Bio-Inspired Nitrile Hydration by Peptidic Ligands Based on L-Cysteine, L-Methionine or L-Penicillamine and Pyridine-2,6-dicarboxylic Acid

Abstract
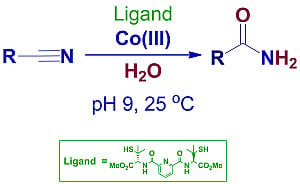
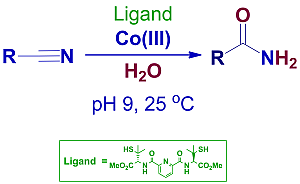
:
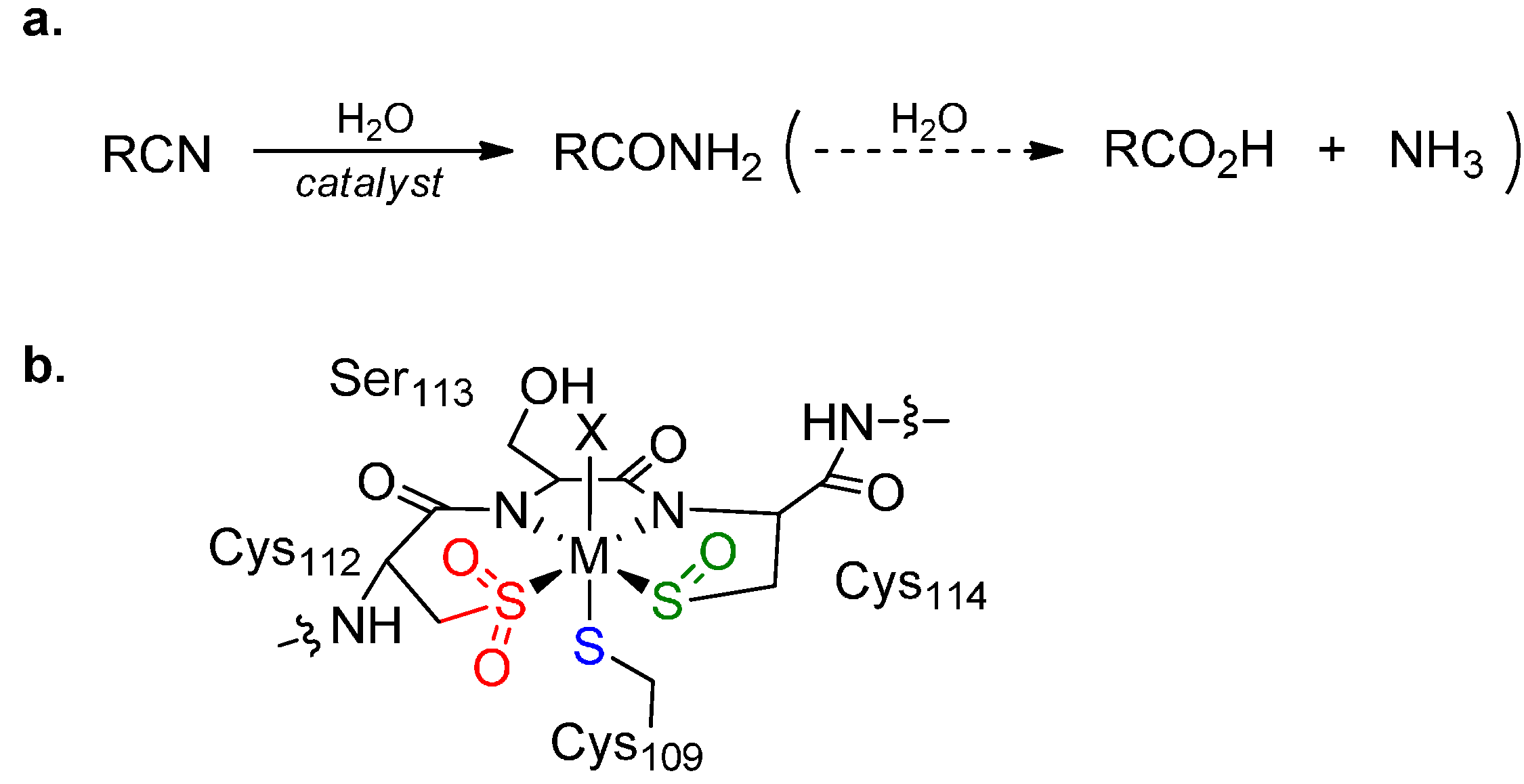
1. Introduction


2. Results and Discussion
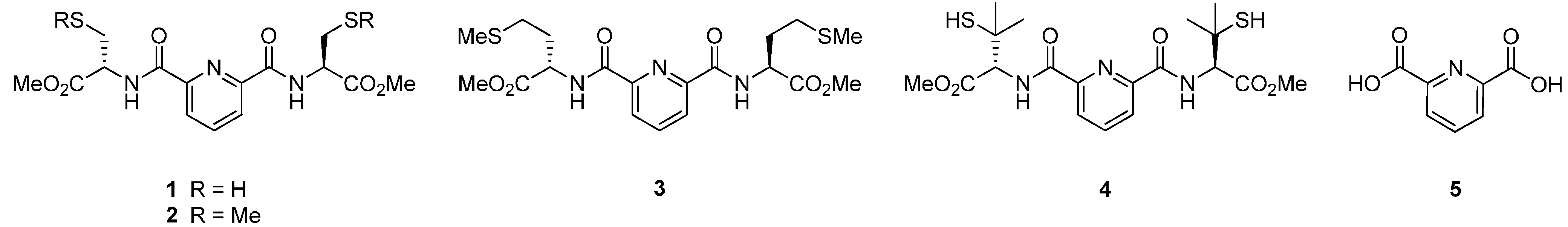
2.1. Synthesis of Ligands


2.2. Nitrile Hydration Experiments

{kind=link}
{kind=link}
{kind=link}
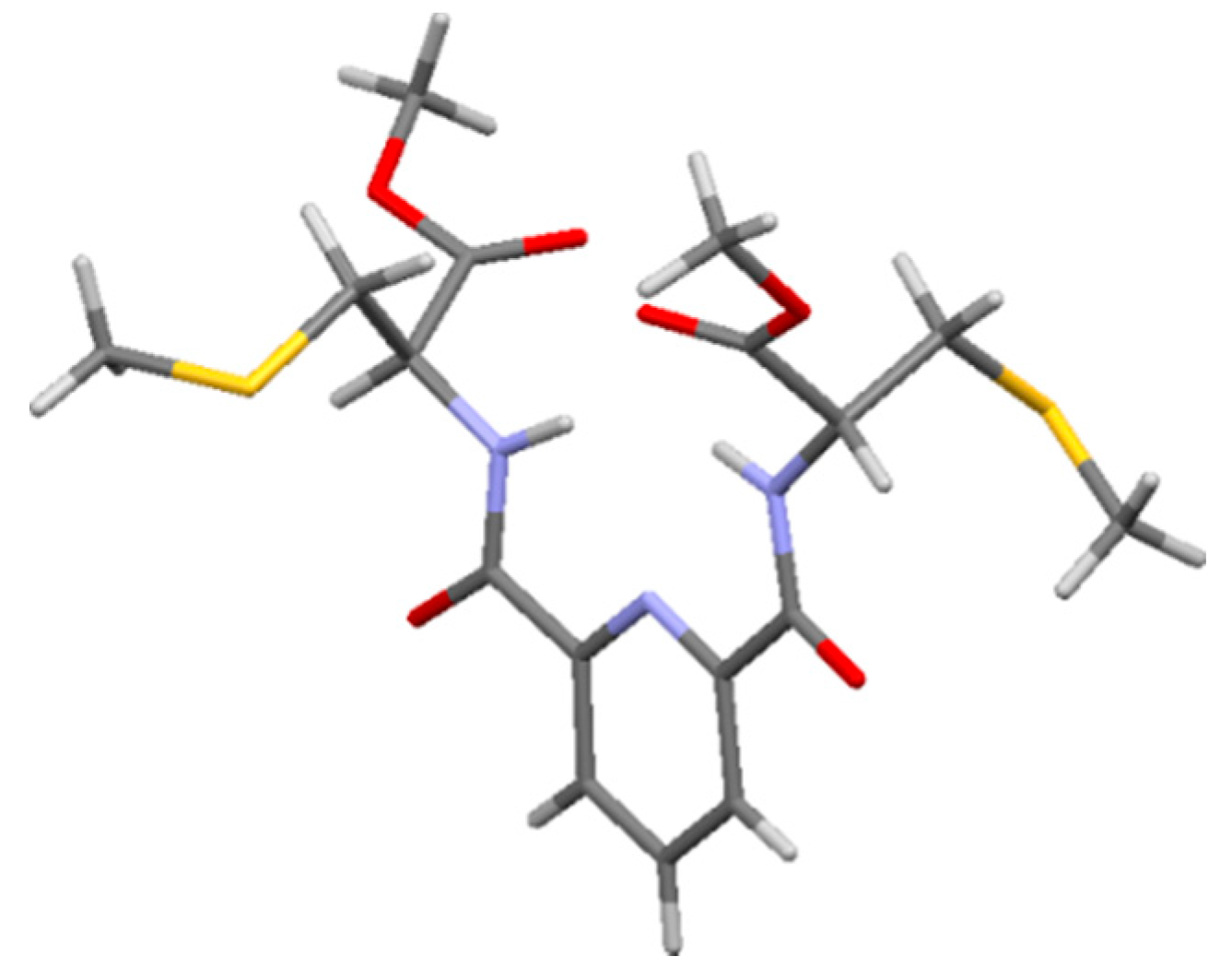{kind=link}
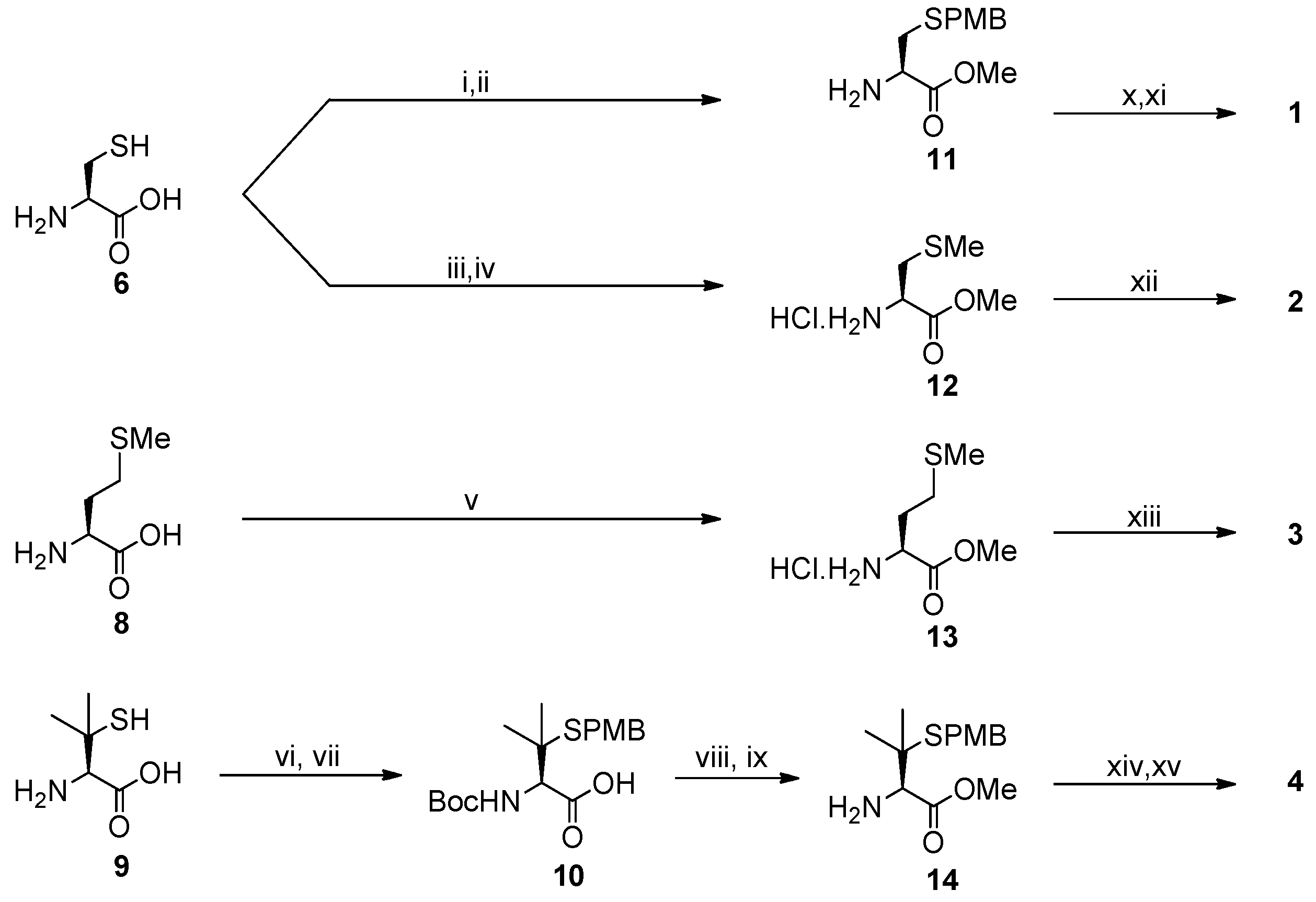{kind=link}
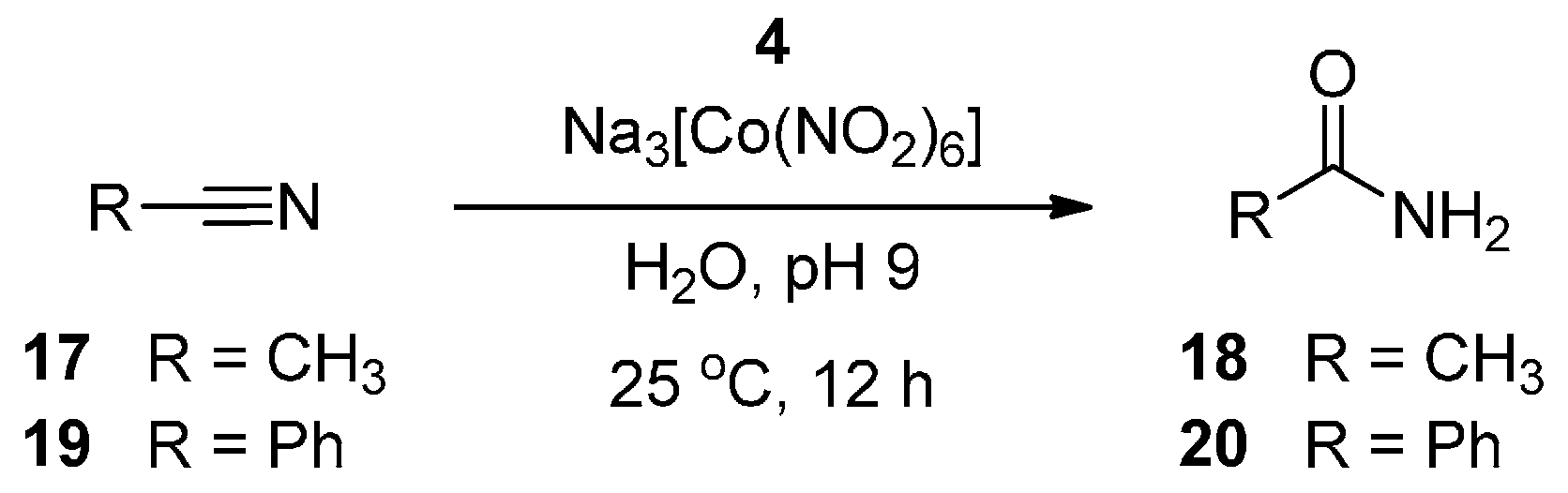{kind=link}
| Ligand | Metal Salt | Temperature (°C) | pH | Oxidant |
|---|---|---|---|---|
| 1 2 3 4 | Fe(OAc)2 | 0 20 50 | 2 5 9 | +H2O2 –H2O2 |
| Fe(NO3)3 | ||||
| Na3[Co(NO2)6] | ||||
| CoCl2.6H2O | ||||
| [Co(NH3)5Cl]Cl2 |
| Ligand | Optimal Turnover Conditions | Turnovers | ||||
|---|---|---|---|---|---|---|
| 1 | Na3[Co(NO2)]6 | 50 °C | pH 9 | 72 h | −H2O2 | 0.20 |
| 2 | Fe(NO3)3 | 20 °C | pH 9 | 72 h | +H2O2 | 0.40 |
| 3 | – | – | – | – | – | – |
| 4 | Na3[Co(NO2)]6 | 25 °C | pH 9 | 12 h | −H2O2 | 1.25 |

3. Experimental Section
3.1. General Information
3.2. Preparation of Protected Amino Acids
General Methyl Esterification Procedure [50]
3.3. Peptide Coupling Method
3.4. Ligand Synthesis
3.5. Crystallography
3.6. Turnover Experiments
3.6.1. General Procedure
3.6.2. GC Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Garcia-Alvarez, R.; Crochet, P.; Cadierno, V. Metal-catalyzed amide bond forming reactions in an environmentally friendly aqueous medium: Nitrile hydrations and beyond. Green Chem. 2013, 15, 46–66. [Google Scholar] [CrossRef]
- Ahmed, T.J.; Knapp, S.M.M.; Tyler, D.R. Frontiers in catalytic nitrile hydration: Nitrile and cyanohydrin hydration catalyzed by homogeneous organometallic complexes. Coord. Chem. Rev. 2011, 255, 949–974. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Metal-mediated and metal-catalyzed hydrolysis of nitriles. Inorg. Chim. Acta 2005, 358, 1–21. [Google Scholar] [CrossRef]
