Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-species

Abstract
:1. Introduction
2. Ruthenium Tetroxide Chemistry
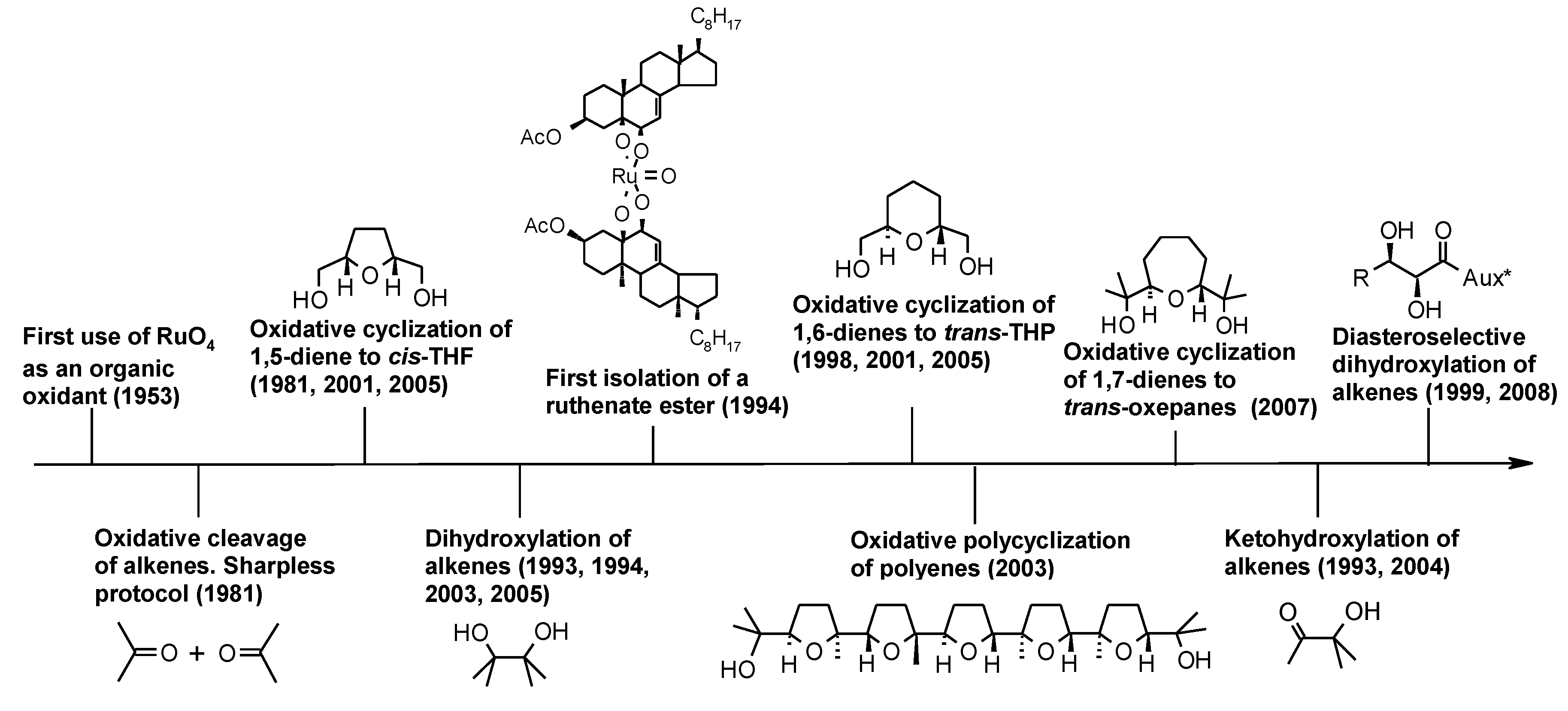
2.1. An Historical Overview


