Atomic Model and Micelle Dynamics of QS-21 Saponin

Abstract
:
1. Introduction

2. Results and Discussion
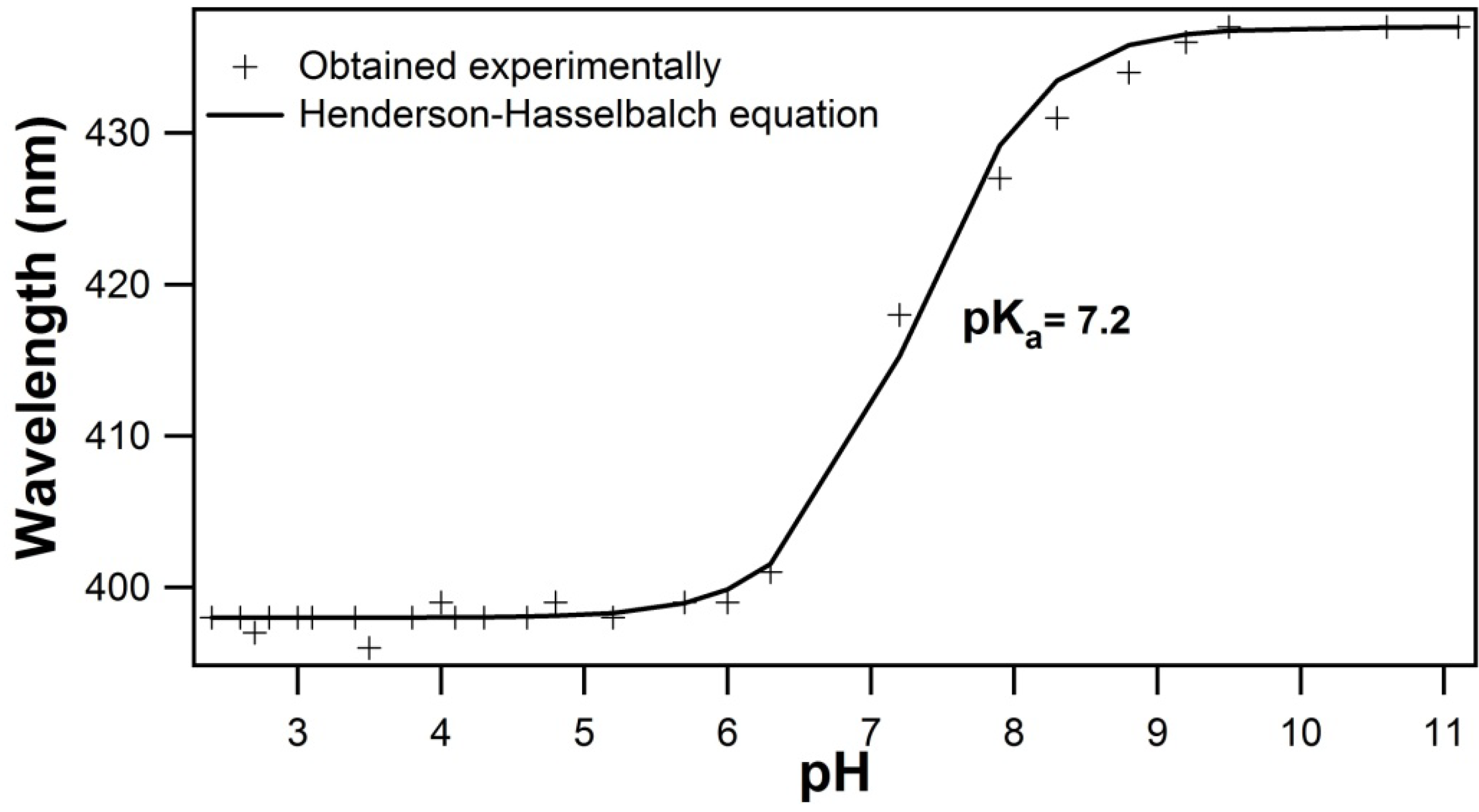
2.1. Titration by Fluorescence

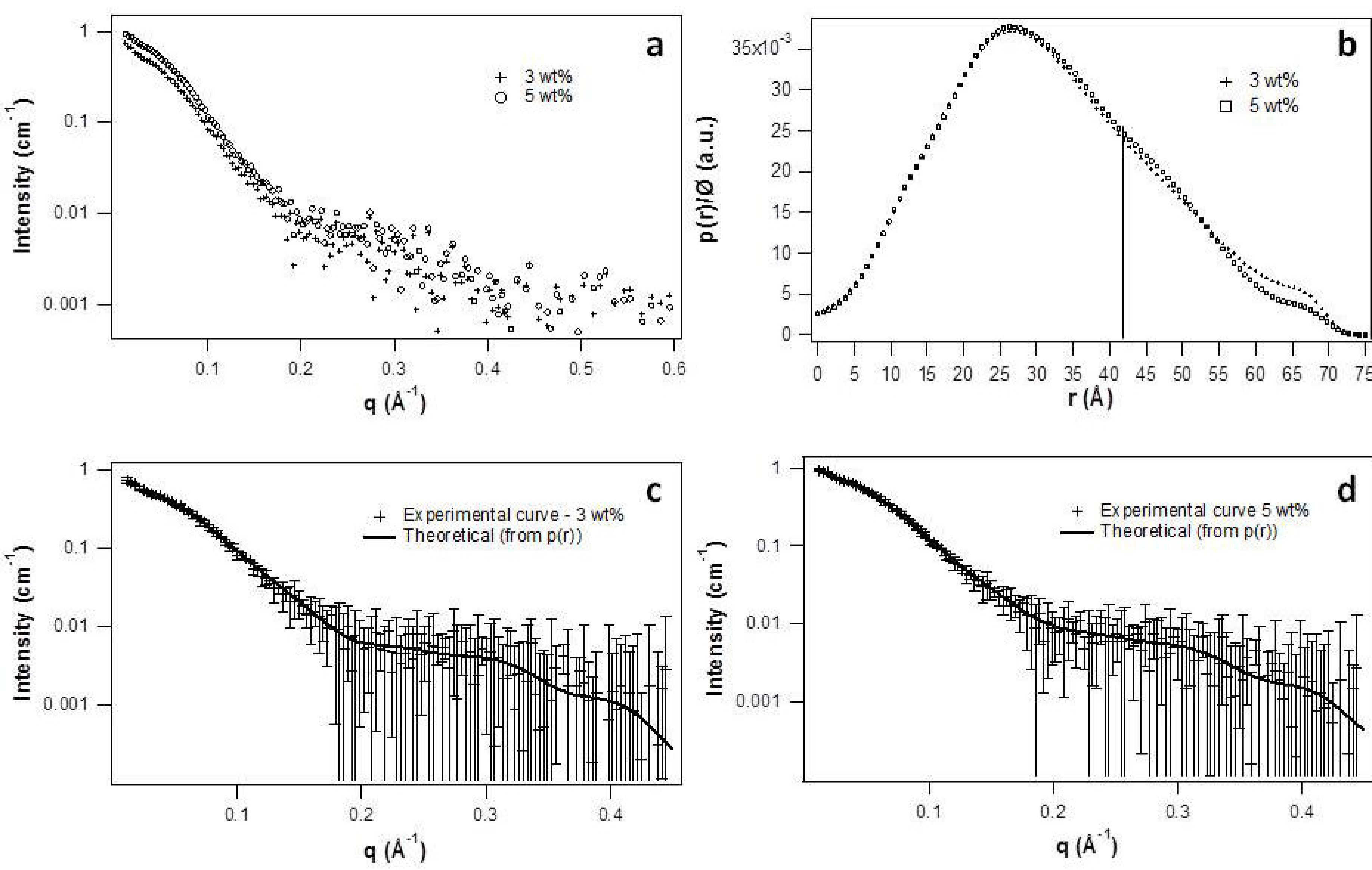
2.2. Micellar Structure by SAXS

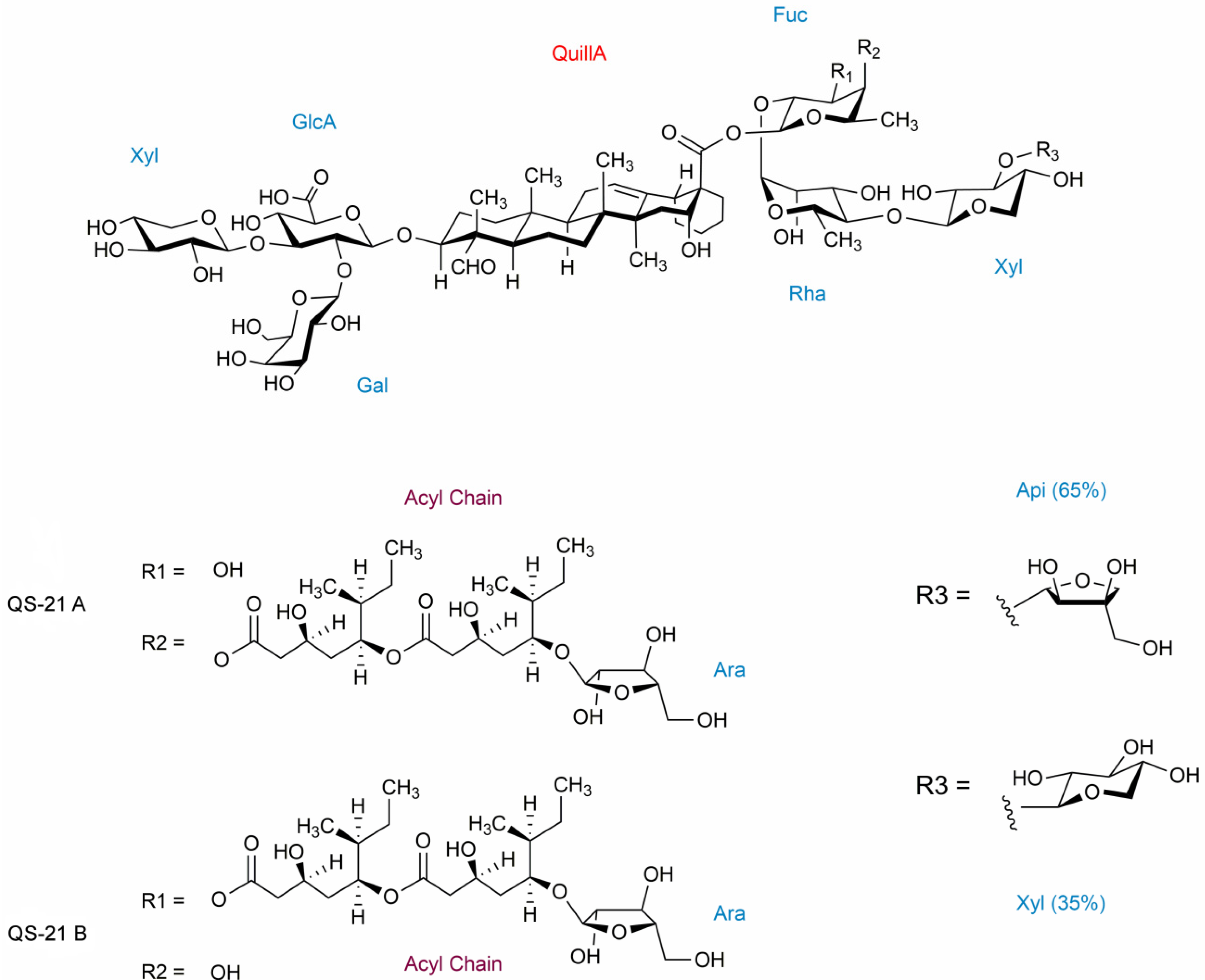
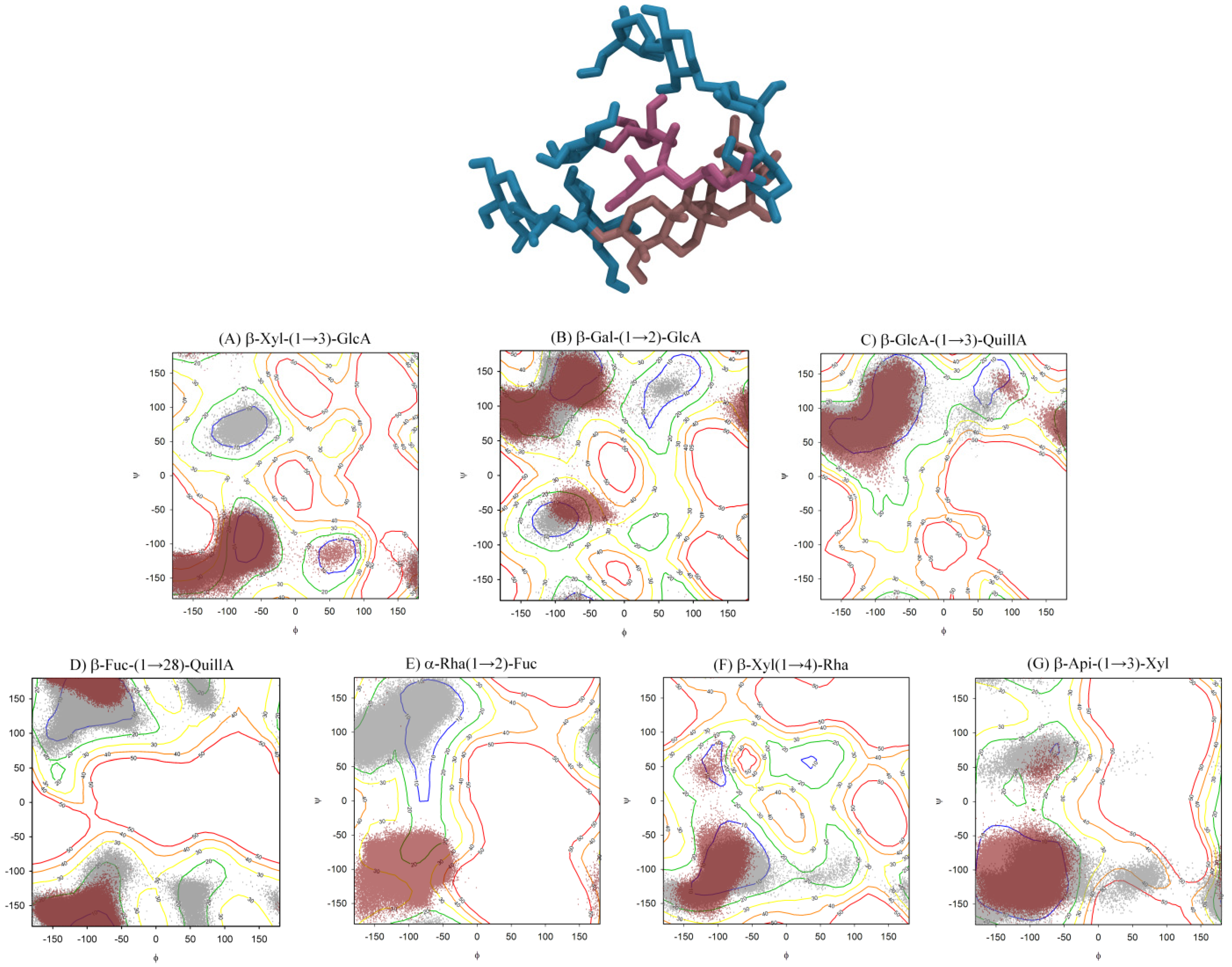
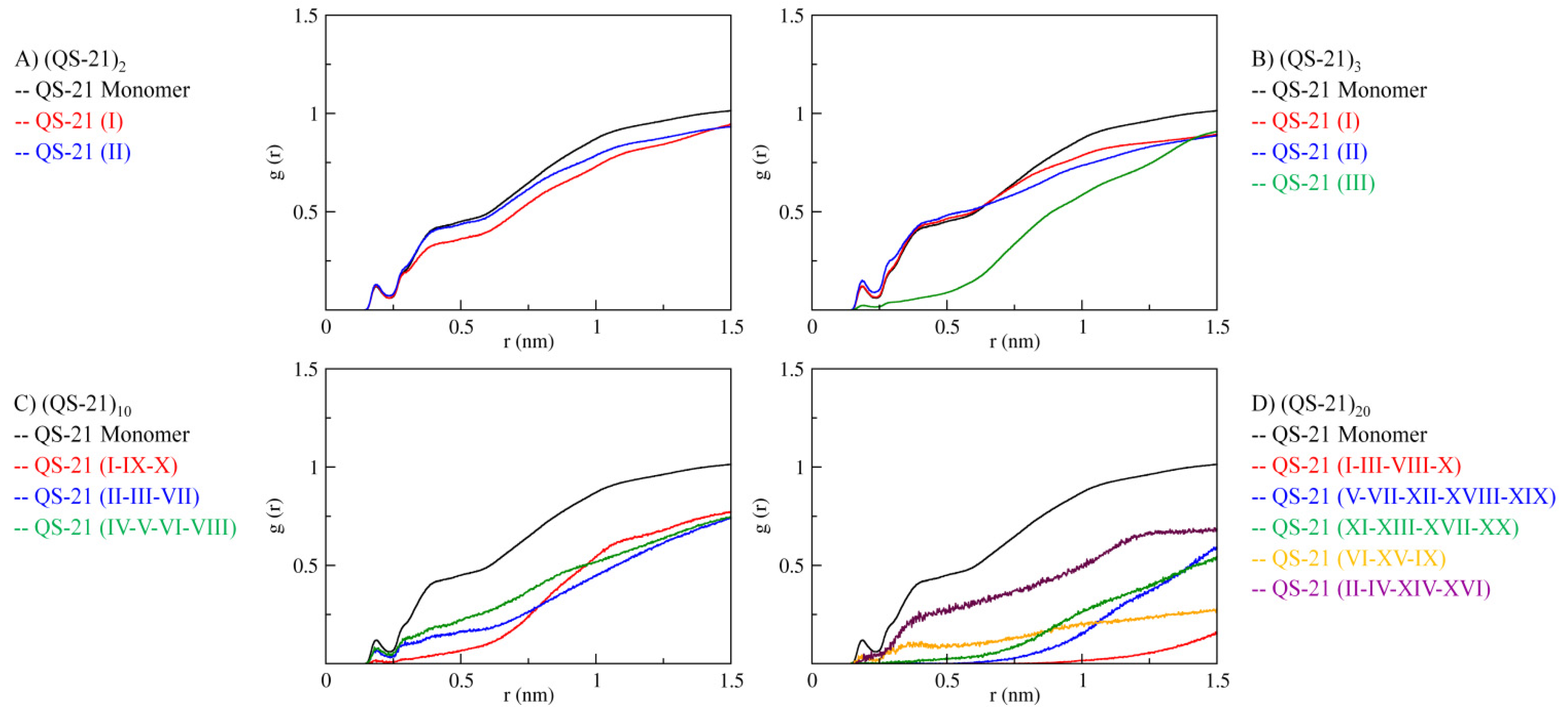
2.3. QS-21 Conformational Characterization

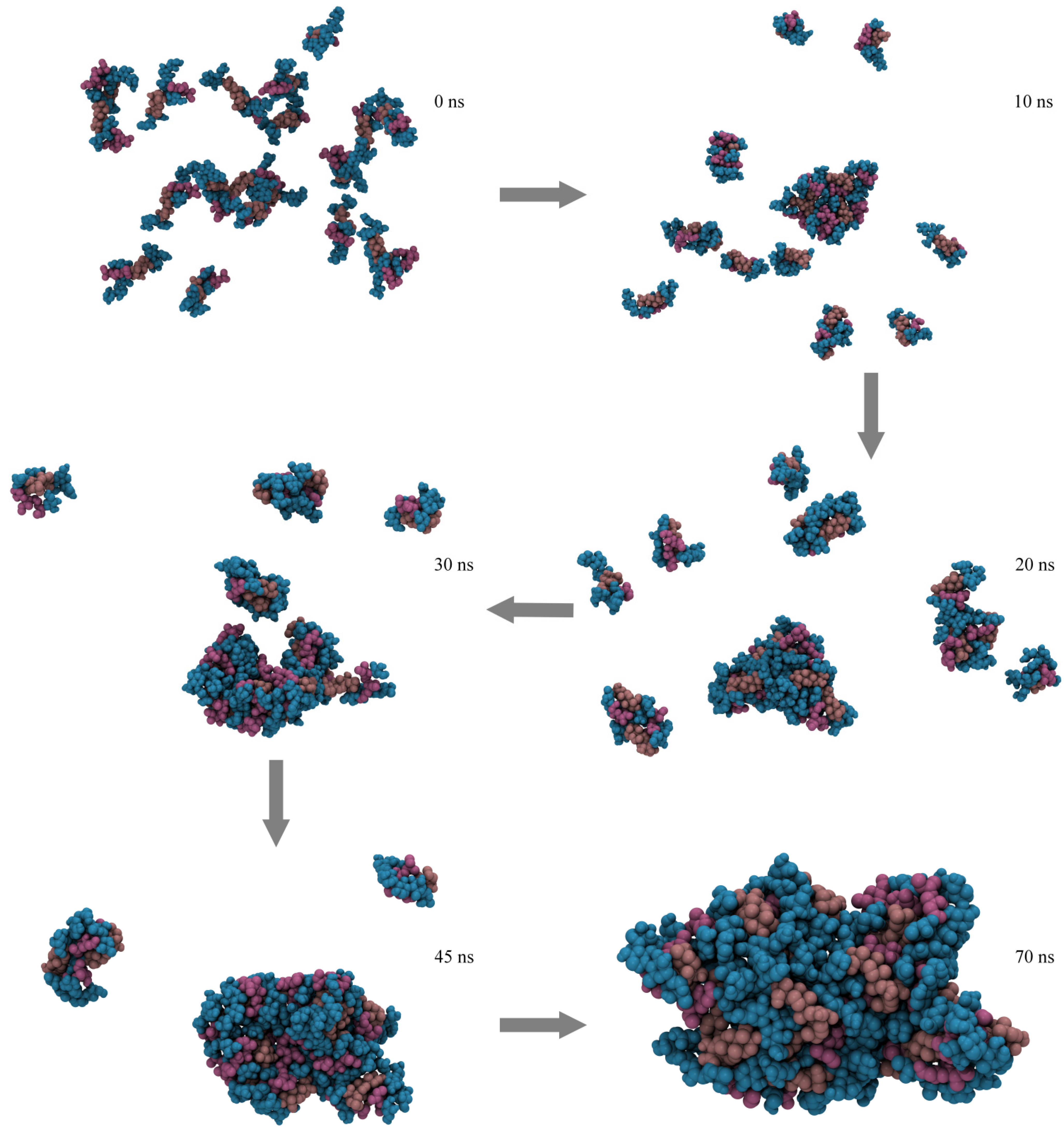
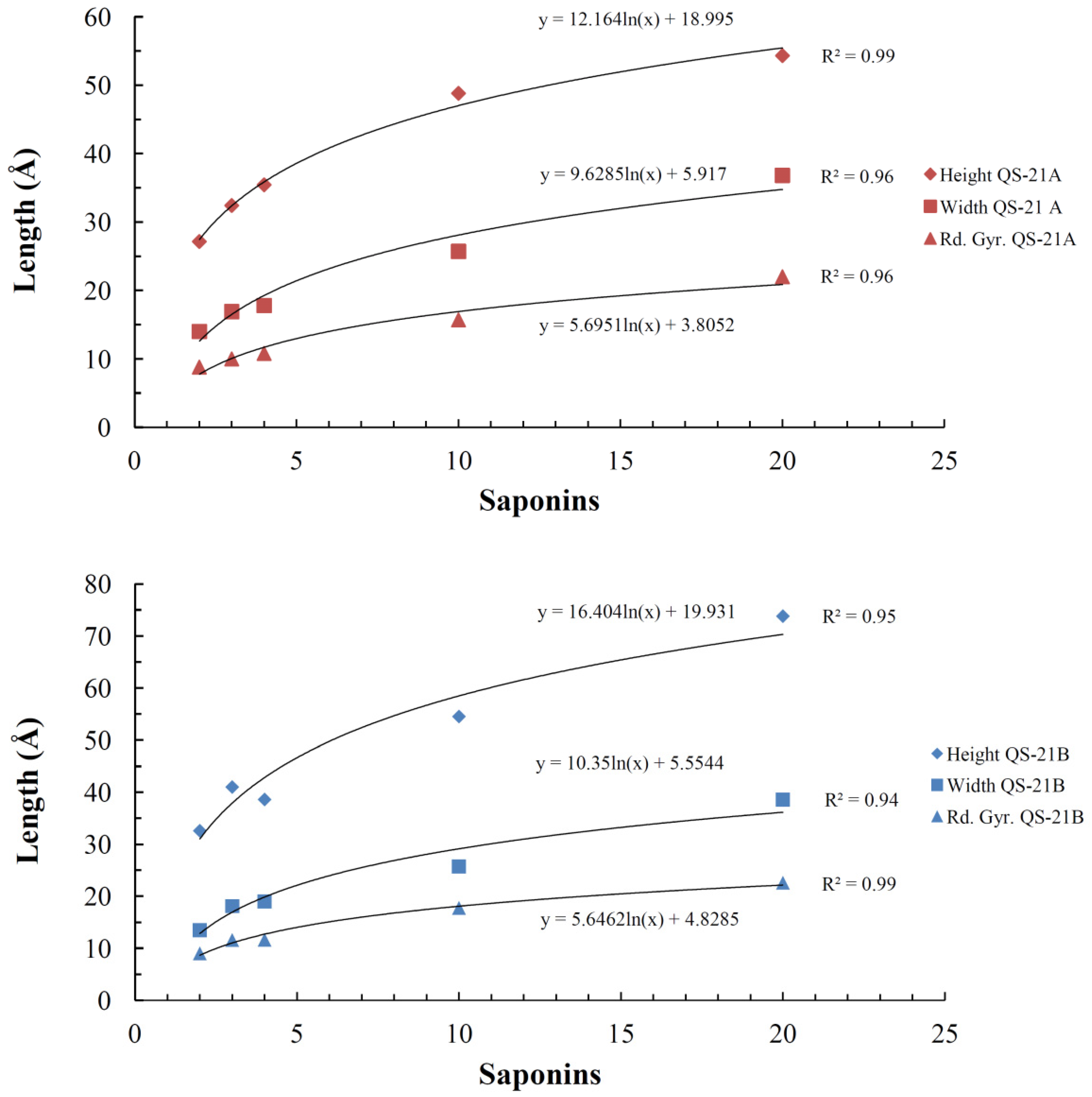
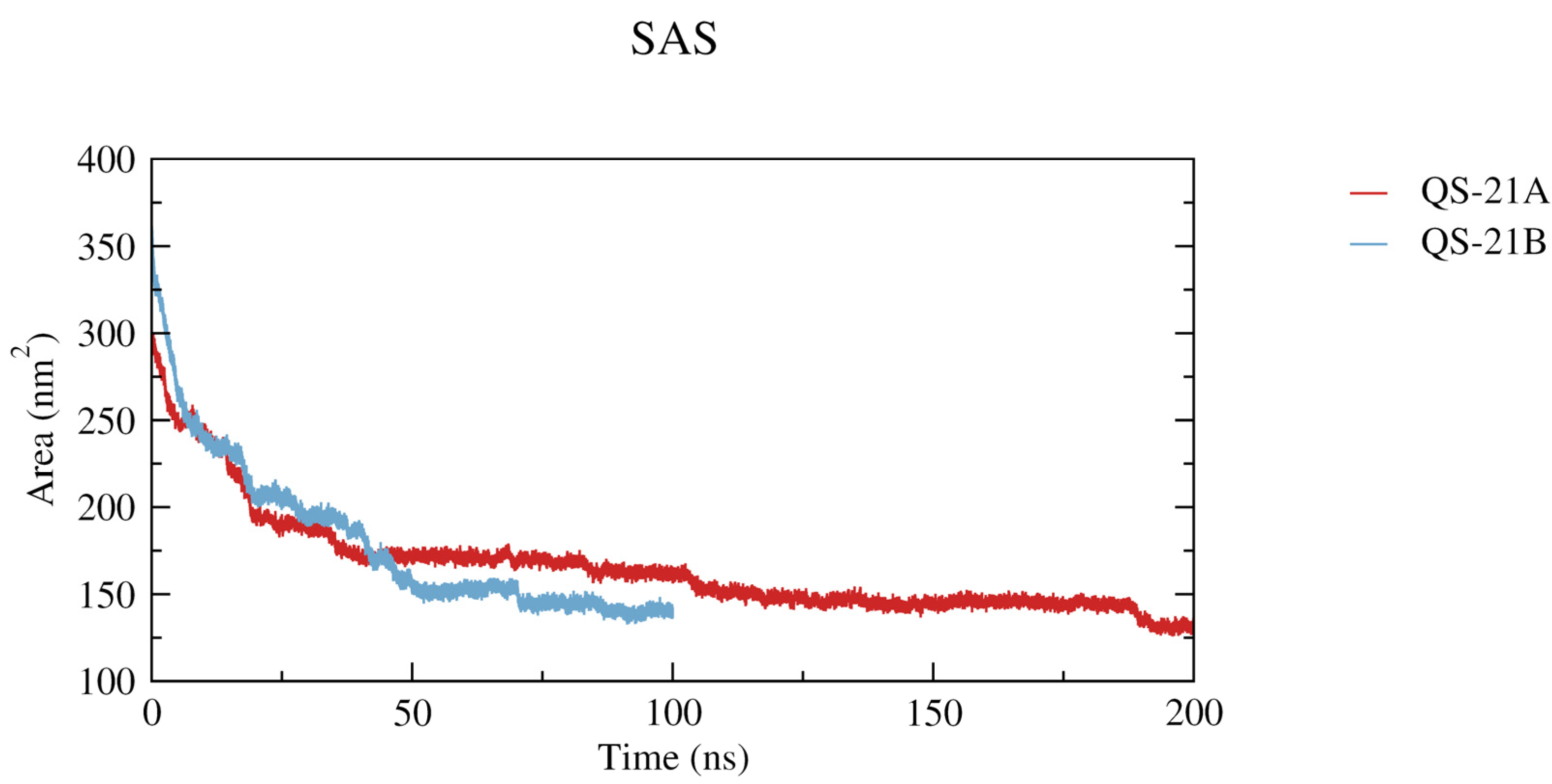
2.4. Micelle Formation and Dimensions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System (QS-21 molecules) | Saponin | |||||||
|---|---|---|---|---|---|---|---|---|
| QS-21A | QS-21B | |||||||
| Rg | Height | Width | Axial Ratio | Rg | Height | Width | Axial Ratio | |
| 2 | 8.8 ± 0.5 | 27.1 ± 2.1 | 14.0 ± 1.0 | 1.9 ± 0.2 | 9.0 ± 0.5 | 32.6 ± 0.5 | 13.5 ± 1.0 | 2.4 ± 0.2 |
| 3 | 10.0 ± 0.2 | 32.4 ± 1.4 | 16.9 ± 0.4 | 1.9 ± 0.1 | 11.6 ± 0.2 | 41.0 ± 0.2 | 18.1 ± 0.2 | 2.3 ± 0.1 |
| 4 | 10.8 ± 0.2 | 35.4 ± 1.9 | 17.8 ± 0.7 | 2.0 ± 0.1 | 11.6 ± 0.3 | 37.2 ± 0.3 | 19.0 ± 0.8 | 2.00 ± 0.03 |