- Kumar, D.; Masitas, C.A.; Nguyen, T.N.; Grapperhaus, C.A. Bioinspired catalytic nitrile hydration by dithiolato, sulfinato/thiolato, and sulfenato/sulfinato ruthenium complexes. Chem. Commun. 2013, 49, 294–296. [Google Scholar] [CrossRef]
- Cadierno, V.; Diez, J.; Francos, J.; Gimeno, J. Bis(allyl)ruthenium(IV) complexes containing water-soluble phosphane ligands: Synthesis, structure, and application as catalysts in the selective hydration of organonitriles into amides. Chem. Eur. J. 2010, 16, 9808–9817. [Google Scholar] [CrossRef] [PubMed]
- Polshettiwar, V.; Varma, R.S. Nanoparticle-supported and magnetically recoverable ruthenium hydroxide catalyst: Efficient hydration of nitriles to amides in aqueous medium. Chem. Eur. J. 2009, 15, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Cadierno, V.; Francos, J.; Gimeno, J. Selective ruthenium-catalyzed hydration of nitriles to amides in pure aqueous medium under neutral conditions. Chem. Eur. J. 2008, 14, 6601–6605. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Endo, K.; Saito, S. Rh(I)-catalyzed hydration of organonitriles under ambient conditions. Angew. Chem. Int. Edit. 2008, 47, 3607–3609. [Google Scholar] [CrossRef]
- Djoman, M.; Ajjou, A.N. The hydration of nitriles catalyzed by water-soluble rhodium complexes. Tetrahedron Lett. 2000, 41, 4845–4849. [Google Scholar] [CrossRef]
- Hirano, T.; Uehara, K.; Kamata, K.; Mizuno, N. Palladium(II) containing gamma-keggin silicodecatungstate that efficiently catalyzes hydration of nitriles. J. Am. Chem. Soc. 2012, 134, 6425–6433. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Kim, H.S.; Kim, J.N. An efficient Pd-catalyzed hydration of nitrile with acetaldoxime. Tetrahedron Lett. 2009, 50, 2973–2975. [Google Scholar] [CrossRef]
- Ghaffar, T.; Parkins, A.W. The catalytic hydration of nitriles to amides using a homogeneous platinum phosphinito catalyst. J. Mol. Catal. A Chem. 2000, 160, 249–261. [Google Scholar] [CrossRef]
- Ghaffar, T.; Parkins, A.W. A new homogeneous platinum-containing catalyst for the hydrolysis of nitriles. Tetrahedron Lett. 1995, 36, 8657–8660. [Google Scholar] [CrossRef]