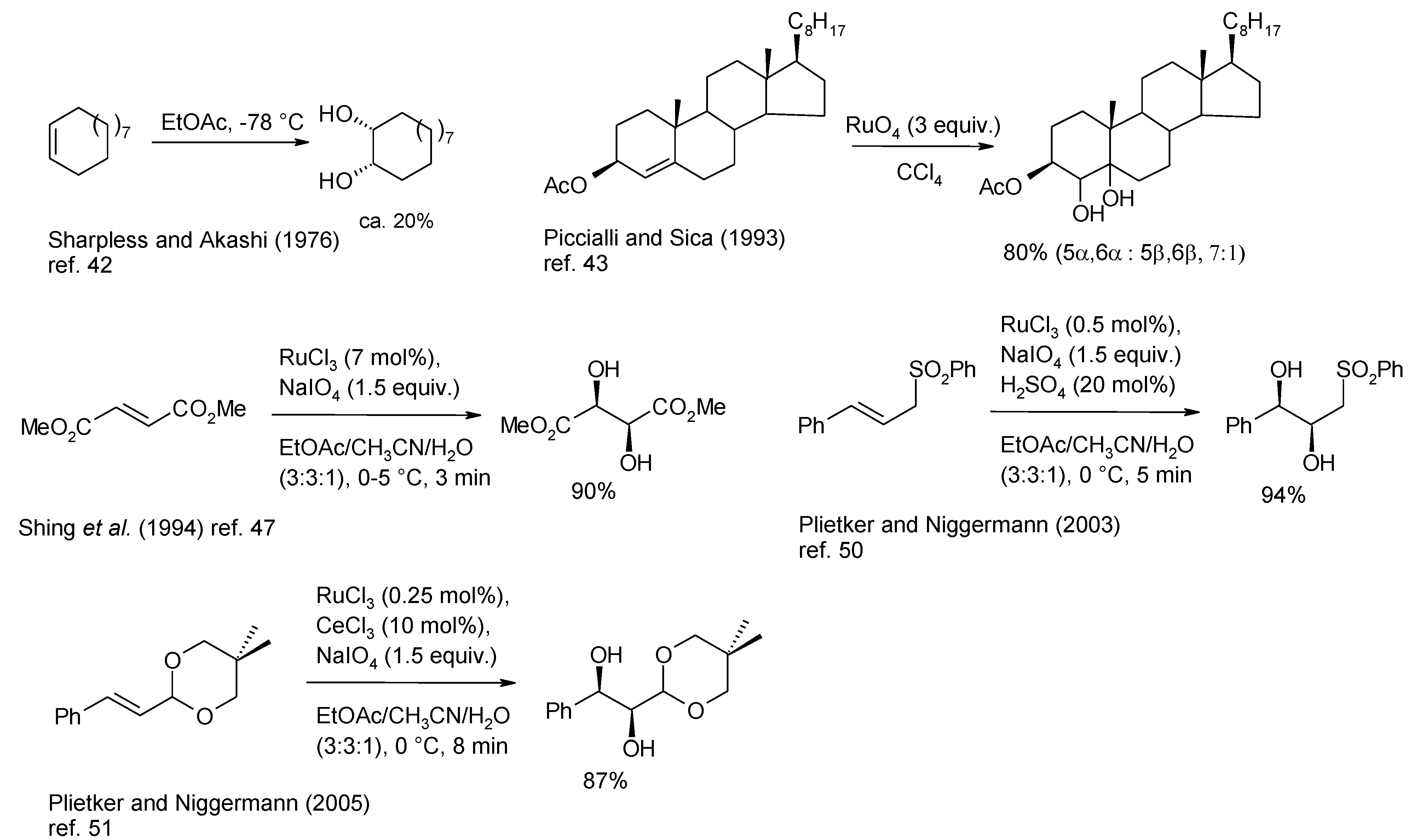
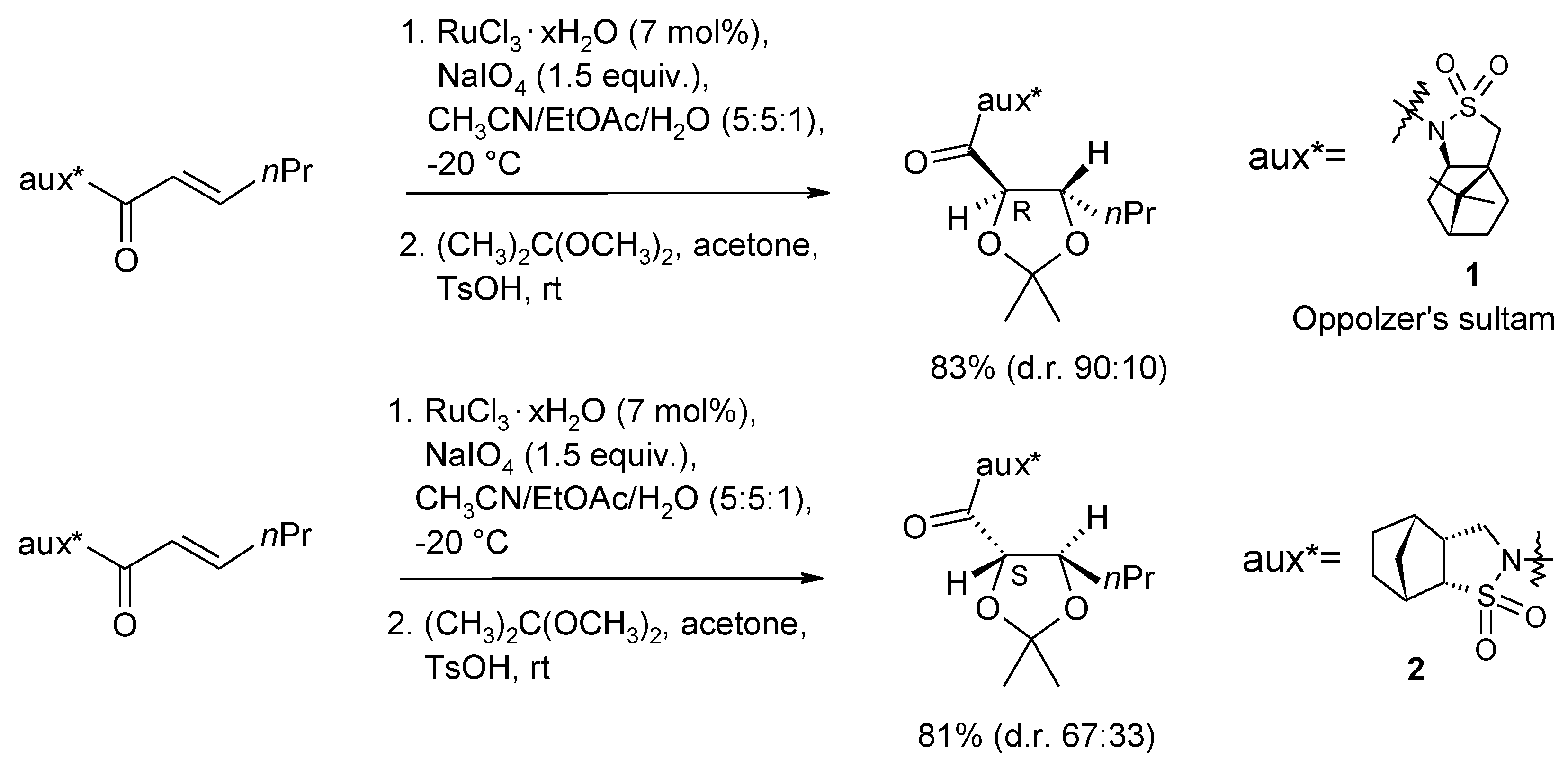
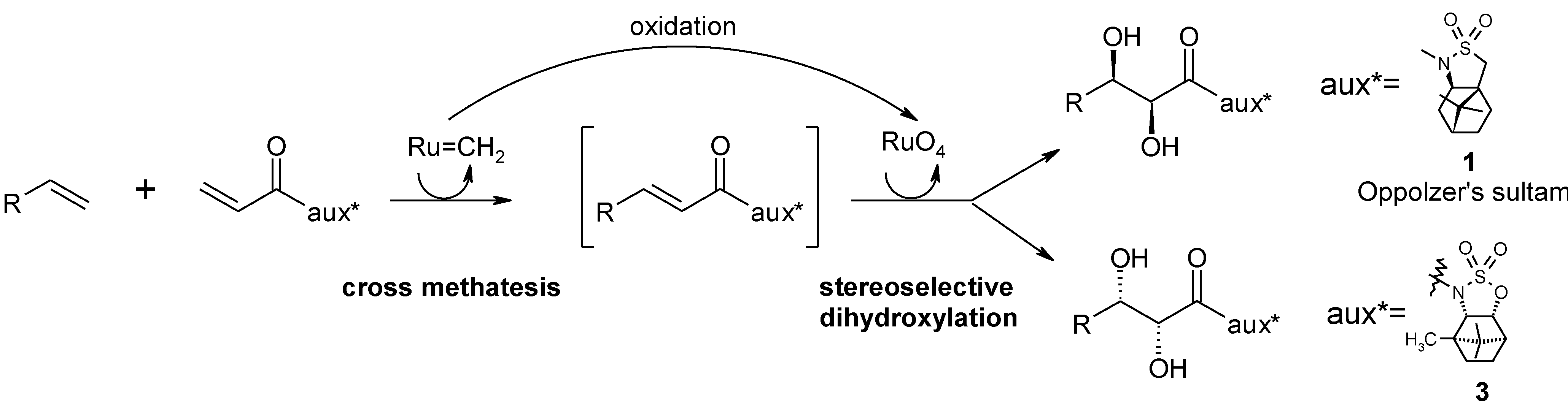
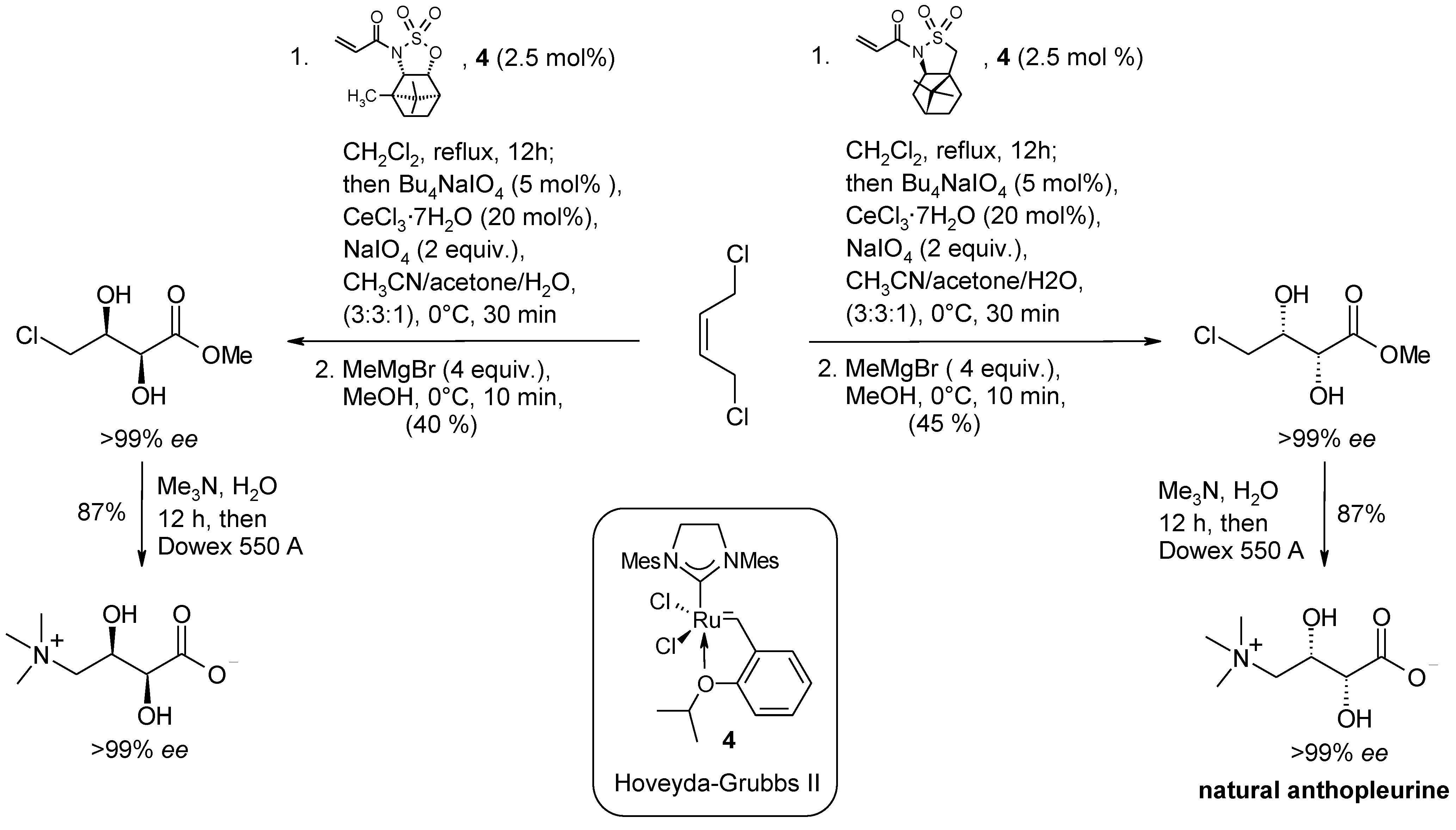
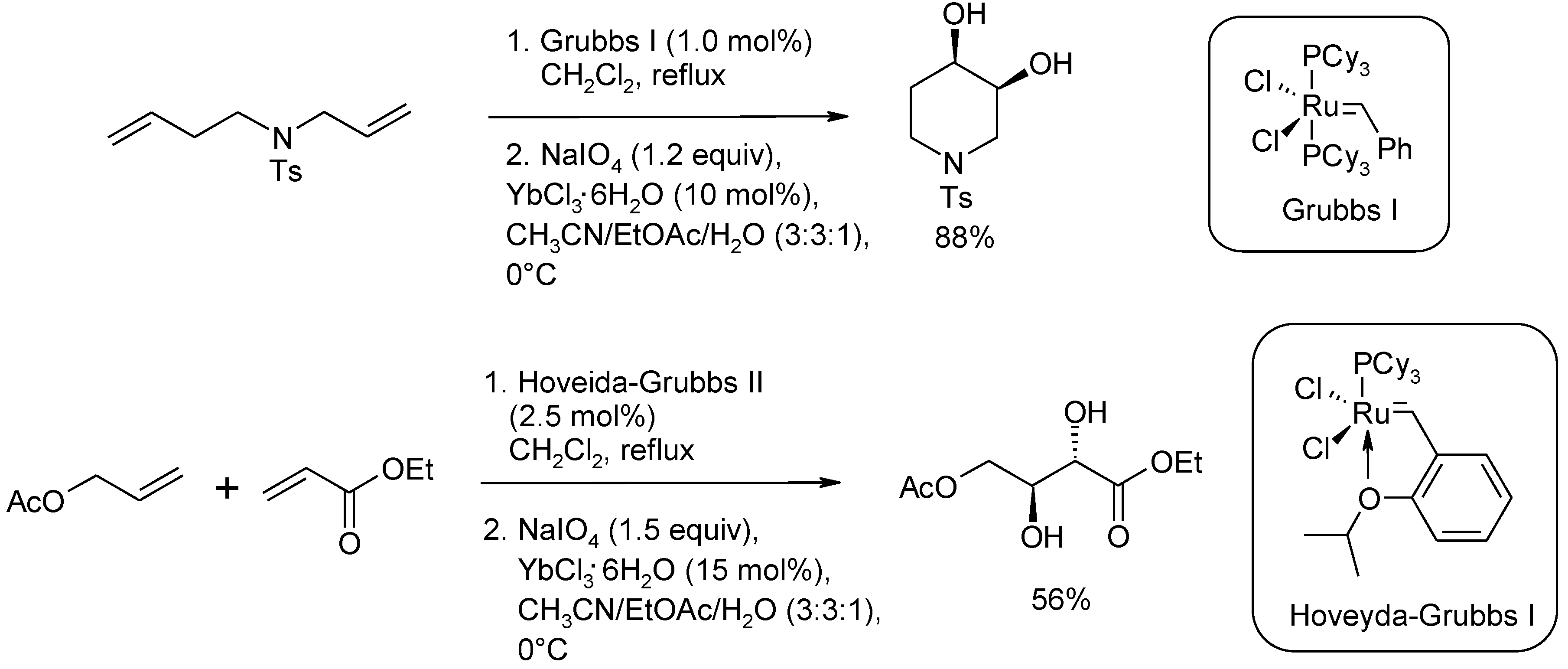