| 10 | 15.7 ± 0.1 | 48.8 ± 1.6 | 25.7 ± 0.8 | 1.9 ± 0.1 | 17.8 ± 0.3 | 54.5 ± 0.3 | 25.7 ± 1.2 | 2.1 ± 0.1 |
| 20 | 22.0 ± 1.4 | 54.3 ± 1.1 | 36.8 ± 1.6 | 1.5 ± 0.1 | 22.0 ± 1.3 | 72.0 ± 1.2 | 39.2 ± 1.1 | 1.8 ± 0.1 |


2.5. Ester Linkage Solvation
2.6. Comparison of Both Methods in the Micelle Structure Analysis


| System | Saponin | SAXS | |||||||
|---|---|---|---|---|---|---|---|---|---|
| QS-21A | QS-21B | ||||||||
| Rg | Height | Width | Rg | Height | Width | Rg | Height | Width | |
| 20 | 22.0 ± 1.4 | 54.3 ± 1.1 | 36.8 ± 1.6 | 22.6 ± 2.1 | 73.8 ± 2.9 | 38.6 ± 1.7 | 25.0 ± 3.9 | 75.0 ± 3.0 | 42.0 ± 3.0 |
3. Experimental
3.1. Saponin Preparation
3.2. Titration
3.3. Small-Angle X-ray Scattering (SAXS)
3.4. Analysis Method


3.5. Nomenclature, Topologies, and Software
3.6. Saponin Construction and Topologies Refinement
3.7. Contour Plots
3.8. MD Simulations
4. Conclusions
Acknowledgments
Author Contributions
Conflictts of Interest
References
