated at 80 °C (temp. of oil bath) for 8 h. The compound was purified by chromatography on silica gel (EtOAc-hexane, 1:3). Brown oil. Yield 0.150 g (31%). 1H-NMR (CDCl3) δ: 3.44 (s, 2H C4-H); 5.12 (s, 2H, OCH2Ph); 5.10 and 5.20 (AB, J = 12.1 Hz, 4H, CH2PH); 6.76 (d, J = 8.7 Hz, 1H, C7-H); 7.48 (d, J = 8.7 Hz, 1H, C6-H); 7.14–7.43 (m, 15H, Ar); 8.95 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 30.8; 67.8; 70.3; 70.6; 113.2; 114.5; 118.8; 126.9; 127.1; 128.2; 128.3; 128.4; 128.5; 128.7; 134.4; 135.1; 135.8; 136.5; 155.7; 157.7; 168.4. MS ESI calculated for C32H27BrNO5 (M+H) 586.1. Found 586.1.
Dibenzyl 8-benzyloxy-7-bromo-3,4-dihydroisoquinolin-3,3-dicarboxylate (4b). The reaction mixture was heated at 73 °C for 15 h. The product was purified on silica gel (EtOAc-hexane 1:6). Brown oil. Yield 100 mg (26%). 1H-NMR (CDCl3) δ: 3.44 (s, 2H C4-H); 5.11 (s, 2H, OCH2Ph); 5.10 and 5.21 (AB, J = 12.5 Hz, 4H, CH2PH); 6.76 (d, J = 8.7 Hz, 1H, C5-H); 7.19–7.40 (m, 15H, Ar); 7.47 (d, J = 8.7 Hz, 1H, C6-H); 8.94 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 30.8; 67.8; 70.3; 70.6; 113.2; 114.5; 118.8; 127.1; 128.0; 128.2; 128.3; 128.4; 128.7; 134.4; 135.1; 135.8; 136.5; 155.7; 157.7; 168.4. MS ESI calculated for C32H27NO5Br (M+H) 584.1. Found 583.9.
Dibenzyl 6.8-dibenzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4c). The reaction mixture was heated at 70 °C for 8 h. Product was purified by extraction with hot cyclohexane. Orange solid. Yield 94%. 1H-NMR (CDCl3) δ: 3.31 (s, 2H C4-H); 5.00 (s, 2H, OCH2Ph); 5.07 (s, 2H, OCH2Ph); 5.10 and 5.19 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.33 (d, J = 2.1 Hz, 1H, C8-H); 6.42 (d, J = 2.1 Hz, 1H, C5-H); 7.20–7.44 (m, 20H, Ar); 8.87 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.7; 67.8; 68.3; 70.3; 99.1; 106.0; 109.1; 127.1; 127.6; 128.0; 128.2; 128.4; 128.7; 128.74; 134.4; 135.1; 135.8; 136.9; 158.0; 158.1. HR MS EI calculated for C39N33NO6 611.2308. Found 611.2292.
Dibenzyl 6-methoxy-7-benzyloxy-8-iodo-3,4-dihydroisoquinolin-3,3-dicarboxylate (4d). The reaction mixture was heated at 80 °C for 9 h. The product was purified on silica gel (EtOAc-hexane 1:3). Brown oil. Yield 94%. 1H-NMR (CDCl3) δ: 3.30 (s, 2H, C4-H); 3.86 (s, 3H, OCH3); 4.97 (s, 2H, OCH2Ph); 5.14 and 5.19 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.63 (s, 1H); 7.17–7.21 (m, 4H, Ar); 7.26–7.29 (m, 5H, Ar); 7.34–7.42 (m, 4H); 7.55–7.58 (m, 2H); 8.65 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.3; 56.1; 67.8; 70.3; 74.5; 98.3; 111.6; 122.4; 128.0; 128.18; 128.2; 128.3; 128.43; 128.44; 132.6; 135.1; 136.7; 147.0; 155.4; 165.2; 168.6. HR MS EI calculated for C33H29INO6 662.1034. Found 662.1037.
Dibenzyl 8-bromo-5,6-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4f). The reaction mixture was heated at 95 °C for 4 h. The product was purified on silica gel (EtOAc-hexane 1:3). Beige oil. Yield 58%. 1H-NMR (CDCl3) δ: 3.36 (s, 2H, C4-H); 3.67 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 5.14 and 5.17 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.95 (s, 1H, C7-H); 7.18–7.30 (m, 10H, Ar); 8.72 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 25.4; 56.0; 60.6; 67.7; 70.1; 114.9; 119.0; 119.7; 128.0; 128.2; 128.4; 129.3; 135.1; 145.2; 155.9; 160.8; 168.5. MS ESI calculated for C27H25BrNO6 (M+H) 538.1. Found 538.1.
Dibenzyl 8-bromo-6,7-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4g). The reaction mixture was heated at 80 °C for 9 h. Product was used in the next step without purification. Brown oil. Yield 94%. 1H-NMR (CDCl3) δ: 3.30 (s, 2H, C4-H); 3.83 (s, 3H, OCH3); 3.86 (s, 3H, OCH3); 5.14 and 5.19 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.59 (s, 1H, C5-H); 7.15–7.36 (m, 10H, Ar); 8.78 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.2; 56.1; 60.6; 67.8; 70.2; 110.5; 119.7; 119.8; 128.0; 128.2; 128.4; 132.1; 135.1; 145.7; 156.3; 160.8; 168.6. HR MS EI calculated for C27H24BrNO6 537.0787. Found 537.0774.
Dibenzyl 8-chloro-6,7-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4h). The reaction mixture was heated at 70 °C for 8 h. Product was used in the next step without purification. Brown oil. Yield 95%. 1H-NMR (CDCl3) δ: 3.31 (s, 2H, C4-H); 3.84 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 5.14 and 5.20 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.56 (s, 1H, C5-H); 7.19–7.38 (m, 10H, Ar); 8.85 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.1; 56.1; 60.7; 67.8; 70.2; 109.8; 118.4; 128.0; 128.2; 128.3; 128.4; 131.4; 135.1; 144.6; 156.5; 158.5; 168.6. HR MS ESI calculated for C27H25ClNO6 (M+H) 494.1365. Found 494.1360.
