Photochemical Aryl Radical Cyclizations to Give (E)-3-Ylideneoxindoles

Abstract
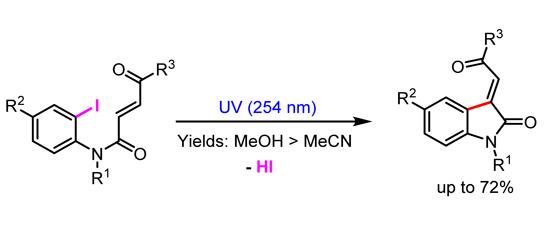
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction


2. Results and Discussion




3. Experimental Section
3.1. General
3.2. Experimental Procedures
3.2.1. Synthesis of Radical Precursors
3.2.2. Photochemical Radical Cyclizations
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Cao, Y.; Jiang, X.; Liu, L.; Shen, F.; Zhang, F.; Wang, R. Enantioselective Michael/cyclization reaction sequence: Scaffold-inspired synthesis of spirooxindoles with multiple stereocenters. Angew. Chem. Int. Ed. 2011, 50, 9124–9127. [Google Scholar] [CrossRef]
- Palumbo, C.; Mazzeo, G.; Mazziotta, A.; Gambacorta, A.; Loreto, M.A.; Migliorini, A.; Superchi, S.; Tofani, D.; Gasperi, T. Noncovalent organocatalysis: A powerful tool for nucleophilic epoxidation of α-ylideneoxindoles. Org. Lett. 2011, 13, 6248–6251. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, M.; Pal, A.; Sharma, R.; Nair, V.A. Quinine as an organocatalytic dual activator for diastereoselective synthesis of spiro-epoxyoxindoles. Tetrahedron Lett. 2013, 54, 7119–7123. [Google Scholar] [CrossRef]
- Cao, S.H.; Zhang, X.C.; Wei, Y.; Shi, M. Chemoselective reduction of isatin-derived electron-deficient alkenes using alkylphosphanes as reduction agents. Eur. J. Org. Chem. 2011, 2668–2672. [Google Scholar]
- Zou, Y.Q.; Duan, S.W.; Meng, X.G.; Hu, X.Q.; Gao, S.; Chen, J.R.; Xiao, W.J. Visible light induced intermolecular [2+2]-cycloaddition reactions of 3-ylideneoxindoles through energy transfer pathway. Tetrahedron 2012, 68, 6914–6919. [Google Scholar] [CrossRef]
- Duan, S.W.; Li, Y.; Liu, Y.Y.; Zou, Y.Q.; Shi, D.Q.; Xiao, W.J. An organocatalytic Michael-aldol cascade: Formal [3+2] annulations to construct enantioenriched spirocyclic oxindole derivatives. Chem. Commun. 2012, 48, 5160–5162. [Google Scholar] [CrossRef]
- Li, T.R.; Duan, S.W.; Ding, W.; Liu, Y.Y.; Chen, J.R.; Lu, L.Q.; Xiao, W.J. Synthesis of CF3-containing 3,3'-cyclopropyl spirooxindoles by sequential [3+2] cycloaddition/ring contraction of ylideneoxindoles with 2,2,2-trifluorodiazoethane. J. Org. Chem. 2014, 79, 2296–2302. [Google Scholar] [CrossRef] [PubMed]
- Azizian, J.; Mohammadizadeh, M.R.; Kazemizadeh, Z.; Karimi, N.; Mohammadi, A.A.; Karimi, A.R.; Alizadeh, A. A rapid and highly efficient one-pot methodology for preparation of alkyl oxindolideneacetates. Lett. Org. Chem. 2006, 3, 56–57. [Google Scholar] [CrossRef]
- Majik, M.S.; Rodrigues, C.; Mascarenhas, S.; D’Souza, L. Design and synthesis of marine natural product-based 1H-indole-2,3-dione scaffold as a new antifouling/antibacterial agent against fouling bacteria. Bioorg Chem. 2014, 54, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Millemaggi, A.; Taylor, R.J.K. 3-Alkenyl-oxindoles: Natural products, pharmaceuticals, and recent synthetic advances in tandem/telescoped approaches. Eur. J. Org. Chem. 2010, 2010, 4527–4547. [Google Scholar] [CrossRef]
- Wright, C.; Shulkind, M.; Jones, K.; Thompson, M. A formal total synthesis of geneserine. Tetrahedron Lett. 1987, 28, 6389–6390. [Google Scholar] [CrossRef]
- Bowman, W.R.; Heaney, H.; Jordan, B.M. Synthesis of oxindoles by radical cyclisations. Tetrahedron Lett. 1988, 29, 6657–6660. [Google Scholar] [CrossRef]
- Jones, K.; McCarthy, C. Chiral induction in aryl radical cyclisations. Tetrahedron Lett. 1989, 30, 2657–2660. [Google Scholar] [CrossRef]