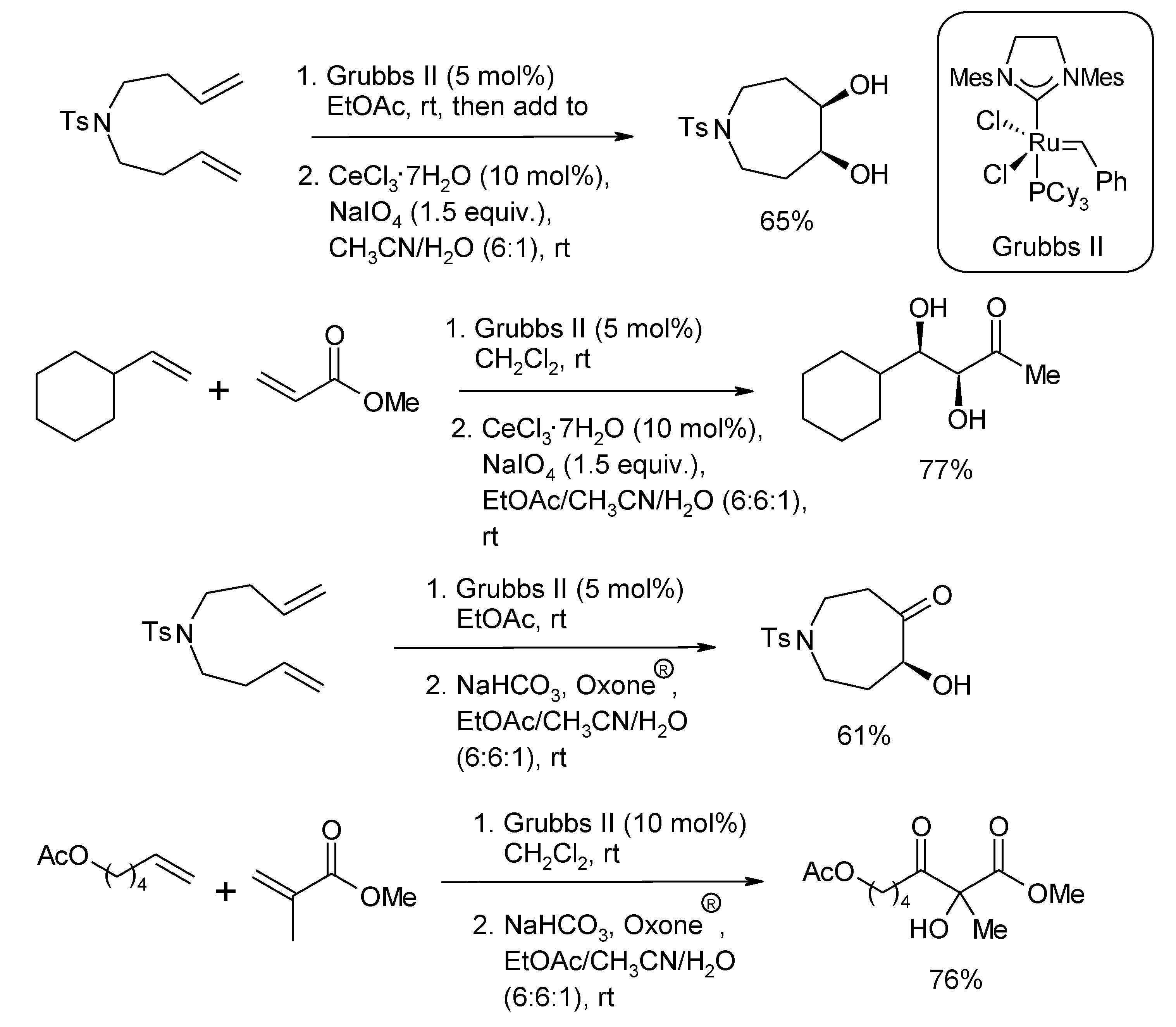
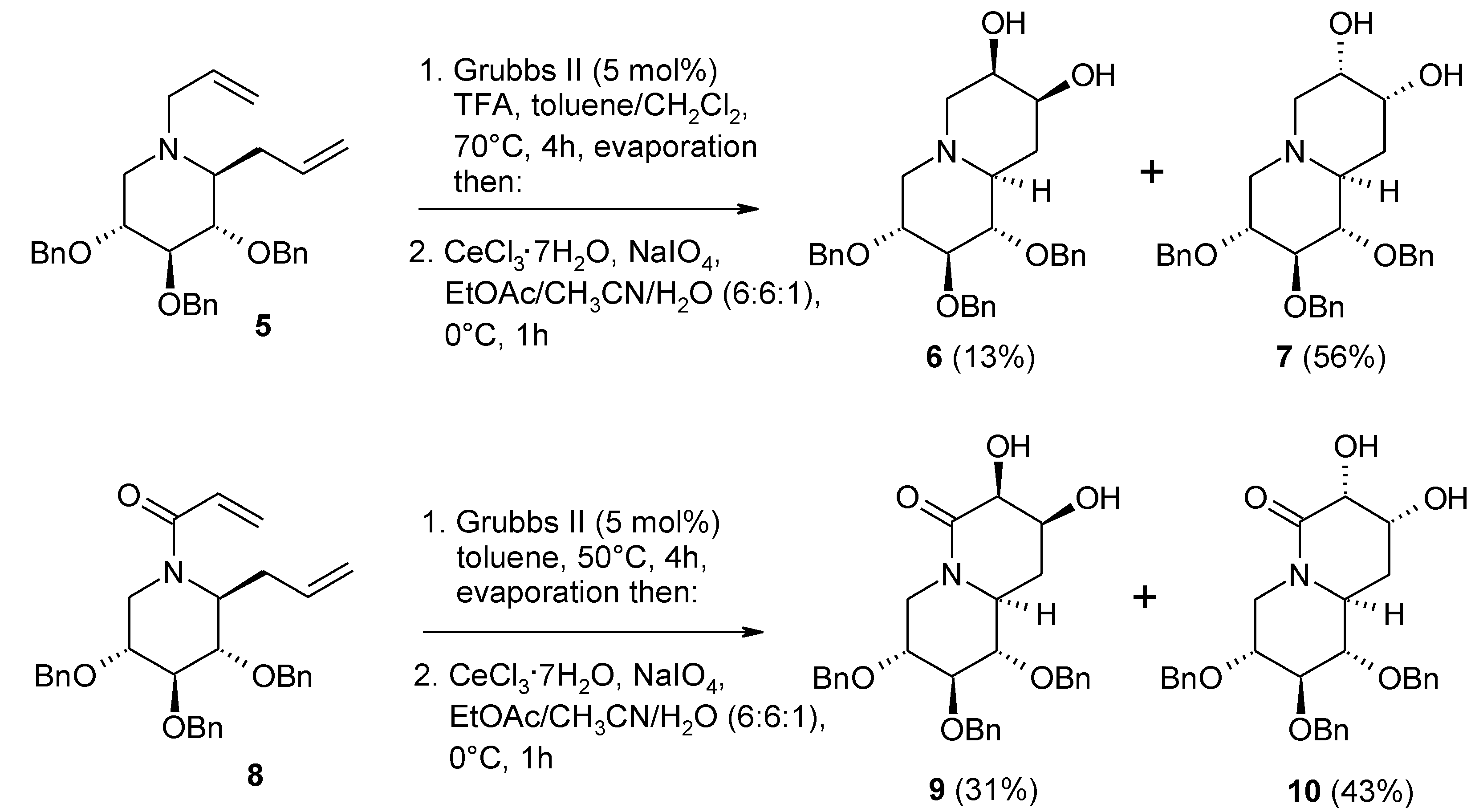
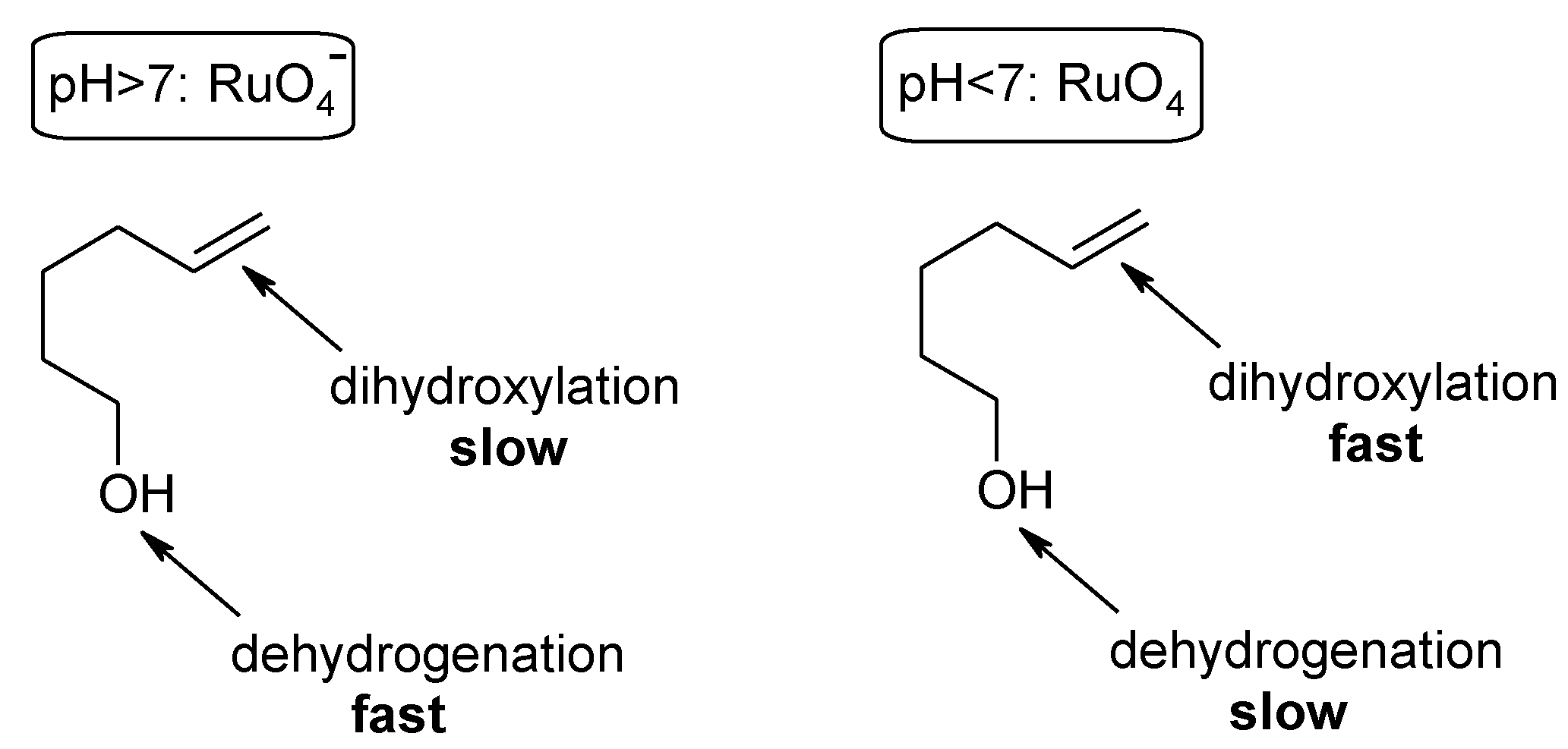
2.2. Diastereoselective Dihydroxylation of Alkenes and Tandem RCM/ Oxidation or CM/Oxidation Processes