- Ragupathi, G.; Gardner, J.R.; Livingston, P.O.; Gin, D.Y. Natural and synthetic saponin adjuvant QS-21 for vaccines against cancer. Expert Rev. Vaccines 2011, 10, 463–470. [Google Scholar] [CrossRef]
- Marciani, D.J.; Press, J.B.; Reynolds, R.C.; Pathak, A.K.; Pathak, V.; Gundy, L.E.; Farmer, J.T.; Koratich, M.S.; May, R.D. Development of semisynthetic triterpenoid saponin derivatives with immune stimulating activity. Vaccine 2000, 18, 3141–3151. [Google Scholar] [CrossRef]
- Adams, M.M.; Damani, P.; Perl, N.R.; Won, A.; Hong, F.; Livingston, P.O.; Ragupathi, G.; Gin, D.Y. Design and synthesis of potent Quillaja saponin vaccine adjuvants. J. Am. Chem. Soc. 2010, 132, 1939–1945. [Google Scholar]
- Chea, E.K.; Fernández-Tejada, A.; Damani, P.; Adams, M.M.; Gardner, J.R.; Livingston, P.O.; Ragupathi, G.; Gin, D.Y. Synthesis and Preclinical Evaluation of QS-21 Variants Leading to Simplified Vaccine Adjuvants and Mechanistic Probes. J. Am. Chem. Soc. 2012, 134, 13448–13457. [Google Scholar] [CrossRef]
- Jacobsen, N.E.; Fairbrother, W.J.; Kensil, C.R.; Lim, A.; Wheeler, D.A.; Powell, M.F. Structure of the saponin adjuvant QS-21 and its base-catalyzed isomerization product by 1H and natural abundance 13C NMR spectroscopy. Carbohydr. Res. 1996, 280, 1–14. [Google Scholar] [CrossRef]