- Li, Z.K.; Wang, L.X.; Zhou, X.G. An efficient and practical protocol for catalytic hydrolysis of nitriles by a copper(I) complex in water. Adv. Synth. Catal. 2012, 354, 584–588. [Google Scholar] [CrossRef]
- Mitsudome, T.; Mikami, Y.; Mori, H.; Arita, S.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Supported silver nanoparticle catalyst for selective hydration of nitriles to amides in water. Chem. Commun. 2009, 3258–3260. [Google Scholar]
- Liu, Y.M.; He, L.; Wang, M.M.; Cao, Y.; He, H.Y.; Fan, K.N. A general and efficient heterogeneous gold-catalyzed hydration of nitriles in neat water under mild atmospheric conditions. ChemSusChem 2012, 5, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Ramon, R.S.; Marion, N.; Nolan, S.P. Gold activation of nitriles: Catalytic hydration to amides. Chem. Eur. J. 2009, 15, 8695–8697. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Wang, Z.X.; Liu, Z.L.; Feng, X.K.; Wang, Q.Y. Efficient and practical transition metal-free catalytic hydration of organonitriles to amides. Green Chem. 2012, 14, 921–924. [Google Scholar] [CrossRef]
- Verma, P.K.; Sharma, U.; Bala, M.; Kumar, N.; Singh, B. Transition metal-free 1,3-dimethylimidazolium hydrogen carbonate catalyzed hydration of organonitriles to amides. RSC Adv. 2013, 3, 895–899. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.; Chang, S.B.; Lee, H.Y. Anhydrous hydration of nitriles to amides using aldoximes as the water source. Org. Lett. 2009, 11, 5598–5601. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Lee, H.S.; Kim, S.H.; Kim, J.N. An efficient InCl3-catalyzed hydration of nitriles to amides: Acetaldoxime as an effective water surrogate. Tetrahedron Lett. 2010, 51, 1589–1591. [Google Scholar] [CrossRef]
- Miyanaga, A.; Fushinobu, S.; Ito, K.; Wakagi, T. Crystal structure of cobalt-containing nitrile hydratase. Biochem. Biophys. Res. Commun. 2001, 288, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Shimizu, S. Metalloenzyme nitrile hydratase: Structure, regulation, and application to biotechnology. Nat. Biotechnol. 