2.3. Synthetic Applications of the Ru-Catalyzed Dihydroxylation of Olefins












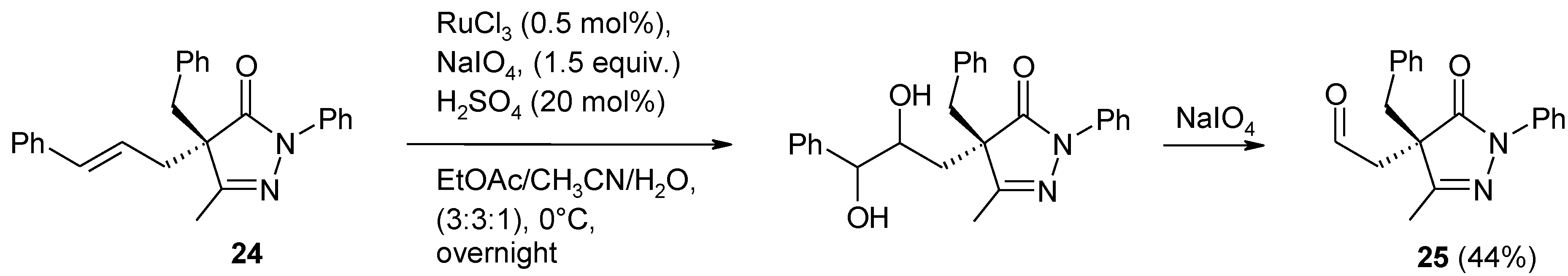
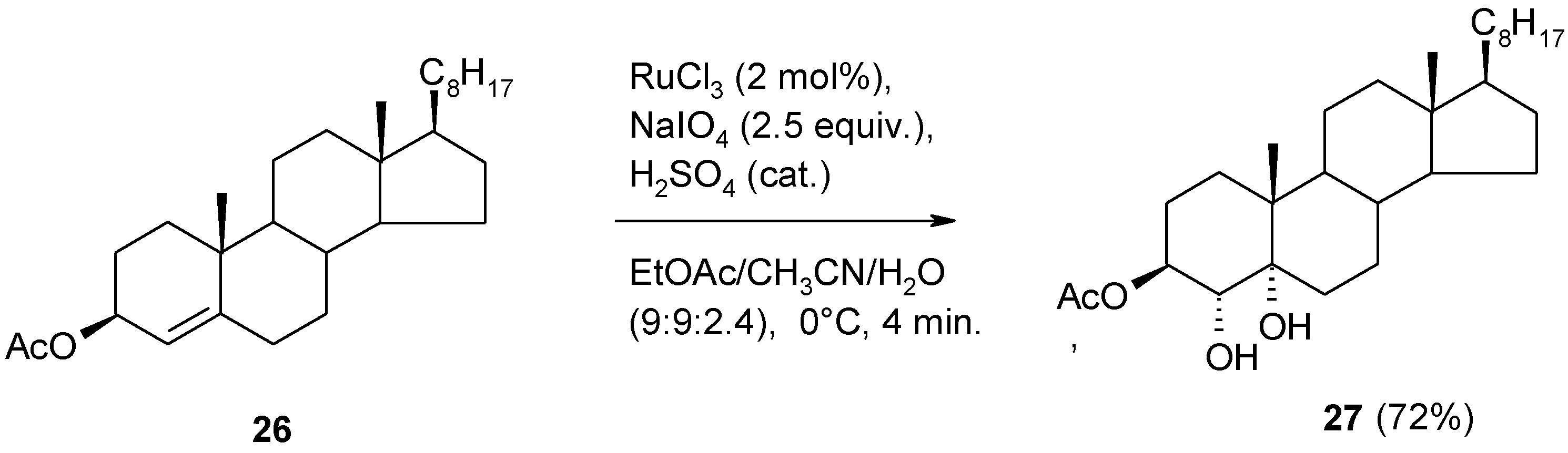
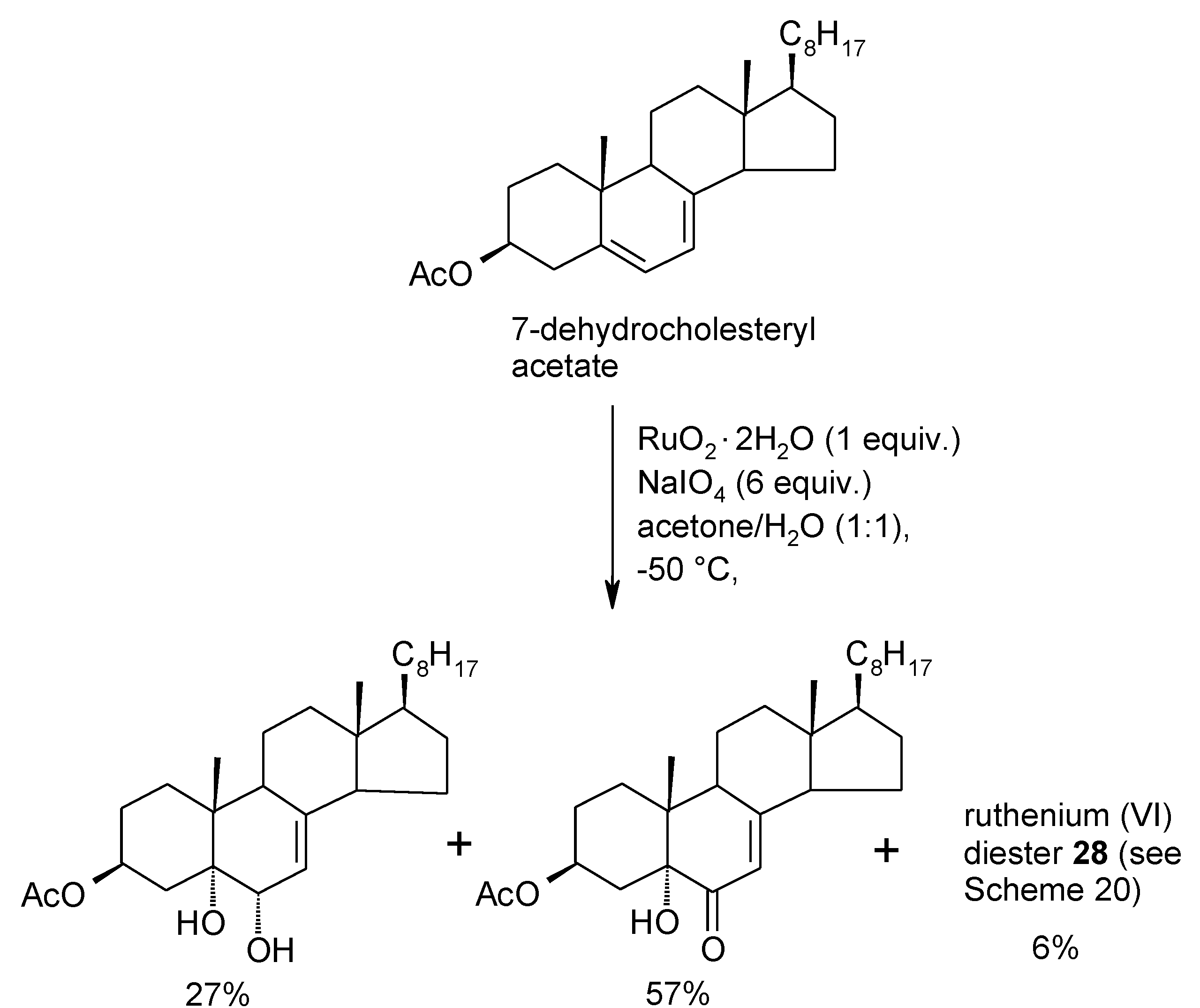