Dibenzyl 6,7,8-trimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4i). The reaction mixture was heated at 70 °C for 9 h. Product was used in the next step without purification. Brown oil. Yield 95%. 1H-NMR (CDCl3) δ: 3.66 (s, 2H, C4-H); 3.83 (s, 3H, OCH3), 3.99 (s, 3H, OCH3); 4.17 (s, 3H, OCH3); 5.20 and 5.27 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.53 (s, 1H, C5-H); 7.21–7.39 (m, 10H, Ar); 9.02 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 32.4; 57.3; 61.5; 61.6; 66.3; 69.9; 107.4; 110.3; 128.6; 128.7; 128.9; 133.2; 133.8; 139.7; 157.0; 161.5; 163.8; 165.0. HR MS ESI calculated for C28H28NO7 (M+H) 490.1860. Found 490.1841.
Dibenzyl 7-bromo-6,8-dibenzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4j). The reaction mixture was heated at 80 °C for 7 h. Product was used in the next step without purification. Brown oil. Yield 85%. 1H-NMR (CDCl3) δ: 3.29 (s, 2H C4-H); 4.84 (s, 2H, OCH2Ph); 5.15 (s, 2H, OCH2Ph); 5.15 and 5.18 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.55 (s, 1H, C5-H); 7.10–7.55 (m, 20H, Ar); 8.69 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.4; 53.4; 67.8; 70.3; 71.1; 106.5; 108.5; 116.4; 127.0; 128.1; 128.26; 128.3; 128.4; 128.44; 128.57; 128.61; 128.7; 135.0; 135.1; 135.6; 135.8; 156.1; 157.8; 159.0; 168.6. HR MS ESI calculated for C39H33BrNO6 (M+H) 690.1485. Found 690.1464.
Dibenzyl 5-chloro-6,8-dibenzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4k). The reaction mixture was heated at 75 °C for 10 h. Product was used in the next step without purification. Brown oil. Yield of crude product 94%. 1H-NMR (CDCl3) δ: 3.75 (s, 2H, C4-H); 5.18 and 5.26 (AB, J = 12.1 Hz, 4H, CH2Ph); 5.20 (s, 2H, OCH2Ph); 5.21 (s, 2H, OCH2Ph); 6.49 (s, 1H, C7-H); 7.22–7.46 (m, 20H, Ar); 9.07 (s, 1H, C1-H). HR MS EI calculated for C39H33ClNO6 646.1991. Found 646.1978.
Dibenzyl 5-chloro-6,7-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4l). The reaction mixture was heated at 75 °C for 10 h. Product was used in the next step without purification. Brown oil. Yield of crude product 86%. 1H-NMR (CDCl3) δ: 3.44 (s, 2H, C4-H); 3.88 (s, 3H, OCH3); 3.89 (s, 3H, OCH3); 5.09 and 5.22 (AB, J = 12.1 Hz, 2H, CH2Ph); 6.84 (s, 1H, C8-H); 7.15–7.40 (m, 10H, Ar); 8.43 (s, 1H, C1-H). HR MS ESI calculated for C27H25ClNO6 (M+H) 494.1365. Found 494.1352.
3.2.4. General Procedure for Removal of Benzylic Groups. Preparation of 3,4-Dihydroisoquinolin-3-carboxylic Acids 5a–5l
BBr3 (0.2 mL, 2.13 mmol) was added dropwise under argon to a solution of respective dibenzyl 3,4-dihydroisoquinolin-3,3-dicarboxylate 4a–4l (0.17 mmol) in CH2Cl2 (10 mL) at −20 °C. The reaction mixture was stirred for 0.5 h at the same temperature and quenched with H2O (25 mL). The resultant mixture was then stirred at rt for 0.5 h. The water layer was separated and washed 3 times with CH2Cl2. The solution was concentrated under vacuum at rt and the residue was loaded on a column packed with ion exchange resin Dowex 50W X4 (ca. 30 mL, pretreated subsequently with water, 2 M HCl, and water to pH 6–7). The column was washed with water (200 mL), and the product was eluted with 2 M aqueous NH3. Brown to yellow colored fractions were collected. Ammonia was removed under water pump vacuum at rt and the solution was evaporated (water bath temp. 35 °C). The residue was acidified with 2 M aqueous HCl to pH 2, and purified on polyacrylamide (Biogel P-2) using 0.1% aqueous TFA.
5-Bromo-8-hydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5a). Beige powder. Yield 10 mg (22%). 1H-NMR (D2O/DCl) δ: 3.25 and 3.35 (ABX, J = 7.1 Hz, 8.8 Hz, 17.5 Hz, 2H, C4-H); 4.48 (t, J = 7.1 Hz, 1H, C3-H); 6.58 (d, J = 9.6 Hz, 1H, C7-H); 7.59 (d, J = 9.6 Hz, 1H, C6-H); 8.73 (s, 1H, C1-H). MS ESI calculated for C10H9NO3Br (M+H) 269.9. Found 269.9.
7-Bromo-8-hydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5b). Beige powder. Yield 87%. 1H-NMR (D2O/DCl) δ: 3.27 and 3.36 (ABX, J = 7.1 Hz, 8.8 Hz, 17.5 Hz, 2H, C4-H), 4.50 (t, J = 7.9 Hz, C3-H); 6.62 (d, J = 9.1, 1H, C5-H), 7.62 (d, J = 9.1, 1H, C6-H), 8.77 (s, 1H, C1-H). IR: 579; 617; 665; 698; 723; 750; 781; 804; 841; 897; 937; 993; 1040; 1084; 1136; 1184; 1234; 1277; 1292; 1310; 1344; 1364; 1400; 1452; 1601; 1634; 2250–3500 br; 2806; 3044; 3129. MS ESI calculated for C10H9NO3Br (M+H) 270.0. Found 269.9.
6,8-Dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5c). Beige powder. Yield 62%. 1H-NMR (D2O-H2O/DCl) δ: 3.19 and 3.31 (ABX, J = 7.0 Hz, 9.2 Hz, 16.9 Hz, 2H, C3-H); 4.44 (m, 1H, C3-H); 6.13 (d, J = 1.5 Hz, 1H, C7-H); 6.26 (s, 1H, C6-H); 8.64 (s, 1H, C1-H). IR: 586; 619; 633; 662; 704; 733; 770; 808; 843; 870; 901; 957; 986; 1015; 1069; 1113; 1169; 1188; 1202; 1250; 1314; 1348; 1385; 1404; 1504; 1541; 1582; 1609; 2250–3300 br; 2519; 2612; 2689; 2722; 2793; 3071; 3250. MS ESI calculated for C10H10NO4 (M+H) 208.0. Found 208.0.