- Jones, K.; Storey, J.M.D. Aryl radical cyclisation approach to highly substituted oxindoles related to mitomycins. Tetrahedron Lett. 1993, 34, 7797–7798. [Google Scholar] [CrossRef]
- Jones, K.; Wilkinson, J.; Ewin, R. Intramolecular reactions using amide links: Aryl radical cyclisation of silylated acryloylanilides. Tetrahedron Lett. 1994, 35, 7673–7676. [Google Scholar] [CrossRef]
- Curran, D.P.; Liu, W.; Hui-Tung Chen, C. Transfer of chirality in radical cyclizations. cyclization of o-haloacrylanilides to oxindoles with transfer of axial chirality to a newly formed stereocenter. J. Am. Chem. Soc. 1999, 121, 11012–11013. [Google Scholar] [CrossRef]
- Akamatsu, H.; Fukase, K.; Kusumoto, S. Solid-Phase synthesis of indol-2-ones by microwave-assisted radical cyclization. Synlett 2004, 2004, 1049–1053. [Google Scholar]
- Petit, M.; Geib, S.J.; Curran, D.P. Asymmetric reactions of axially chiral amides: Use of removable ortho-substituents in radical cyclizations of o-iodoacrylanilides and N-allyl-N-o-iodoacrylamides. Tetrahedron 2004, 60, 7543–7552. [Google Scholar] [CrossRef]
- Nishio, T.; Iseki, K.; Araki, N.; Miyazaki, T. Synthesis of indolones via radical cyclization of N-(2-halogenoalkanoyl)-substituted anilines. Helv. Chim. Acta 2005, 88, 35–41. [Google Scholar] [CrossRef]
- Pudlo, M.; Gérard, S.; Mirand, C.; Sapi, J. A tandem radical cyclizations approach to 3-(2-oxopyrrolidin-3-yl)indolin-2-ones, potential intermediates toward complex indole-heterocycles. Tetrahedron Lett. 2008, 49, 1066–1070. [Google Scholar] [CrossRef]
- Pudlo, M.; Allart-Simon, I.; Tinant, B.; Gérard, S.; Sapi, J. First domino radical cyclisation/Smiles rearrangement combination. Chem. Commun. 2012, 48, 2442–2444. [Google Scholar]
- Clyne, M.A.; Aldabbagh, F. Photochemical intramolecular aromatic substitutions of the imidazol-2-yl radical are superior to those mediated by Bu3SnH. Org. Biomol. Chem. 2006, 4, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Aldabbagh, F.; Clyne, M.A. Intramolecular aromatic substitutions of the imidazol-5-yl radical to form tricyclic imidazo[5,1-a] heterocycles. Lett. Org. Chem. 2006, 3, 510–513. [Google Scholar] [CrossRef]
- O’Connell, J.M.; Moriarty, E.; Aldabbagh, F. Access to aromatic ring-fused benzimidazoles using photochemical substitutions of the benzimidazol-2-yl radical. Synthesis 2012, 44, 3371–3377. [Google Scholar] [CrossRef]
- CCDC 1019237 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-Mail: [email protected]).
- Harwood, L.M. Dry-column flash chromatography. Aldrichim. Acta 1985, 18, 25. [Google Scholar]
- Sample Availability: Samples of some compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurry, M.; Allart-Simon, I.; McArdle, P.; Gérard, S.; Sapi, J.; Aldabbagh, F. Photochemical Aryl Radical Cyclizations to Give (E)-3-Ylideneoxindoles. Molecules 2014, 19, 15891-15899. https://doi.org/10.3390/molecules191015891
Gurry M, Allart-Simon I, McArdle P, Gérard S, Sapi J, Aldabbagh F. Photochemical Aryl Radical Cyclizations to Give (E)-3-Ylideneoxindoles. Molecules. 2014; 19(10):15891-15899. https://doi.org/10.3390/molecules191015891
Chicago/Turabian StyleGurry, Michael, Ingrid Allart-Simon, Patrick McArdle, Stéphane Gérard, Janos Sapi, and Fawaz Aldabbagh. 2014. "Photochemical Aryl Radical Cyclizations to Give (E)-3-Ylideneoxindoles" Molecules 19, no. 10: 15891-15899. https://doi.org/10.3390/molecules191015891