1998, 16, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nagasawa, T.; Yamada, H. Enzymatic synthesis of acrylamide: A success story not yet over. Trends Biotechnol. 1992, 10, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, R.; Oriel, P. Bioconversion of acrylonitrile to acrylamide using a thermostable nitrile hydratase. Appl. Biochem. Biotechnol. 1999, 77–79, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-X. Enantioselective biotransformations of nitriles in organic synthesis. Top. Catal. 2005, 35, 117–130. [Google Scholar] [CrossRef]
- Prasad, S.; Bhalla, T.C. Nitrile hydratases (NHases): At the interface of academia and industry. Biotechnol. Adv. 2010, 28, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Shigehiro, S.; Nakasako, M.; Dohmae, N.; Tsujimura, M.; Tokoi, K.; Odaka, M.; Yohda, M.; Kamiya, N.; Endo, I. Novel non-heme iron center of nitrile hydratase with a claw setting of oxygen atoms. Nat. Struct. Biol. 1998, 5, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Mascharak, P.K. Structural and functional models of nitrile hydratase. Coord. Chem. Rev. 2002, 225, 201–214. [Google Scholar] [CrossRef]
- Kovacs, J.A. Synthetic analogues of cysteinate-ligated non-heme iron and non-corrinoid cobalt enzymes. Chem. Rev. 2004, 104, 825–848. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.; Wu, R.; Sanishvili, R.; Liu, D.; Holz, R. The active site sulfenic acid ligand in nitrile hydratases can function as a nucleophile. J. Am. Chem. Soc. 2014, 136, 1186–1189. [Google Scholar] [CrossRef]
- Hopmann, K.H.; Himo, F. Theoretical investigation of the second-shell mechanism of nitrile hydratase. Eur. J. Inorg. Chem. 2008, 2008, 1406–1412. [Google Scholar] [CrossRef]
- Hopmann, K.H. Full reaction mechanism of nitrile hydratase: A cyclic intermediate and an unexpected disulfide switch. Inorg. Chem. 2014, 53, 2760–2762. [Google Scholar] [CrossRef] [PubMed]
- Tyler, L.A.; Noveron, J.C.; Olmstead, M.M.; Mascharak, P.K. Modulation of the pKa of metal-bound water via oxidation of thiolato sulfur in model complexes of Co(III) containing nitrile hydratase: Insight into possible effect of cysteine oxidation in Co-nitrile hydratase. Inorg. Chem. 2003, 42, 5751–5761. [Google Scholar] [CrossRef] [PubMed]