- Cleland, J.L.; Kensil, C.R.; Lim, A.; Jacobsen, N.E.; Basa, L.; Spellman, M.; Wheeler, D.A.; Wu, J.-Y.; Powell, M.F. Isomerization and formulation stability of the vaccine adjuvant QS-21. J. Pharm. Sci. 1996, 85, 22–28. [Google Scholar] [CrossRef]
- Liu, G.; Anderson, C.; Scaltreto, H.; Barbon, J.; Kensil, C.R. QS-21 structure/function studies: Effect of acylation on adjuvant activity. Vaccine 2002, 20, 2808–2815. [Google Scholar] [CrossRef]
- Marciani, D.J.; Pathak, A.K.; Reynolds, R.C.;Seitz, L; May, R.D. Altered immunomodulating and toxicological properties of degraded Quillaja saponaria Molina saponins. Int. Immunopharmacol. 2001, 1, 813–818. [Google Scholar] [CrossRef]
- Press, J.B.; Reynolds, R.C.; May, R.D.; Marciani, D.J. Structure/function relationships of immunostimulating saponins. Stud. Nat. Prod. Chem. 2000, 24, 131–174. [Google Scholar] [CrossRef]
- Morein, B.; Sundqvist, B.; Hölund, S.; Dasgaard, K.; Osterhaus, A. Iscom, a novel structure for antigenic presentation of membrane proteins from enveloped viruses. Nature 1984, 308, 457–460. [Google Scholar] [CrossRef]
- Marciani, D.J.; Kensil, C.R.; Beltz, G.A.; Hung, C.-H.; Cronier, J.; Aubert, A. Genetically-engineered subunit vaccine against feline leukaemia virus: Protective immune response in cats. Vaccine 1991, 9, 89–96. [Google Scholar] [CrossRef]
- Fernandes, C.L.; Sachett, L.G.; Pol-Fachin, L.; Verli, H. GROMOS96 43a1 performance in predicting oligosaccharide conformational ensembles within glycoproteins. Carbohydr. Res. 2010, 345, 663–671. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Serrato, R.V.; Verli, H. Solution conformation and dynamics of exopolysaccharides from Burkholderia species. Carbohydr. Res. 2010, 13, 1922–1931. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Becker, C.F.; Guimarães, J.A.; Verli, H. Effects of glycosylation on heparin binding and antithrombin activation by heparin. Proteins 2011, 79, 2735–2745. [Google Scholar] [CrossRef]