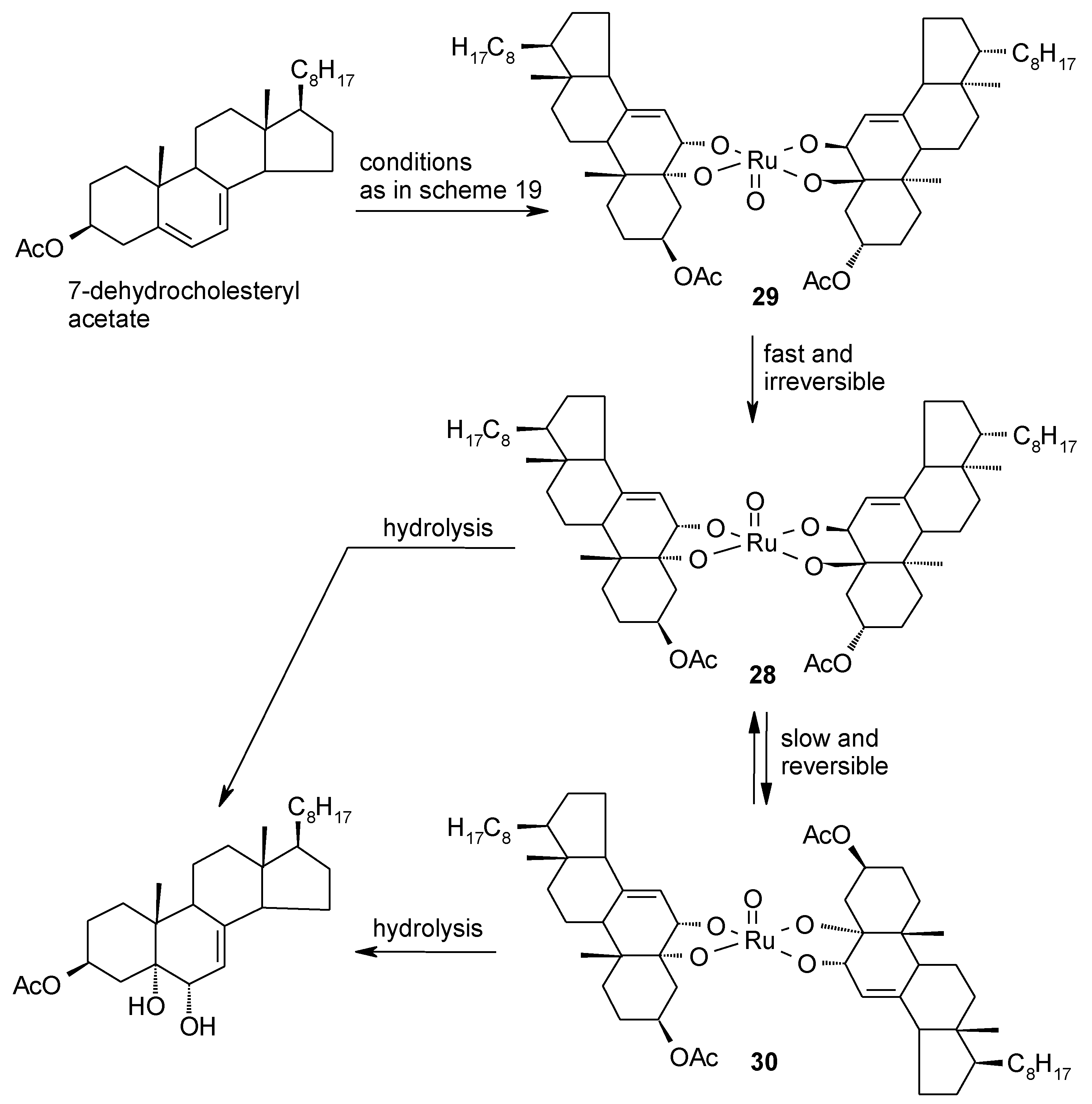
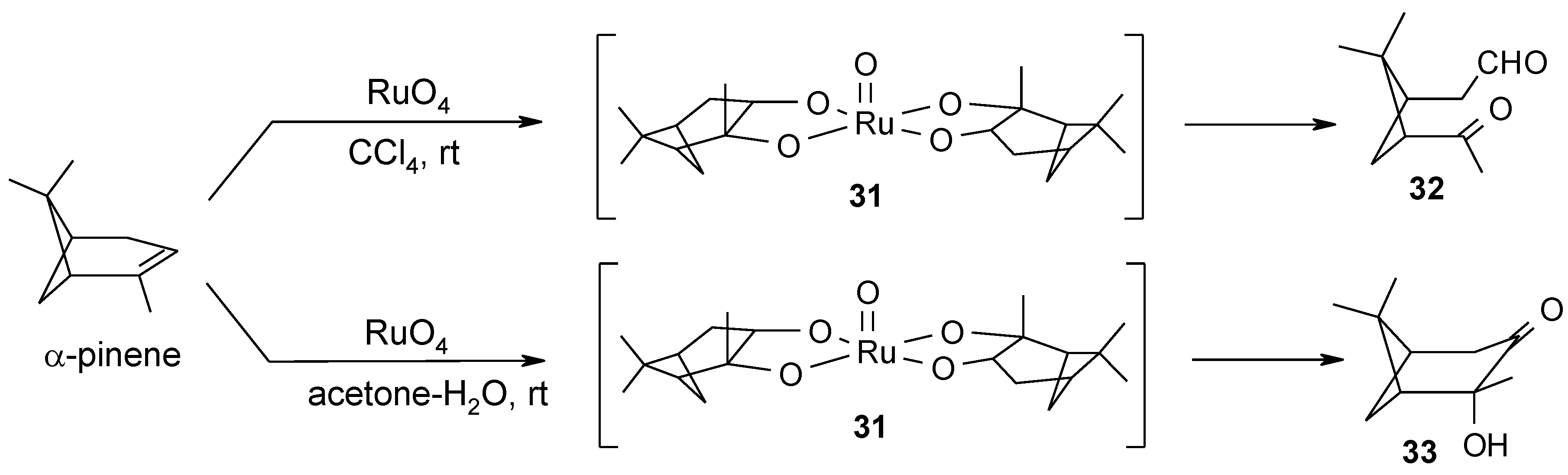
2.4. The Ruthenium Diester Intermediates in the Oxidation of Alkenes




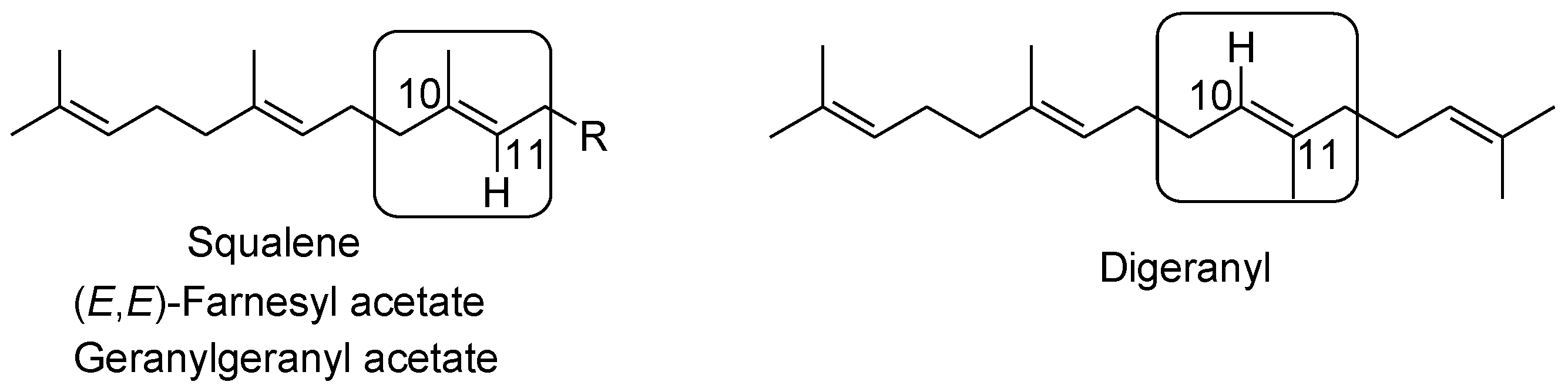