6,7-Dihydroxy-8-iodo-3,4-dihydroisoquinolin-3-carboxylic acid (5d). The reaction mixture was kept at rt for 24 h. Beige powder. Yield 22%. 1H-NMR (DMSO) δ: 3.23 and 3.31 (ABX, J = 7.5; 8.3; 17.5 Hz, 2H, C4-H); 4.82 (t, J = 7.9 Hz, 1H, C3-H); 6.59 (s, 1H, C5-H); 8.48 (s, 1H, C1-H); 11.9 (bs, 1H, OH). IR: 598; 648; 667; 692; 741; 768; 795; 822; 870; 885; 908; 943; 966; 1013; 1032; 1057; 1128; 1177; 1192; 1219; 1256; 1281; 1298; 1371; 1485; 1508; 1557; 1587; 1634; 1721; 2250–3650 br; 2826; 2911; 2959; 3146. MS ESI calculated for C10H9INO4 (M+H) 333.9. Found: 333.9.
6,7-Dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5e). This compound was obtained from l-DOPA according to published procedure [9]. 1H-NMR (D2O-H2O/DCl) δ: 3.28 and 3.38 (ABX, J = 17.0, 8.6, 7.7, Hz, 1H, C4-H); 4.56 (dd, J = 8.6 Hz, 7.7 Hz, 1H, C3-H); 6.91 (s, 1H, C5-H); 7.28 (s, 1H, C8-H); 8.64 (s, 1H, C1-H). IR: 586; 629; 648; 691; 725; 752; 773; 802; 843; 853; 883; 908; 937; 982; 1013; 1130; 1177; 1196; 1258; 1288; 1371; 1393; 1429; 1531; 1557; 2250–3500 br; 3038.
8-Bromo-5,6-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5f). The reaction mixture was kept at rt for 24 h. Beige powder. Yield 21%. 1H-NMR (D2O-H2O/DCl) δ: 3.13 and 3.21 (ABX, J = 16.9, 8.8, 7.0 Hz, 2H, C4-H); 4.30 (t, J = 8.1 Hz, 1H, C3-H) 6.73 (s, 1H, C7-H); 8.42 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 23.9; 54.9; 108.8; 120.1; 121.4; 126.2; 143.2; 158.4; 175.9. IR: 615; 669; 694; 735; 770; 797; 824; 853; 891; 962; 1009; 1096; 1128; 1202; 1219; 1234; 1298; 1348; 1377; 1404; 1420; 1474; 1499; 1560; 1595; 1719; 2250–3500 br; 2970; 3038; 3134. MS ESI calculated for C10H8BrNO4 (M+H) 286.0. Found 286.0.
8-Bromo-6,7-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5g). The reaction was kept at rt for 24 h. Beige powder. Yield 34%. 1H NMR (D2O-H2O/DCl) δ: 3.32 and 3.43 (ABX, J = 16.5, 8.8, 7.0 Hz, 2H, C4-H); 4.53 (t, J = 8.8 Hz, 1H, C3-H); 6.59 (s, 1H, C5-H); 8.64 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 32.7; 114.1; 114.9; 115.8; 133.9; 142.4; 155.7; 163.9; 170.3. IR: 600; 667; 696; 721; 762; 799; 839; 893; 1040; 1088; 1136; 1182; 1236; 1258; 1279; 1292; 1344; 1368; 1393; 1557; 1593; 2250–3500 br; 2806; 3044; 3125. MS ESI calculated for C10H8BrNO4 (M+H) 286.0. Found 286.0.
8-Chloro-6,7-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5h). The reaction mixture was kept at rt for 16 h. Beige powder. Yield 15%. 1H-NMR (D2O-H2O/DCl) δ: 3.15 and 3.25 (ABX, J = 7.1 Hz, J = 9.2 Hz, J = 17.1 Hz, 2H, C4-H); 4.37 (t, J = 7.9 Hz, 1H, C3-H); 6.41 (s, 1H, C5-H); 8.50 (s, 1H, C1-H). IR: 600; 640; 665; 696; 719; 735; 824; 851; 876; 908; 943; 968; 989; 1032; 1047; 1105; 1134; 1179; 1194; 1215; 1234; 1256; 1265; 1325; 1379; 1393; 1454; 1470; 1487; 1557; 1595; 1611; 1682; 1730; 2851; 2916; 2957; 3225; 3294. HR MS ESI calculated for C10H9ClNO4 (M+H) 242.0215. Found: 242.0213.
6,8-Dihydroxy-7-methoxy-3,4-dihydroisoquinolin-3-carboxylic acid (5i). The reaction mixture was kept at rt for 24 h. Product 5i could not be purified from 6- and 8-methoxy isomers. Beige powder. Yield 61 mg. Major isomer (60%). 1H-NMR (D2O-H2O/DCl) δ: 3.10 (m, 2H, C4-H), 3.82 (s, 3H, OCH3), 4.30 (m, 1H, C3-H), 6.33 (s, 1H, C5-H), 8.39 (s, 1H, C1-H). IR: 554; 600; 635; 671; 756; 777; 800; 901; 949; 991; 1080; 1098; 1138; 1202; 1265; 1304; 1354; 1373; 1454; 1522; 1589; 2250–3650 br; 2835; 3030; 3179. HR MS ESI calculated for C11H12NO5 (M+H) 238.0709. Found: 238.0702.
7-Bromo-6,8-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5j). Beige powder. Yield 53%. 1H-NMR (D2O-H2O/DCl) δ: 2.99 and 3.11 (ABX, J = 6.7, 9.6, 16.2 Hz, 2H, C4-H); 4.25 (dd, J = 9.6, 6.7 Hz; 1H, C3-H); 6.00 (s, 1H, C5-H); 8.33 (s, 1H, C1-H). 13C-NMR (D2O-H2O/DCl) δ: 27.7; 53.8; 97.5; 106.4; 109.0; 137.8; 159.6; 159.7; 164.5; 171.3. IR: 590; 660; 691; 746; 764; 797; 826; 854; 872; 945; 1011; 1030; 1061; 1096; 1123; 1173; 1229; 1246; 1283; 1329; 1369; 1514; 1589; 2250–3500 br; 2970; 3144. MS ESI calculated for C10H9NO4Br (M+H) 286.0. Found 285.9.