- Pedebos, C.; Pol-Fachin, L.; Verli, H. Unrestrained conformational characterization of Stenocereus eruca saponins in aqueous and nonaqueous solvents. J. Nat. Prod. 2012, 75, 1196–1200. [Google Scholar] [CrossRef]
- Mortensen, K.; Pedersen, J.S. Structural study on the micelle formation of poly (ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) triblock copolymer in aqueous solution. Macromolecules 1993, 26, 805–812. [Google Scholar] [CrossRef]
- Glatter, O. Small-Angle X-ray Scattering; Glatter, O., Kratky, O., Eds.; Academic Press: London, UK, 1982. [Google Scholar]
- Svergun, D.I.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Singh, M.A.; Ghosh, S.S.; Shannon, R.F. A direct method of beam-height correction in small-angle X-ray scattering. J. Appl. Crystallogr. 1993, 26, 787–794. [Google Scholar] [CrossRef]
- Zemb, T.N.; Hyde, S.T.; Derian, P.J.; Barnes, I.S.; Ninham, B.W. Microstructure from X-ray scattering: The disordered open connected model of microemulslons. J. Phys. Chem. 1987, 91, 3814–3820. [Google Scholar] [CrossRef]
- Orthaber, D.; Bergmann, A.; Glatter, O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Appl. Crystallogr. 2000, 33, 218–225. [Google Scholar] [CrossRef]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Cryst. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Glatter, O. Convolution square root of band-limited symmetrical functions and its application to small-angle scattering data. J. Appl. Cryst. 1981, 14, 101–108. [Google Scholar] [CrossRef]
- IUPAC; IUB. Nomenclature of carbohydrates. Comm. Biochem. Nomencl. Pure Appl. Chem. 1996, 68, 1919–2008. [Google Scholar]
- Schuettelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughputcrystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. Available online: Available online: https://www.zotero.org/groups/ncdirtrd3/items/itemKey/HJ2H2FHT (accessed on 10 March 2014).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J. An Efficient Library for Parallel Ray Tracing and Animation; Computer Science Department, University of Missouri-Rolla: Rolla, MO, USA, 1998. [Google Scholar]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Fraga, C.A.M.; Barreiro, E.J.; Verli, H. Characterization of the conformational ensemble from bioactive N-acylhydrazone derivatives. J. Mol. Graph. Model. 2010, 28, 446–454. [Google Scholar] [CrossRef]
- Verli, H.; Guimarães, J.A. Molecular dynamics simulation of a decasaccharide fragment of heparin in aqueous solution. Carbohydr. Res. 2004, 339, 281–290. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Sahu, N.P.; Achari, B. Advances in Structural Determination of Saponins and Terpenoid Glycosides. Curr. Org. Chem. 2001, 5, 315–334. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pedebos, C.; Pol-Fachin, L.; Pons, R.; Teixeira, C.V.; Verli, H. Atomic Model and Micelle Dynamics of QS-21 Saponin. Molecules 2014, 19, 3744-3760. https://doi.org/10.3390/molecules19033744
Pedebos C, Pol-Fachin L, Pons R, Teixeira CV, Verli H. Atomic Model and Micelle Dynamics of QS-21 Saponin. Molecules. 2014; 19(3):3744-3760. https://doi.org/10.3390/molecules19033744
Chicago/Turabian StylePedebos, Conrado, Laércio Pol-Fachin, Ramon Pons, Cilâine V. Teixeira, and Hugo Verli. 2014. "Atomic Model and Micelle Dynamics of QS-21 Saponin" Molecules 19, no. 3: 3744-3760. https://doi.org/10.3390/molecules19033744