- Noveron, J.C.; Olmstead, M.M.; Mascharak, P.K. Co(III) complexes with carboxamido N and thiolato S donor centers: Models for the active site of Co-containing nitrile hydratases. J. Am. Chem. Soc. 1999, 121, 3553–3554. [Google Scholar] [CrossRef]
- Heinrich, L.; Li, Y.; Vaissermann, J.; Chottard, G.; Chottard, J.C. A pentacoordinated di-N-carboxamido-dithiolato-O-sulfinato-iron(III) complex related to the metal site of nitrile hydratase. Angew. Chem. Int. Ed. 1999, 38, 3526–3528. [Google Scholar] [CrossRef]
- Heinrich, L.; Mary-Verla, A.; Li, Y.; Vaissermann, J.; Chottard, J.C. Cobalt(III) complexes with carboxamido-N and sulfenato-S or sulfinato-S ligands suggest that a coordinated sulfenate-S is essential for the catalytic activity of nitrile hydratases. Eur. J. Inorg. Chem. 2001, 2001, 2203–2206. [Google Scholar] [CrossRef]
- Artaud, I.; Chatel, S.; Chauvin, A.S.; Bonnet, D.; Kopf, M.A.; Leduc, P. Nitrile hydratase and related non-heme iron sulfur complexes. Coord. Chem. Rev. 1999, 192, 577–586. [Google Scholar] [CrossRef]
- Bourles, E.; Alves de Sousa, R.; Galardon, E.; Giorgi, M.; Artaud, I. Direct synthesis of a thiolato-S and sulfinato-S Co(III) complex related to the active site of nitrile hydratase: A pathway to the post-translational oxidation of the protein. Angew. Chem. Int. Ed. 2005, 44, 6162–6165. [Google Scholar] [CrossRef]
- Lugo-Mas, P.; Dey, A.; Xu, L.; Davin, S.D.; Benedict, J.; Kaminsky, W.; Hodgson, K.O.; Hedman, B.; Solomon, E.I.; Kovacs, J.A. How does single oxygen atom addition affect the properties of an Fe-nitrile hydratase analogue? The compensatory role of the unmodified thiolate. J. Am. Chem. Soc. 2006, 128, 11211–11221. [Google Scholar] [CrossRef] [PubMed]
- Shearer, J.; Jackson, H.L.; Schweitzer, D.; Rittenberg, D.K.; Leavy, T.M.; Kaminsky, W.; Scarrow, R.C.; Kovacs, J.A. The first example of a nitrile hydratase model complex that reversibly binds nitriles. J. Am. Chem. Soc. 2002, 124, 11417–11428. [Google Scholar] [CrossRef] [PubMed]