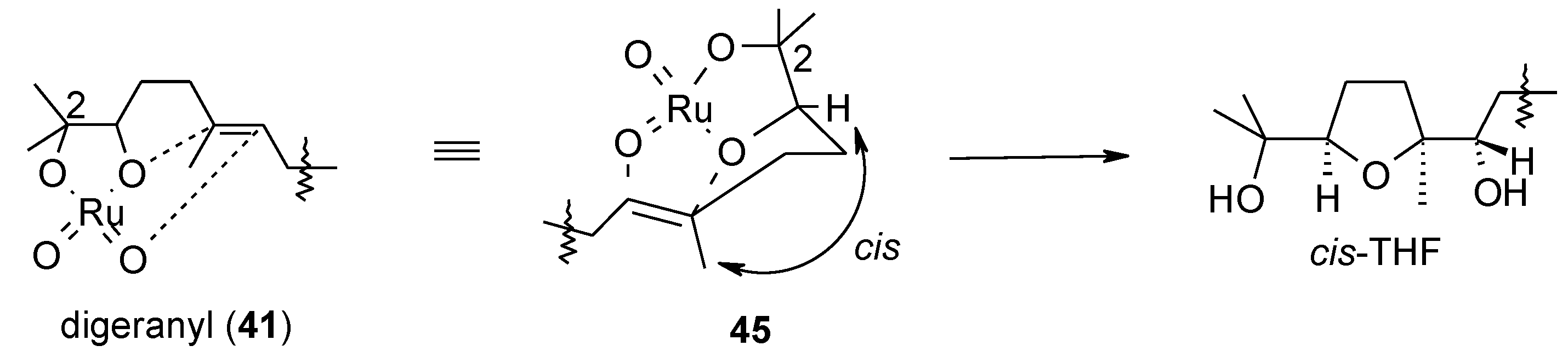
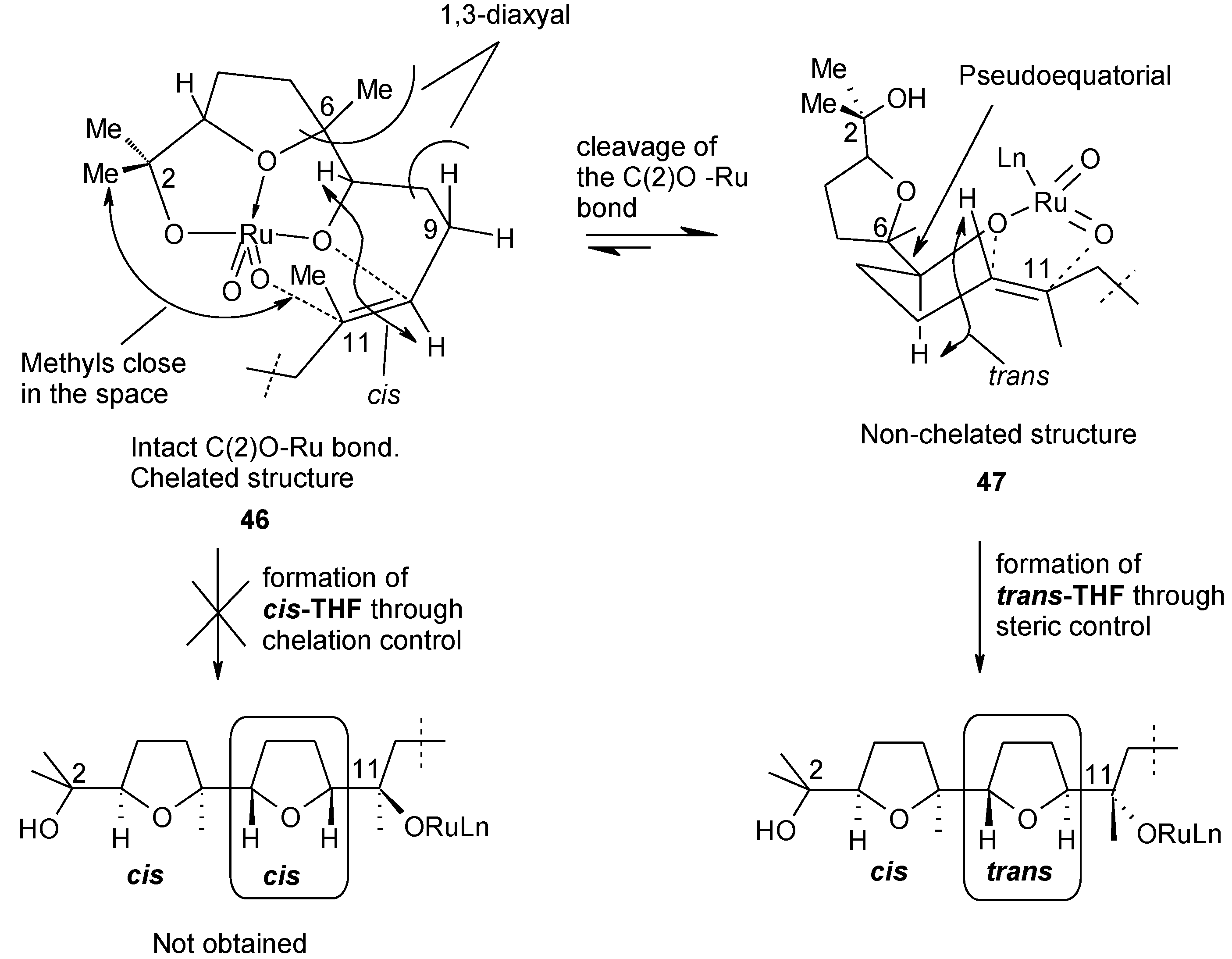
2.5. Oxidative Cyclization of Dienes and Polyenes






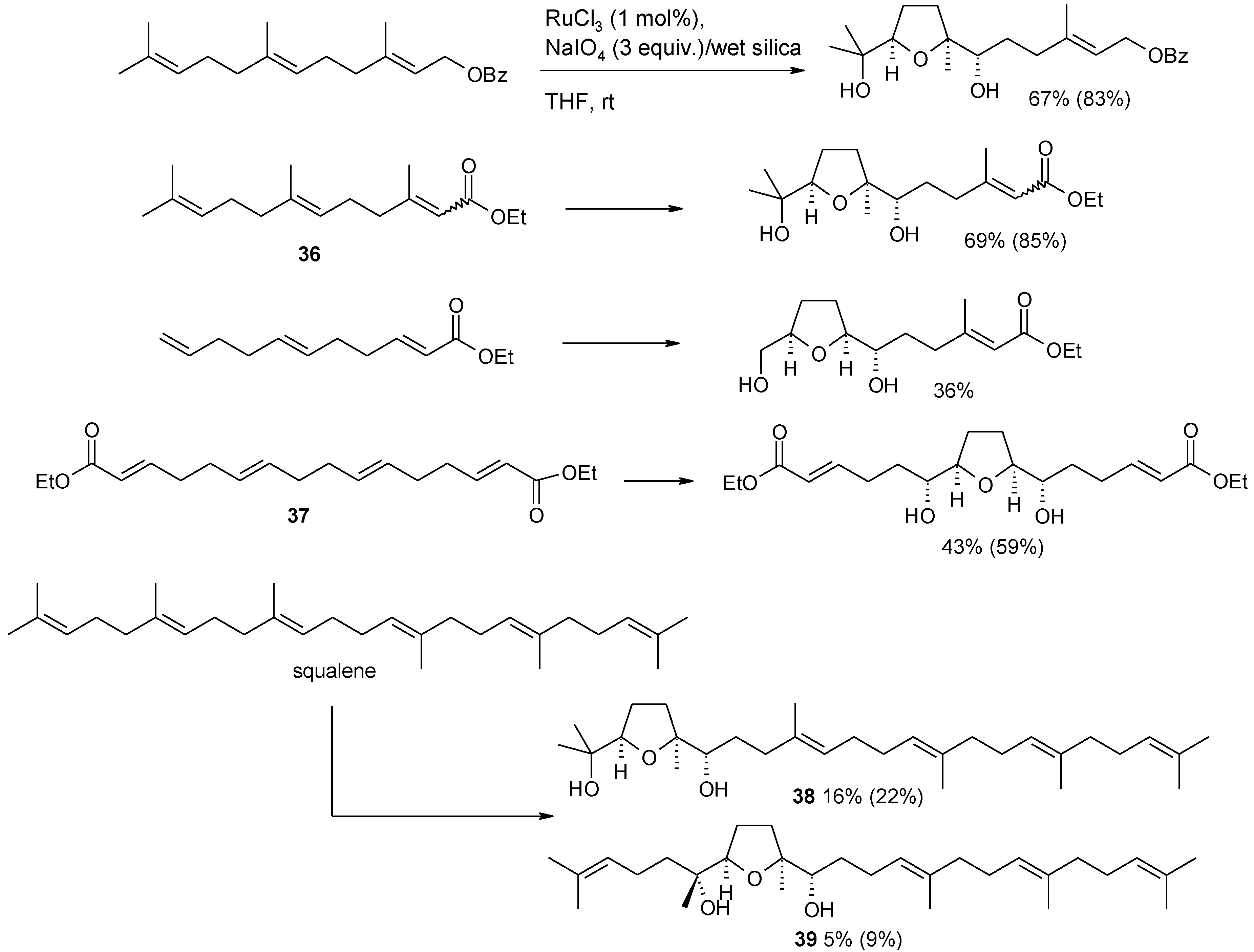