5-Chloro-6,8-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5k). The reaction mixture was kept at −20 °C for 1 h. Beige powder. Yield 17%. 1H-NMR (D2O-H2O/DCl) δ: 3.22 and 3.29 (ABX, J = 7.8, J = 8.6, 18.0 Hz, 2H, C4-H); 4.69 (ddd, J = 1.3, 7.8, 8.6 Hz, 1H, C3-H); 6.25 (s, 1H, C7-H); 8.68 (d, J = 1.3 Hz, 1H, C1-H). IR: 598; 633; 650; 671; 743; 781; 829; 858; 939; 1001; 1036; 1094; 1186; 1215; 1248; 1285; 1319; 1337; 1398; 1420; 1504; 1531; 1557; 2250–3650 br; 2602; 2745; 3065. MS ESI calculated for C10H9ClNO4 (M+H) 242.0. Found: 241.9.
5-Chloro-6,7-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5l). The reaction mixture was kept at rt for 21 h. Beige powder. Yield 8%. 1H-NMR (D2O-H2O/DCl) δ: 2.77 and 2.92 (ABX. J = 7.9 Hz, J = 10.0, 17.5 Hz, 2H, C4-H); 4.42 (ddd, J = 7.9, 10.0, 1.7 Hz, 1H, C3-H); 6.64 (s, 1H, C8-H); 8.20 (d, J = 1.5 Hz, 1H, C1-H). 13C-NMR (D2O-H2O/DCl) δ: 53.4; 114.8; 119.9; 127.6; 143.8; 150.6; 150.7; 164.3; 169.7. IR: 606; 631; 654; 689; 718; 731; 758; 800; 826; 853; 868; 907; 930; 951; 976; 1020; 1142; 1171; 1254; 1265; 1308; 1342; 1396; 1429; 1454; 1472; 1514; 1557; 1593; 1639; 2250–3500 br; 2731; 2920; 3063; 3146; 3566. MS ESI calculated for C10H9ClNO4 (M+H) 242.0. Found 241.9.
3.2.5. Preparation of Dibenzyl 2-[2-(3,4-Dimethoxyphenyl)ethyl]-2-formamidomalonate (7)
Solution of dibenzyl formamidomalonate (1.629 g, 4.98 mmol) in anh. DMF (15 mL) was added to a suspension of NaH (299 mg, 7.46 mmol, 60% in oil) in anh. DMF (10 mL). The reaction mixture was stirred for 1 h at rt. Next, a solution of 4-(2-iodoethyl)-1,2-dimethoxybenzene (1.53 g, 4.98 mmol) in anh. DMF (10 mL) was added and the reaction mixture was heated at 60 °C for 20 h. The solvent was distilled off under vacuum. Water (100 mL) was carefully added and the mixture was extracted with CH2Cl2 (3 × 50 mL). Collected extracts were dried over MgSO4 and evaporated. Product was purified by crystallization from diethyl ether cooling the solution to −20 °C. Orange solid. Yield 1.05 g (43%). 1H-NMR (CDCl3) δ: 2.34 (m, 2H, ArCH2CH2); 2.70 (m, 2H, ArCH2CH2); 3.79 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 5.08 and 5.14 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.51–6.55 (m, 2H, C2,6-H); 6.72 (d, J = 8.0 Hz, C5-H); 6.95 (s, 1H, NH); 7.22–7.35 (m, 10H, Ar), 8.18 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 29.4; 33.9; 55.8; 55.9; 65.9; 68.3; 111.1; 111.6; 120.3; 128.3; 128.5; 128.6; 132.6; 134.6; 147.4; 148.8; 159.7; 167.2. MS ESI calculated for C28H29NO7 (M+H) 492.1. Found: 492.2.
3.2.6. Preparation of Dibenzyl 7,8-Dimethoxy-4,5-dihydro-3H-2-benzazepine-3,3-dicarboxylate (8)
This compound was obtained according to the procedure 3.2.3. The reaction mixture was heated at 80 °C for 20 h. The product was purified by dissolving in boiling cyclohexane and cooling to 10 °C. Brown oil. Yield 52%. 1H-NMR (CDCl3) δ: 2.68 (m, 2H, C5-H); 2.85 (m, 2H, C4-H); 3.86 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 5.09 and 5.18 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.58 (s, 1H, C7-H); 6.84 (s, 1H, C10-H); 7.20–7.30 (m, 10H, Ar); 8.41 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 30.6; 33.1; 55.9; 56.0; 67.5; 75.8; 112.2; 116.4; 125.4; 128.0; 128.1; 128.3; 134.8; 135.2; 147.2; 150.8; 163.7; 169.1. HR MS EI calculated for C28H27NO6 473.1838. Found: 473.1852.
3.2.7. Preparation of 7,8-Dihydroxy-4,5-dihydro-3H-2-benzazepine-3-carboxylic Acid (9)
This compound was obtained according to the procedure 3.2.4. The reaction mixture was kept at rt for 24 h. Brown solid. Yield 58%. 1H-NMR (D2O-H2O/DCl) δ: 2.07–2.18 (m, 2H, C4-H); 2.70 and 2.86 (m, 2H, C5-H); 4.62 (t, J = 5.3 Hz, 1H, C3-H); 6.61 (s, 1H, C6-H); 6.99 (s, 1H, C9-H); 8.11 (s, 1H, C1-H). IR: 552; 565; 594; 667; 694; 752; 804; 843; 881; 941; 1038; 1103; 1138; 1171; 1238; 1252; 1281; 1373; 1456; 1506; 1558; 1576; 1601; 2250–3500 br; 2805; 2870; 3042; 3123. MS ESI calculated for C11H12NO4 (M+H) 222.0. Found 222.0.
3.2.8. Preparation of (S)-Benzyl 2-(3,4-Dibenzyloxy)benzyl-2-formamidopropanoate (11)
Formylation of l-methyl-DOPA. Formyloxyacetonitrile (1.326 g, 15.6 mmol) was added to a solution of l-methyl-DOPA hydrate, (1.38 g, 5.8 mmol) in DMSO (10 mL) and the mixture was stirred at rt for 3 days. Solvent was removed under vacuum and the residue was dissolved in acetonitrile (5 mL). Dichloromethane (150 mL) and hexane (10 mL) were added to precipitate product. After filtration white solid was obtained (0.88 g). Yield 64%. 1H-NMR (DMSO) δ: 1.32 (s, 3H, CH3), 2.94 and 2.98 (AB, J = 13.3 Hz, 2H, CH2), 6.36 (dd, J1=1.9, 7.9 Hz, 1H, C6-H), 6.51 (d, J = 1.9 Hz, 1H, C2-H), 6.60 (d, J = 7.9 Hz, 1H, C5-H), 7.90 (br, 1H, NH), 7.93 (d, J = 1.7 Hz, 1H, CHO); 8.70 (br, 1H, OH). 13C-NMR (DMSO) δ: 22.7; 58.9; 115.1; 117.8; 117.8; 121.2; 127.2; 144.0; 144.7; 160.8; 174.6.