- Krall, J.A.; Rutledge, P.J.; Baldwin, J.E. Design and synthesis of an isopenicillin N synthase mimic. Tetrahedron 2005, 61, 137–143. [Google Scholar] [CrossRef]
- Barry, S.M.; Rutledge, P.J. Cis-dihydroxylation of alkenes by a non-heme iron enzyme mimic. Synlett 2008, 2172–2174. [Google Scholar]
- Dungan, V.J.; Ortin, Y.; Mueller-Bunz, H.; Rutledge, P.J. Design and synthesis of a tetradentate “3-amine-1-carboxylate” ligand to mimic the metal binding environment at the non-heme iron(II) oxidase active site. Org. Biomol. Chem. 2010, 8, 1666–1673. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.M.; Mueller-Bunz, H.; Rutledge, P.J. Investigating the cis-dihydroxylation of alkenes by non-heme iron enzyme mimics. Org. Biomol. Chem. 2012, 10, 7372–7381. [Google Scholar] [CrossRef] [PubMed]
- Dungan, V.J.; Wong, S.M.; Barry, S.M.; Rutledge, P.J. L-Proline-derived ligands to mimic the “2-his-1-carboxylate” triad of the non-heme iron oxidase active site. Tetrahedron 2012, 68, 3231–3236. [Google Scholar] [CrossRef]
- Dungan, V.J.; Poon, B.M.-L.; Barrett, E.; Rutledge, P.J. L-Proline derived mimics of the non-heme iron active site catalyse allylic oxidation in acetonitrile solutions. Tetrahedron Lett. 2013, 54, 1236–1238. [Google Scholar] [CrossRef]
- Hwang, D.R.; Helquist, P.; Shekhani, M.S. Total synthesis of (+)-sparsomycin—Approaches using cysteine and serine inversion. J. Org. Chem. 1985, 50, 1264–1271. [Google Scholar] [CrossRef]
- Bodanzky, M.; Bodanzky, A. The practice of peptide synthesis. In Reactivity and Structure Concepts in Organic Chemistry; Hafner, K., Rees, C.W., Trost, B.M., Lehn, J.M., Schleyer, P.V.R., Zahruchik, R., Eds.; Springer-Verlag: Berlin, Germany, 1984; Volume 21. [Google Scholar]
- McMullen, T.C. The methyl ester of orthobenzoyl-benzoic acid. J. Am. Chem. Soc. 1916, 38, 1228–1230. [Google Scholar] [CrossRef]
- Bocchi, V.; Casnati, G.; Dossena, A.; Marchelli, R. Esterification of amino acids and dipeptides under mild conditions; part ii: Via sodium salts. Synthesis 1979, 961–962. [Google Scholar]