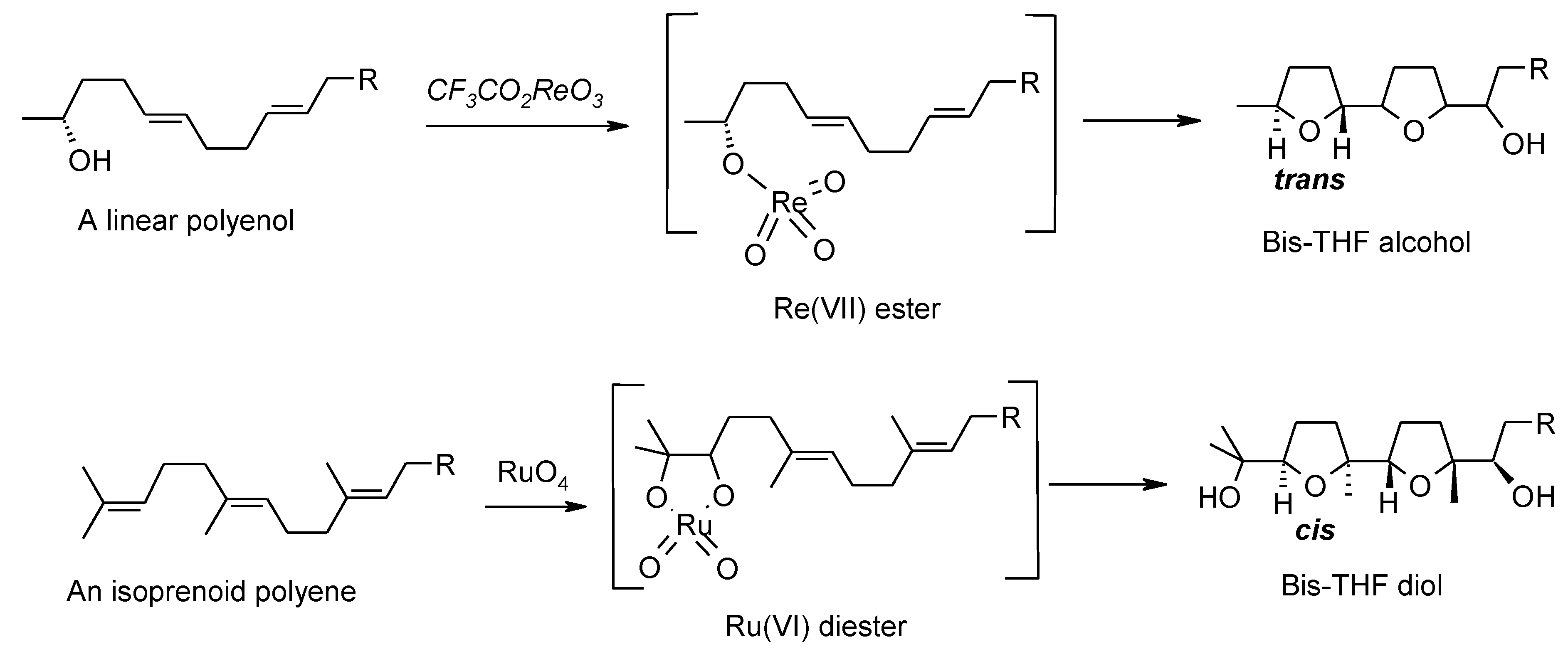
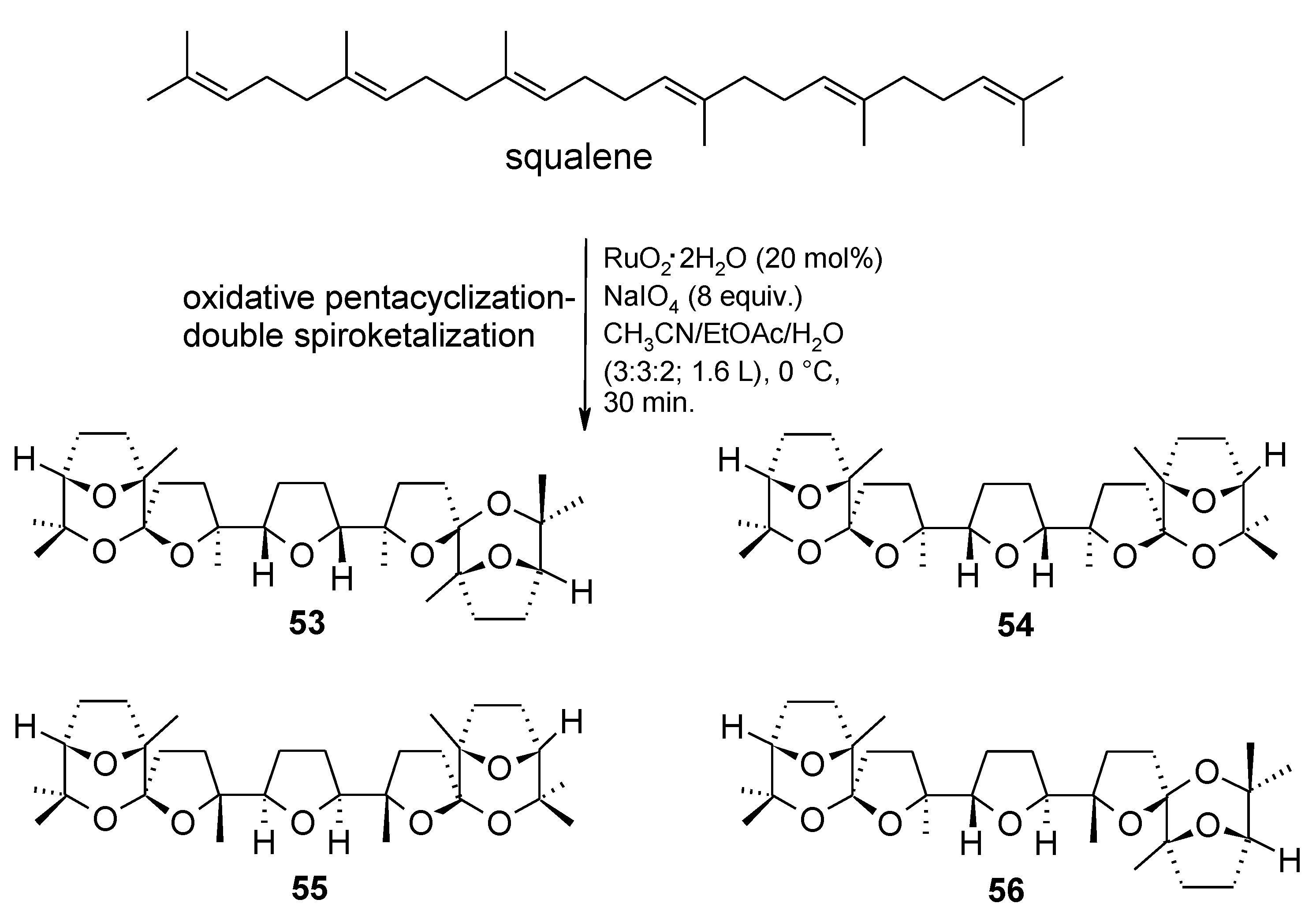
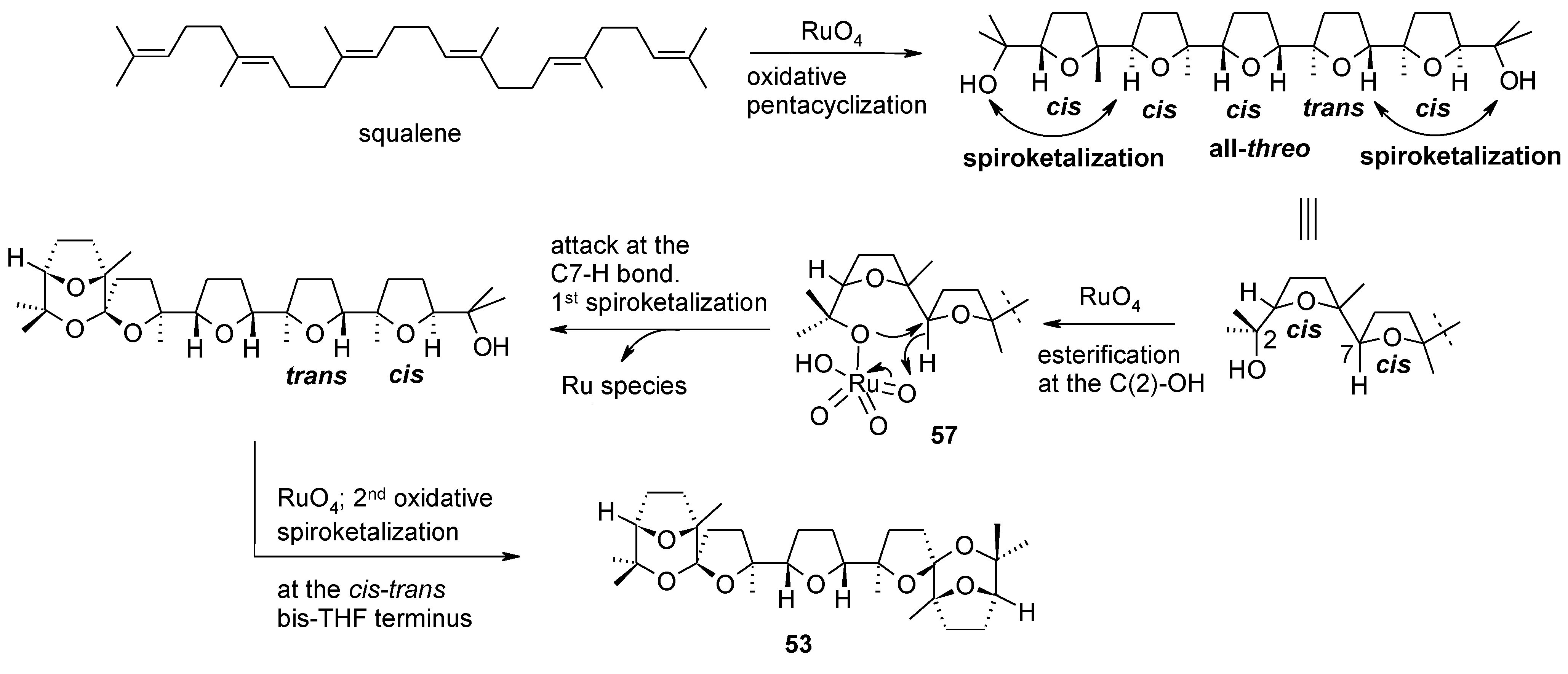
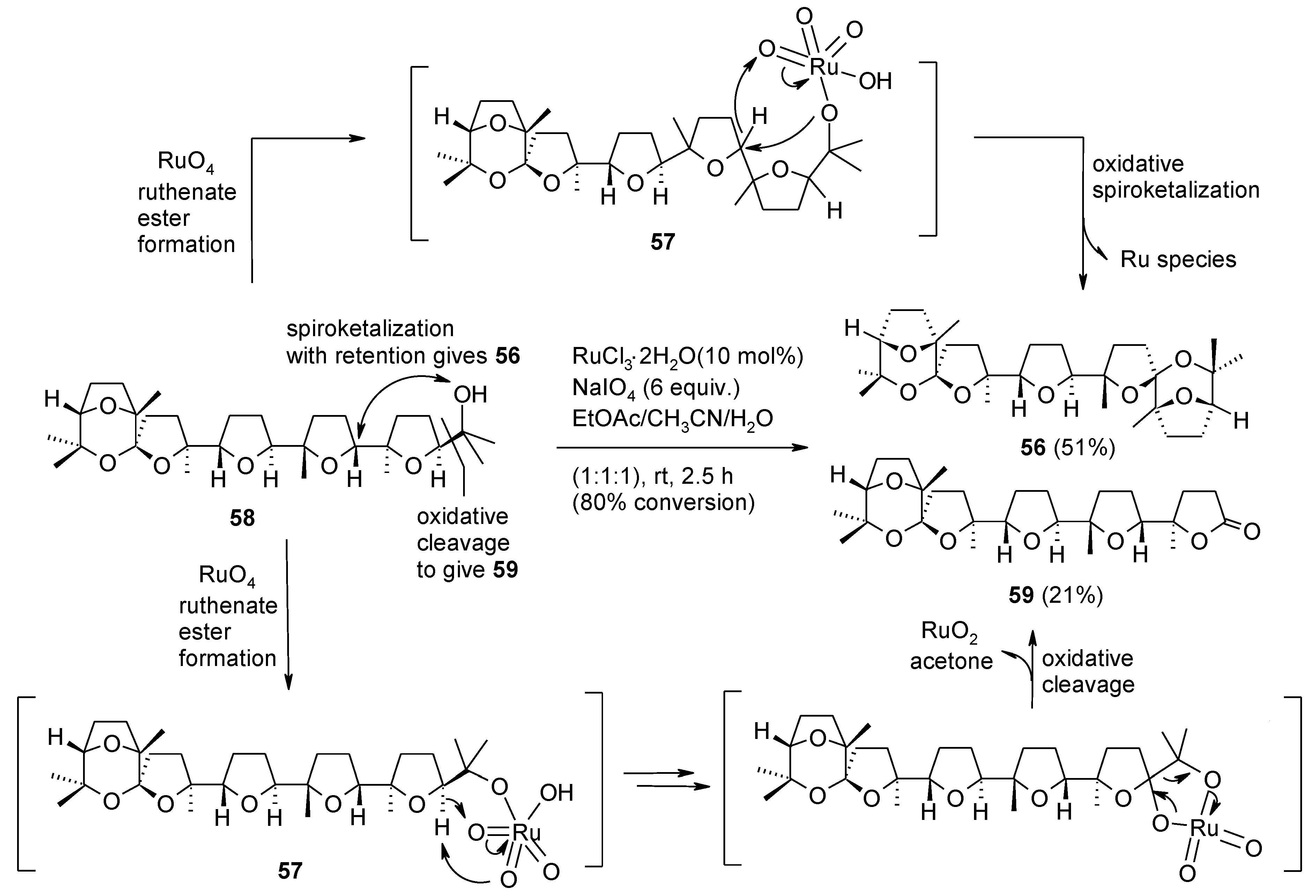
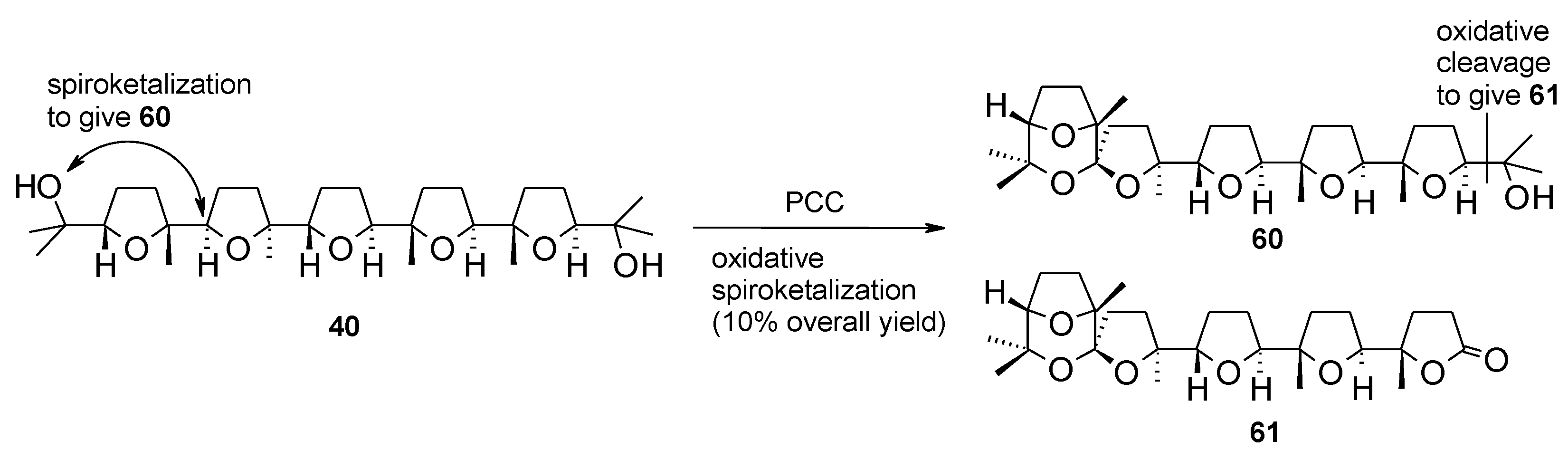