Benzylation of l-N-formylmethyl-DOPA. K2CO3, (12.4 g) and benzyl bromide (10.74 g, 62.8 mmol) were added to a solution of l-N-formylmethyl-DOPA (4.29 g, 17.9 mmol) in anh. acetonitrile (80 mL). The reaction mixture was stirred and heated at 60 °C for 16 h. The solvent was evaporated and the water was added to the residue. The mixture was extracted with CH2Cl2 (3 × 20 mL). Combined extracts were dried with Na2SO4 and evaporated. Product was purified on silica gel (EtOAc-CH2Cl2 1:1). White solid. Yield 244 mg (48%). 1H-NMR (CDCl3) δ: 1.68 (s, 3H, CH3), 3.10 (d, J = 13.8 Hz, 1H, CH2), 3.45 (d, J = 13.8 Hz, 1H, CH2), 5.05–5.14 (m, 6H, CH2Ph), 6.06 (bs, 1H, NH), 6.47 (dd, J = 8.3, 2.1 Hz, 1H, C6-H), 6.63 (d, J = 2.1 Hz, 1H, C2-H), 6.75 (d, J = 8.3 Hz, 1H, C5-H), 7.28–7.46 (m, 15H, Ar), 7.87 (d, J = 2.1 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 23.5; 40.9; 61.4; 67.6; 71.0; 71.2; 114.7; 117.0; 122.7; 127.1; 127.2; 127.3; 127.7; 127.8; 128.3; 128.4; 128.6; 128.7; 129.1; 134.9; 137.3; 148.1; 148.3; 160.4; 173.2. HR MS ESI calculated for C32H32NO5 (M+H) 510.2275. Found 510.2252.
3.2.9. Preparation of (S)-Benzyl 6,7-Dibenzyloxy-3-methyl-3,4-dihydroisoquinolin-3-carboxylate (12)
This compound was obtained according to the procedure 3.2.3. The reaction mixture was heated at 60 °C (oil bath) for 1.5 h. The product was purified on silica gel (first CH2Cl2 then CH2Cl2-EtOAc 1:1). Orange oil. Yield 1.479 g (88%). 1H-NMR (CDCl3) δ: 1.45 (s, 3H, CH3), 2.75 (d, 1H, J = 16.3 Hz, C4-H), 3.16 (d, 1H, J = 16.3 Hz, C4-H), 5.15 (s, 2H, CH2Ph); 5.17 (s, 2H, CH2Ph); 5.18 (s, 2H, CH2Ph); 6.72 (s, 1H, C5-H); 6.92 (s, 1H, C8-H); 7.25–7.45 (m, 15H, Ar); 8.22 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 23.6; 33.8; 62.7; 66.8; 71.0; 71.6; 113.8; 114.5; 120.7; 127.2; 127.3; 127.9; 127. 94; 128.0; 128.0; 128.2; 128.4; 128.5; 128.6; 135.9; 136.5; 136.9; 147.8; 152.0; 159.0; 174.5. HR MS ESI calculated for C32H30NO4 (M+H) 492.2169. Found 492.2169.
3.2.10. Preparation of (S)-6,7-Dihydroxy-3-methyl-3,4-dihydroisoquinolin-3-carboxylic Acid (13)
This compound was obtained according to the procedure 3.2.4. Green powder. Yield 88%. 1H-NMR (D2O-H2O/DCl) δ: 1.62 (s, 3H, CH3); 3.12 and 3.36 (AB, J = 17.1 Hz, 2H, C4-H); 6.79 (s, 1H, C5-H); 7.16 (s, 1H, C8-H); 8.57 (s, 1H, C1-H). 13C-NMR (D2O-H2O/DCl) δ: 21.8; 34.0; 60.8; 115.2; 116.0; 120.7; 131.7; 144.1; 155.8; 164.2; 173.8. IR: 554; 596; 627; 650; 719; 750; 797; 835; 883; 941; 966; 1126; 1180; 1233; 1277; 1331; 1381; 1447; 1516; 1557; 1568; 1614; 1634; 1643; 1667; 1732; 2250–3500 br; 2787; 2945; 3123; 3196. HR MS ESI calculated for C11H11NO4Na (M+Na) 244.0580. Found 244.0589.
3.2.11. Preparation of Benzyl 1-Chloro-7,8-dibenzyloxy-4-methyl-2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]isoquinoline-4-carboxylate (14)
Et3N (654 mg, 6.48 mmol) and solution of chloroacetyl chloride (549 mg, 4.86 mmol) in CH2Cl2 (2 mL) were added to a stirred solution of benzyl 6,7-dibenzyloxy-3,4-dihydroisoquinolin-3-methyl-3-carboxylate (400 mg, 0.81 mmol) in CH2Cl2 (10 mL). The reaction mixture was stirred at rt for 20 h and quenched with aqueous NaHCO3 (2 mL). Stirring was continued for next 1 h. Water layer was separated and extracted with CH2Cl2. Organic layers were dried over MgSO4, evaporated and purified by chromatography on silica gel (acetone/CH2Cl2 150:1). Brown oil. Yield 140 mg (47%). 1H-NMR (CDCl3) δ: 1.88 (s, 3H, CH3); 2.94 and 3.14 (AB, J = 15.8, 2H, C5-H); 4.41 (d, J = 1.7, 1H, C1-H); 4.69 (br, 1H, C9b-H); 5.09 (1/2AB, J = 12.1, 1H, CH2Ph); 5.10 (s, 2H, OCH2Ph); 5.12 (s, 2H, OCH2Ph); 5.13 (1/2AB, J = 12.1, 1H, CH2Ph); 6.64 (s, 1H, C6-H); 6.75 (s, 1H, C9-H); 7.13–7.46 (m, 15H, Ar). 13C-NMR (CDCl3) δ: 22.3; 39.3; 60.3; 60.4; 61.2; 67.3; 71.2; 71.4; 111.6; 115.4; 124. 2; 124.9; 127.2; 127.4; 127.8; 127.9; 128.0; 128.4; 128.5; 135.1; 136.6; 136.7; 148.5; 149.0; 162.9; 171.3. HR MS ESI calculated for C34H31ClNO5 (M+H) 568.1813. Found: 568.1887.