- Han, S.-Y.; Kim, Y.-A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron 2004, 60, 2447–2467. [Google Scholar] [CrossRef]
- Derdau, V.; Laschat, S.; Hupe, E.; König, W.A.; Dix, I.; Jones, P.G. Novel chiral pyridine N-oxide ligands and their application in the enantioselective catalytic reduction of ketones and the addition of diethylzinc to aldehydes. Eur. J. Inorg. Chem. 1999, 1999, 1001–1007. [Google Scholar] [CrossRef]
- Alexie, M.; Dimitru, F.; Razvan, A.; Pasare, L.; Guran, C. New membranes with mercapto-2,6-pyridinediamides as anion recognition fixed sites. Rev. Chim. 2010, 61, 1197–1201. [Google Scholar]
- Loconto, P.R. Trace Environmental Quantitative Analysis—Principles, Techniques and Applications, 2nd ed.; Marcel Dekker Inc.: New York, NY, USA, 2005. [Google Scholar]
- Harris, D.C. Quantitative Chemical Analysis, 6th ed.; W.H. Freeman: New York, NY, USA, 2003. [Google Scholar]
- Heinrich, L.; Li, Y.; Vaissermann, J.; Chottard, J.C. A bis(carboxamido-N)diisocyanidobis(sulfenato-S)cobalt(III) complex, model for the post-translational oxygenation of nitrile hydratase thiolato ligands. Eur. J. Inorg. Chem. 2001, 2001, 1407–1409. [Google Scholar] [CrossRef]
- Cordes, E.H.; Zervos, C. Mercaptoethanol catalysis for hydrolysis of N-benzyl-3-cyanopyridinium bromide. A model for the nitrilase reaction. J. Org. Chem. 1971, 36, 1661–1667. [Google Scholar] [CrossRef]
- Balahura, R.; Cock, P.; Purcell, W. Kinetics studies of the hydrolysis of coordinated nitriles. J. Am. Chem. Soc. 1974, 96, 2739–2742. [Google Scholar] [CrossRef]
- Platen, M.; Steckhan, E. Oxidative deblocking of the 4-methoxybenzyl thioether protecting group: Application to the directed synthesis of poly-cystinyl peptides. Liebigs Ann. Chem. 1984, 1984, 1563–1576. [Google Scholar] [CrossRef]
- Matteucci, M.; Bhalay, G.; Bradley, M. Mild and highly chemoselective oxidation of thioethers mediated by Sc(OTf)3. Org. Lett. 2003, 5, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Lumbroso, A.; Coeffard, V.; le Grognec, E.; Beaudet, I.; Quintard, J.-P. An efficient and scalable synthesis of N-(benzyloxycarbonyl)- and N-(methyloxycarbonyl)-(S)-vinylglycinol. Tetrahedron Lett. 2010, 51, 3226–3228. [Google Scholar] [CrossRef]
- Bruker. Area Detector Control and Data Integration and Reduction Software; Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1995. [Google Scholar]
- Barbour, L.J. X-seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Sadabs. Empirical Absorption Correction Program for Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Shelx97 Programs for Crystal Structure Analysis; University of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Crystallogr. Sect. A Found. Crystallogr. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Bernardinelli, G.; Flack, H.D. Least-squares absolute-structure refinement—Practical experience and ancillary calculations. Acta Crystallogr. Sect. A Found. Crystallogr. 1985, 41, 500–511. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Absolute structure and absolute configuration. Acta Crystallogr. Sect. A Found. Crystallogr. 1999, 55, 908–915. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration determinations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byrne, C.; Houlihan, K.M.; Devi, P.; Jensen, P.; Rutledge, P.J. Bio-Inspired Nitrile Hydration by Peptidic Ligands Based on L-Cysteine, L-Methionine or L-Penicillamine and Pyridine-2,6-dicarboxylic Acid. Molecules 2014, 19, 20751-20767. https://doi.org/10.3390/molecules191220751
Byrne C, Houlihan KM, Devi P, Jensen P, Rutledge PJ. Bio-Inspired Nitrile Hydration by Peptidic Ligands Based on L-Cysteine, L-Methionine or L-Penicillamine and Pyridine-2,6-dicarboxylic Acid. Molecules. 2014; 19(12):20751-20767. https://doi.org/10.3390/molecules191220751
Chicago/Turabian StyleByrne, Cillian, Kate M. Houlihan, Prarthana Devi, Paul Jensen, and Peter J. Rutledge. 2014. "Bio-Inspired Nitrile Hydration by Peptidic Ligands Based on L-Cysteine, L-Methionine or L-Penicillamine and Pyridine-2,6-dicarboxylic Acid" Molecules 19, no. 12: 20751-20767. https://doi.org/10.3390/molecules191220751