3.2.12. Preparation of 1-Chloro-7,8-dihydroxy-4-methyl-2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]-isoquinoline-4-carboxylic Acid (15)
Pd/C (10%, 15 mg) was added to a solution of benzyl 1-chloro-7,8-dibenzyloxy-4-methyl-2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]isoquinoline-4-carboxylate 14 (90 mg, 0.16 mmol) in EtOH (7 mL) and a balloon with hydrogen was connected. The suspension was stirred for 3 h. The catalyst was centrifuged and washed two times with EtOH (3 mL). Collected solution were evaporated and dried under vacuum. White solid. Yield 40 mg (85%). 1H-NMR (acetone-d6) δ: 1.80 (s, 3H, CH3); 2.94 and 3.13 (AB, J = 15.4 Hz, 2H, C5-H); 4.70 (br, 1H, C9b-H); 4.74 (d, J = 2.1 Hz, 1H, C1-H); 6.69 (s, 1H, C6-H); 6.77 (s, 1H, C9-H); 8.10 (br, 1H, OH); 8.15 (br, 1H, OH). 13C-NMR (acetone-d6) δ: 22.5; 39.5; 60.7; 61.1; 62.1; 112.8; 116.8; 124.1; 124.7; 145.5; 146.1; 163.3; 173.3. HR MS ESI calculated for C13H12ClNO5 (M+H) 298.0404. Found: 298.0478.
3.3. Free-Radical Scavenging Properties
3.3.1. DPPH Radical Scavenging Assay
The radical-scavenging activity of tested compounds against DPPH· was determined by Williams’ method [25]. Tested compounds dissolved in water or methanol (30 μL) were added at various concentrations (to final concentration of 2, 4, 8, 16, 32, 64, 128, 256, 512 μg/mL) to freshly prepared 0.22 mM DPPH· in methanol (120 μL). The absorbance was measured against the control in the microplate reader at 517 nm after 30 min of incubation at room temperature (rt). The control contained all reagents without the tested compound. The corresponding blank reading was also taken and the remaining DPPH· was calculated. Each sample was replicated three times. Ascorbic acid was used as a standard.
3.3.2. ABTS Radical Scavenging Assay
The radical-scavenging activity of tested compounds against ABTS·+ and the generation of the ABTS·+ radical was performed according to methods described previously [26,27]. Briefly, an ABTS·+ solution was prepared by mixing 2 mM ABTS (50 mL) with 70 mM potassium persulphate (200 μL), both in deionized water, and used after 2 h of being kept in the dark at rt. The ABTS·+ solution (40 μL) was added to 0.1 M phosphate buffer pH 7.4 (60 μL) and tested compounds (25 μL) were dissolved in water or methanol at various concentrations (to final concentration of 2, 4, 8, 16, 32, 64, 128, 256, 512 μg/mL). The absorbance was measured against the control in the microplate reader at 734 nm after 30 min of incubation at rt. The control contained all reagents without the tested compound. The corresponding blank reading was also taken and the remaining ABTS·+ was calculated. Each sample was replicated three times. Ascorbic acid was used as a standard.
3.3.3. Superoxide Anion Radical Scavenging Assay
Superoxide scavenging assay was based on inhibition of the formazan dye formation. O2·− was generated in alkaline DMSO by the addition of sodium hydroxide to air-saturated pure DMSO [28]. The generated superoxide reacts with NBT to give coloured diformazan. To the reaction mixture containing NBT (10 μL; 1 mg/mL solution in DMSO) and the various concentration of scavenger (30 μL; to final concentration of 2, 4, 8, 16, 32, 64, 128, 256, 512 μg/mL), alkaline DMSO (100 μL) was added to give a final volume of 140 μL per well in a microplate. The absorbance was measured in the microplate reader at 560 nm after 30 min of incubation at rt [29]. The same procedure was repeated for the control in which DMSO was added instead of scavenger solution. Each sample was replicated three times and the corresponding blank reading was also taken. Ascorbic acid was used as a standard.
3.3.4. Nitric Oxide Radical Scavenging Assay
Nitric oxide was generated from sodium nitroprusside and measured by the Griess reaction [26]. Scavengers of ·NO compete with oxygen leading to reduced production of nitric oxide. 10 mM sodium nitroprusside in 0.1 M phosphate buffer pH 7.4 (30 μL) was mixed with tested compounds dissolved in water or methanol (25 μL) at various concentrations (to final concentration of 2, 4, 8, 16, 32, 64, 128, 256, 512 μg/mL) and incubated at 25 °C for 5 h. After incubation, Griess reagent (50 μL) was added. The absorbance was measured in the microplate reader at 546 nm against a control (without tested compounds) treated in the same way with Griess reagent. The corresponding blank reading was also taken. Each sample was replicated three times. Trolox was used as a standard.
3.3.5. Effective Concentration of Scavengers (EC50)
The percentage of radical scavenging was calculated using Equation (1) (DPPH·, ABTS·+ and ·NO scavenging assays). The absorbance for the control is Ac and the absorbance in the presence of the compounds or other scavenger is As:
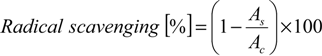
The percentage of O2·− scavenging was calculated using Equation (2). The absorbance for the control is Ac and the absorbance in the presence of the compounds or other scavenger is As:
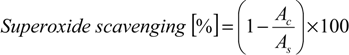
The amount of tested compound needed to scavenge free radicals by 50%, EC50 (effective concentration), was calculated by the linear regression between scavenger concentration and percentage of scavenging and expressed as μM.
3.4. DAAO Inhibitory Activity
Inhibition of DAAO was based on the determination of the α-keto acid obtained from the reaction between a DAAO and d-Ala according to the described method [30]. The method was adapted to microtitre plates and a total volume of 210 μL per well. Inhibitor solution in DMSO (15 μL), 5 mM d-Ala in 0.2 M Tris-HCl buffer pH 8.2 (121 μL) and 10 U/mL DAAO solution in 0.2 M Tris-HCl buffer pH 8.2 (5 μL) were mixed and incubated at 37 °C for 30 min. After incubation, 1 mM 2,4-dinitro phenylhydrazine dissolved in 1 M HCl (14 μL) was added to the well, mixed and further incubated at 37 °C for 10 min. Next, 1.5 M NaOH (55 μL) was added and incubated for 10 min. The absorbance was read at 445 nm against a blank sample consisting the same assay mixture but without the substrate (using Tris-HCl buffer instead of d-Ala). The positive control of the enzyme activity consisted of DMSO (15 μL) instead of inhibitor solution and the analogous blank contained Tris-HCl buffer instead of d-Ala. Sample, control and related blanks were carried out under the same condition. Benzoic acid was used as a standard. Eight different solutions of inhibitor were examined: 0.0005, 0.0025, 0.005, 0.00625, 0.0125, 0.025, 0.0375 and 0.05 M. For each of these inhibitor concentrations percentage of inhibition was calculated by using Equation (3). The absorbance for the control is Ac and the absorbance in the presence of the compounds or other inhibitor is Ai:
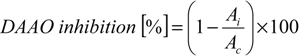
The inhibitor concentration needed to inhibit DAAO by 50% (IC50) was calculated by the linear regression between inhibitor concentration and percentage of inhibition. Each sample was replicated three times.
3.5. AChE Inhibitory Activity
The assay was based on Ellman’s method [31] with modifications. AChE activity was measured by the determination of a yellow colour produced from acetylthiocholine iodide when it reacted with the dithiobisnitrobenzoate ion. The assay was performed using microtitre plates and a total volume of 100 µL per well. In a 96-well plate, 0.1 M pH 7.4 phosphate buffer (32 µL), a serially diluted solution of tested compounds (5 µL), 0.59 U/mL AChE solution (4 µL) and 0.3 mM DTNB (42 µL) were mixed and pre-incubated at 25 °C for 2 min. Then, the substrate, 0.4mM acetylthiocholine iodide (17 µL), was added to the well and mixed. The enzymatic reaction was followed for 10 min, with a measurement every 12 s. Changes in the absorbance at 412 nm were detected in the microplate reader. The absorbance was measured against a blank sample containing the same assay mixture but without an enzyme (using phosphate buffer instead of AChE). The positive control of the enzyme activity consisted of DMSO (1.5 μL) instead of inhibitor solution. The sample, control and related blanks were measured under the same conditions. Galanthamine hydrobromide was used as a standard. Each sample was replicated three times. The concentration of the compounds that caused 50% inhibition of AChE activity (IC50) was calculated using linear regression analysis. At least three concentrations of tested compounds were assayed. For each of these inhibitor concentrations, the percentage of inhibition was calculated using Equation (4). The difference in absorbance for the inhibitor at t = 10 min and t = 0 is denoted ΔAi. The difference in absorbance for the control at t = 10 min and t = 0 is denoted ΔAc:
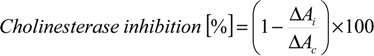
3.6. BuChE Inhibitory Activity
The assay was based on Ellman’s method [31] with modifications. BuChE activity was measured by the determination of the yellow colour produced from butyrylthiocholine iodide when it reacted with the dithiobisnitrobenzoate ion. The assay was performed using microtitre plates and a total volume of 100 µL per well. In a 96-well plate, 0.1 M pH 7.4 phosphate buffer (35 µL), a serially diluted solution of tested compounds (5 µL), 1.485 U/mL BuChE solution (5 µL) and 0.3 mM DTNB (42 µL) were mixed and pre-incubated at 25 °C for 2 min. Then, the substrate, 0.6 mM butyrylthiocholine iodide (13 µL), was added to the well and mixed. The enzymatic reaction was followed for 10 min, with a measurement every 12 s. Changes in absorbance at 412 nm were detected in the microplate reader. The absorbance was measured against a blank sample containing the same assay mixture, but without an enzyme (using phosphate buffer instead of BuChE). The positive control for enzyme activity contained DMSO (1.5 μL) instead of inhibitor solution. The sample, control and related blanks were measured under the same conditions. Galanthamine hydrobromide was used as a standard. Each sample was replicated three times. The concentration of the compounds that caused 50% inhibition of BuChE activity (IC50) was calculated using linear regression analysis. At least three concentrations of the tested compounds were assayed. For each of these inhibitor concentrations, the percentage of inhibition was calculated using Equation (4).
4. Conclusions
We investigated newly synthesised compounds (hydroxy- and halogeno-substituted derivatives of 3,4-dihydroisoquinoline-3-carboxylic acid, 2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]isoquinoline analogue and 4,5-dihydro-3H-2-benzazepine analogues) for their free-radical-scavenging activity and DAAO, AChE, BuChE inhibitory activity. The obtained dihydroxy-substituted derivatives of 3,4-dihydroisoquinoline-3-carboxylic acid were found to be more potent scavengers of DPPH·, ABTS·+, O2·− and ·NO than monohydroxy-substituted derivatives. The presence of ortho-dihydroxy structures determined higher free-radical scavenging activities than the meta location of these substituents. These compounds were shown to be potent free-radical scavengers on at least one free radical used in this study. Obtained results are significant due to the fact that the 3,4-dihydroisoquinoline derivatives have not been described so far as potent free radical scavengers.
In conclusion, the potent free-radical scavengers obtained in the study may be potential candidates for therapeutics used in oxidative-stress-related diseases. The synthesised inhibitors of evaluated enzymes may deserve attention in the design of new antipsychotic drugs. Despite rather low inhibitory activities against DAAO, AChE and BuChE, it seems that compounds with potent free-radical scavenging properties may play a supporting role in the treatment of neurodegenerative and psychiatric diseases.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/19/10/15866/s1.
Supplementary Files
Supplementary File 1Acknowledgments
Financial support by the European Union within the European Regional Development Fund (grant number UDA-POIG.01.03.01-14-136/09/08) is gratefully acknowledged.
Author Contributions
J.S. and R.K. designed research; J.S., R.K., A.G., M.P., J.Z., K.L., A.O., B.P., K.P. and A.W. performed research, analyzed and interpreted the data; J.S, R.K., J.Z., A.G. and M.P wrote the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alvarez, F.N.; Carlson, L.M.; Lindner, I.; Lee, K.P. 6,7-Dihydroxy-3,4-dihydroisoquinoline: A novel inhibitor of nuclear factor-κB and in vitro Invasion in Murine Mammary cancer cells. Chemotherapy 2009, 55, 175–182. [Google Scholar]
- Khalturina, V.V.; Shklyaev, Y.V.; Makhmudov, R.R.; Maslivets, A.N. Synthesis, analgesic and anti-inflammatory activity of (1Z,3Z)-4-aryl-4-hydroxy-1-(3,3-dialkyl-3,4-dihydroisoquinoline-1(2H)-ylidene)-but-3-en-4-ones. Pharm. Chem. J. 2010, 44, 480–482. [Google Scholar]
- Aleksandrov, B.B.; Gavrilov, M.S.; Dautova, R.Z. Biological activity of fluorinated 3,4-dihydroisoquinolines. Pharm. Chem. J. 1992, 26, 57–58. [Google Scholar]
- Diana, G.D.; Hinshaw, W.B.; Lape, H.E. Synthesis and antihypertensive activity of 1-amino-3,4-dihydroisoquinolines. J. Med. Chem. 1977, 20, 449–452. [Google Scholar]
- Harmon, R.E.; Jensen, B.L.; Gupta, S.K.; Hanka, L.J. Synthesis and antibacterial activity of 1-styryl-3,4-dihydroisoquinoline. J. Pharm. Sci. 1970, 59, 576. [Google Scholar]
- Cheng, B.Y.; Origitano, T.C.; Collins, M.A. Inhibition of catechol-O-methyltransferase by 6,7-dihydroxy-3,4-dihydroisoquinolines related to dopamine: demonstration using liquid chromatography and a novel substrate for O-methylation. J. Neurochem. 1987, 48, 779–786. [Google Scholar]
- Christopher, J.A.; Atkinson, F.L.; Bax, B.D.; Brown, M.J.; Champigny, A.C.; Chuang, T.T.; Jones, E.J.; Mosley, J.E.; Musgrave, J.R. 1-Aryl-3,4-dihydroisoquinoline inhibitors of JNK3. Bioorg. Med. Chem. Lett. 2009, 19, 2230–2234. [Google Scholar]
- Solecka, J.; Rajnisz, A.; Laudy, A.E. A novel isoquinoline alkaloid, dd-carboxypeptidase inhibitor, with antibacterial activity isolated from Streptomyces sp. 8812. Part I: Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 2009, 62, 575–580. [Google Scholar]
- Solecka, J.; Sitkowski, J.; Bocian, W.; Bednarek, E.; Kawęcki, R.; Kozerski, L. A novel isoquinoline alkaloid, dd-carboxypeptidase inhibitor, with antibacterial activity isolated from Streptomyces sp. 8812. Part II: Physicochemical properties and structure elucidation. J. Antibiot. 2009, 62, 581–585. [Google Scholar]
- Cassels, B.K.; Asencio, M.; Conget, P.; Speisky, H.; Videla, L.A.; Lissi, E.A. Structure-antioxidative activity relationship in benzylisoquinoline alkaloids. Pharmacol. Res. 1995, 2, 103–107. [Google Scholar]
- Ji, H.F.; Shen, L. Berberine: A Potential multipotent natural product to combat Alzheimer’s disease. Molecules 2011, 16, 6732–6740. [Google Scholar]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Antioxidant properties of phenolic compounds. Trends Plant Sci. 1997, 2, 152–159. [Google Scholar]
- Brewer, M.S. Natural antioxidants: sources, compounds, mechanisms of action, and potential applications. Compr. Rev. Food Sci. Food Saf. 2011, 10, 221–247. [Google Scholar]
- Geldof, N.; Engeseth, N.J. Antioxidant capacity of honeys from various floral sources based on the determination of oxygen radical absorbance capacity and inhibition of in vitro lipoprotein oxidation in human serum samples. J. Agric. Food Chem. 2002, 50, 3050–3055. [Google Scholar]
- Fernandez-Panchon, M.S.; Villano, D.; Troncoso, A.M.; Garcia-Parrilla, M.C. Antioxidant activity of phenolic compounds: From in vitro results to in vivo evidence. Crit. Rev. Food Sci. Nutr. 2008, 48, 649–671. [Google Scholar]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar]
- Ďuračková, Z. Some current insights into oxidative stress. Physiol. Res. 2010, 59, 459–469. [Google Scholar]
- Pham Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Genestra, M. Oxyl radicals, redox-sensitive signaling cascades and antioxidants. Cell Signal. 2007, 19, 1807–1819. [Google Scholar]
- Jomova, K.; Valko, M. Health protective effects of Carotenoids and their interactions with other biological antioxidants. Eur. J. Med. Chem. 2013, 70, 102–110. [Google Scholar]
- Bischler, A.; Napieralski, B. Zur Kenntnis einer neuen Isochinolinsynthese. Chem. Ber. 1893, 26, 1903–1908. [Google Scholar]
- Felix, A.M. Cleavage of protecting groups with boron tribromide. J. Org. Chem. 1974, 39, 1427–1429. [Google Scholar]
- Bose, A.K.; Amin, S.G.; Kapur, J.C.; Manhas, M.S. Studies on lactams. Part 45. Some carbocyclic analogues of cephalosporin. J. Chem. Soc. Perkin Trans. 1 1976, 2193–2197. [Google Scholar]
- Galun, A.B. Preparation of diethyl formamidomalonate. J. Chem. Eng. Data 1976, 21, 241. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar]
- Shirwaikar, A.; Shirwaikar, A.; Rajendran, K.; Punitha, I.S. In vitro antioxidant studies on the benzyl tetra isoquinoline alkaloid berberine. Biol. Pharm. Bull. 2006, 29, 1906–1910. [Google Scholar]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar]
- Hyland, K.; Auclair, C. The formation of superoxide radical anions by a reaction between O2, OH− and dimethyl sulfoxide. Biochem. Biophys. Res. Commun. 1981, 102, 531–537. [Google Scholar]
- Sanja, S.D.; Sheth, N.R.; Patel, N.K.; Patel, D.; Patel, B. Characterization and evaluation of antioxidant activity of Portulaca oleacea. Int. J. Pharm. Pharm.Sci. 2009, 1, 74–84. [Google Scholar]
- Oguri, S.; Watanabe, K.; Nozu, A.; Kamiya, A. Screening of d-amino acid oxidase inhibitor with new multi-assay method. Food Chem. 2007, 100, 616–622. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar]
- Sample Availability: Sample of the compound 5e is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).