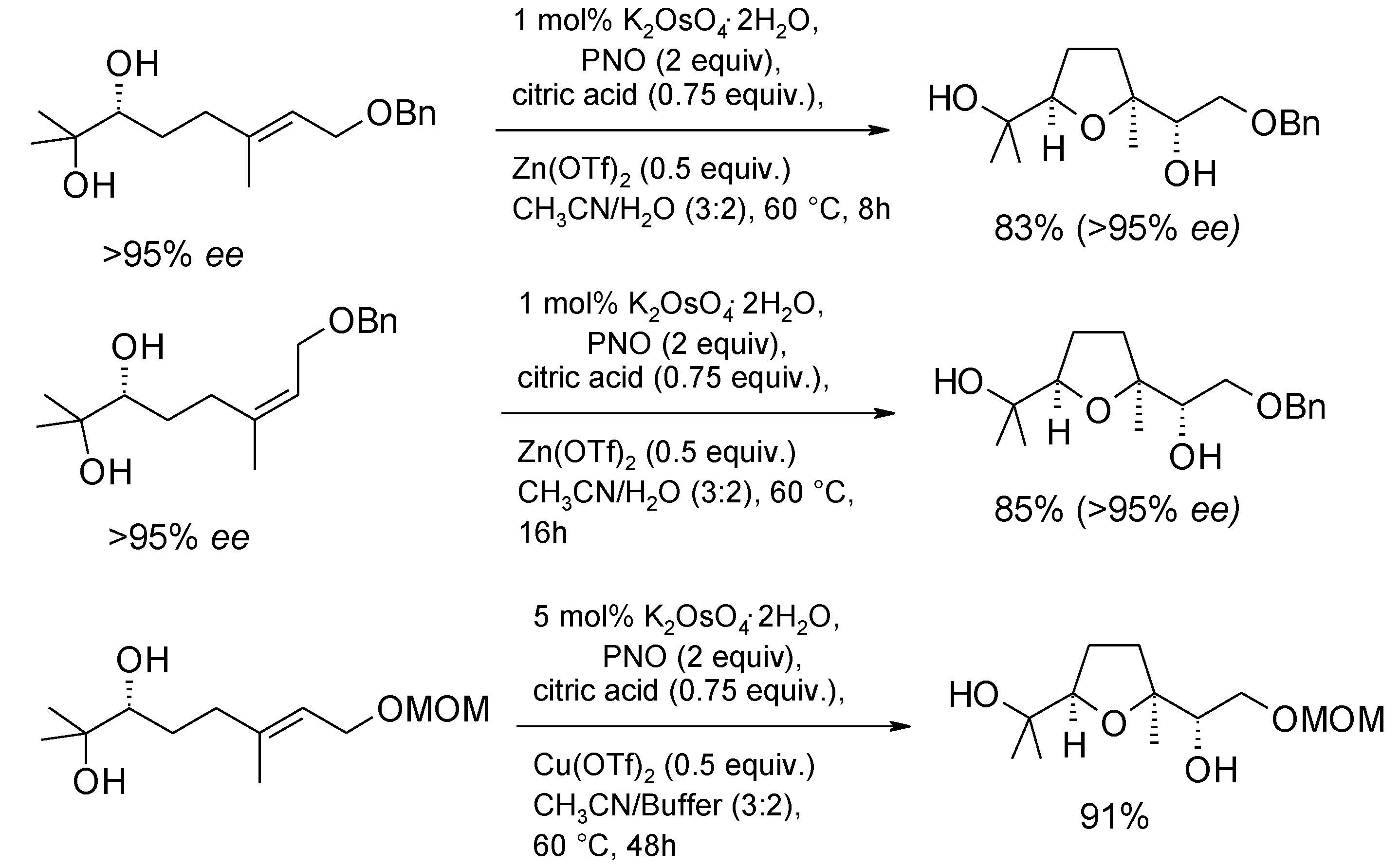
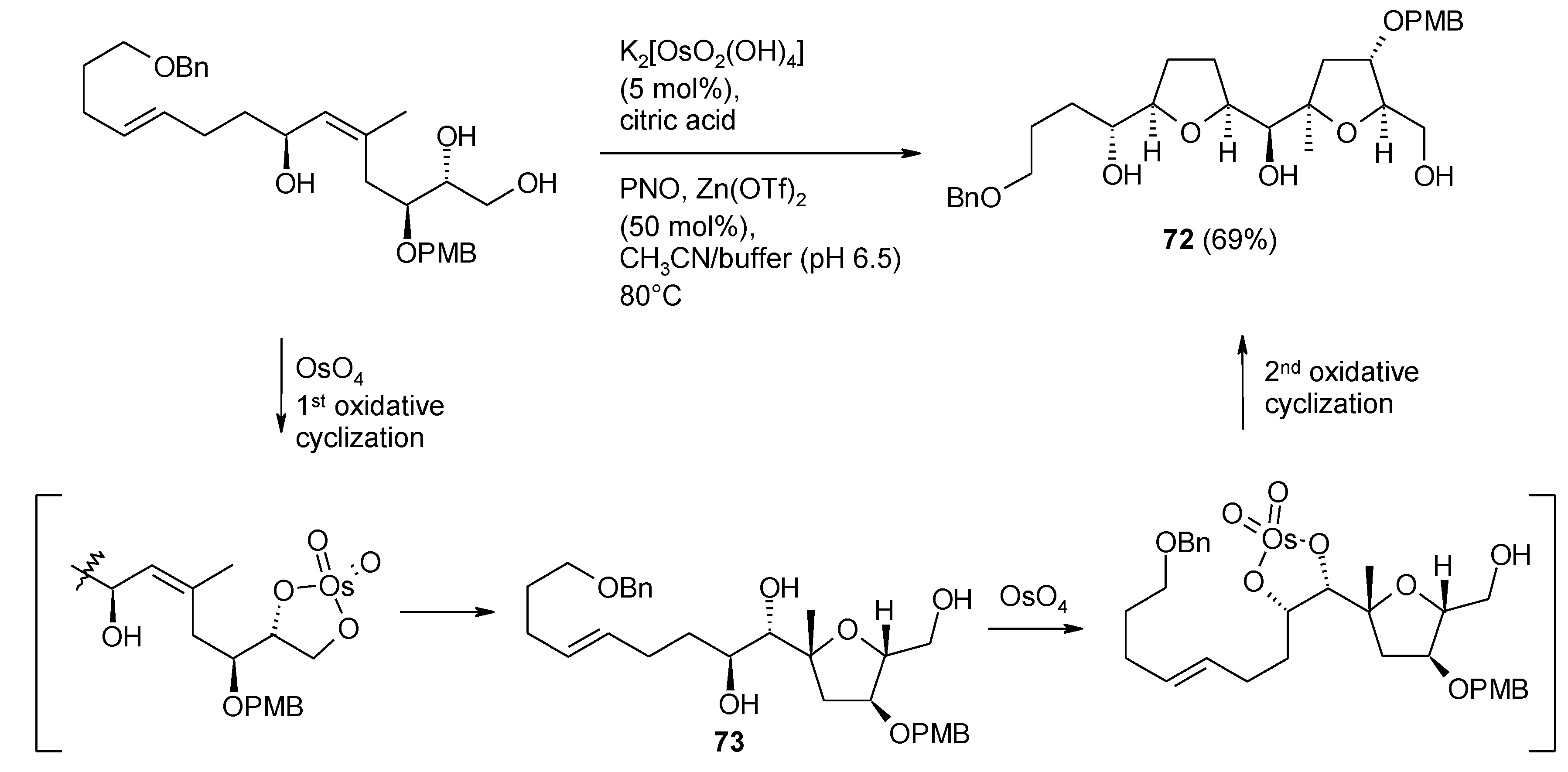
2.5.1. Oxidative Polycyclization of Hydroxypolyenes




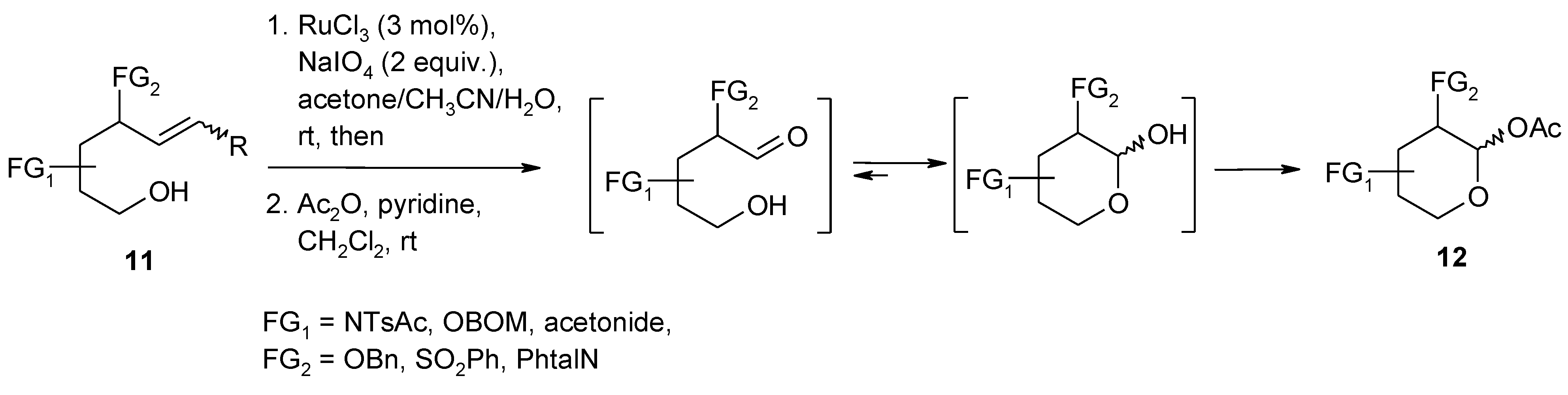
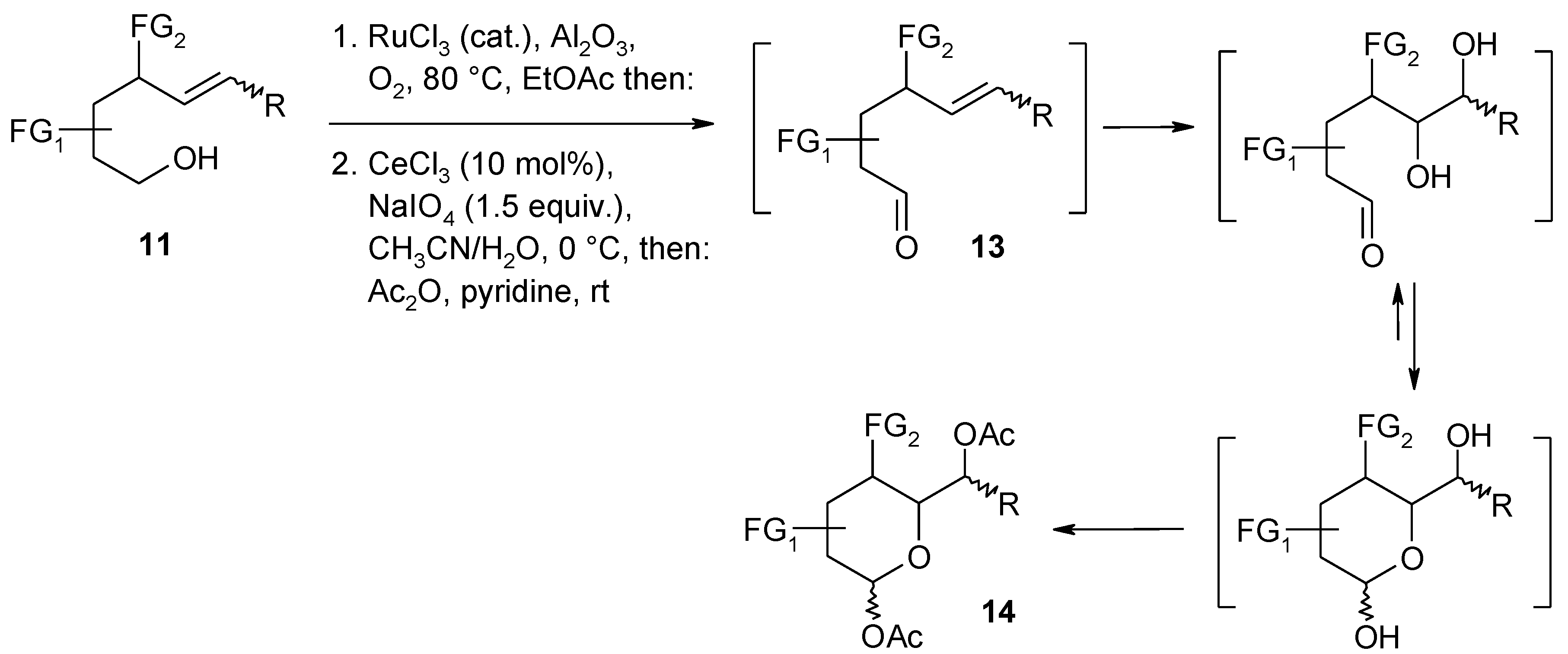
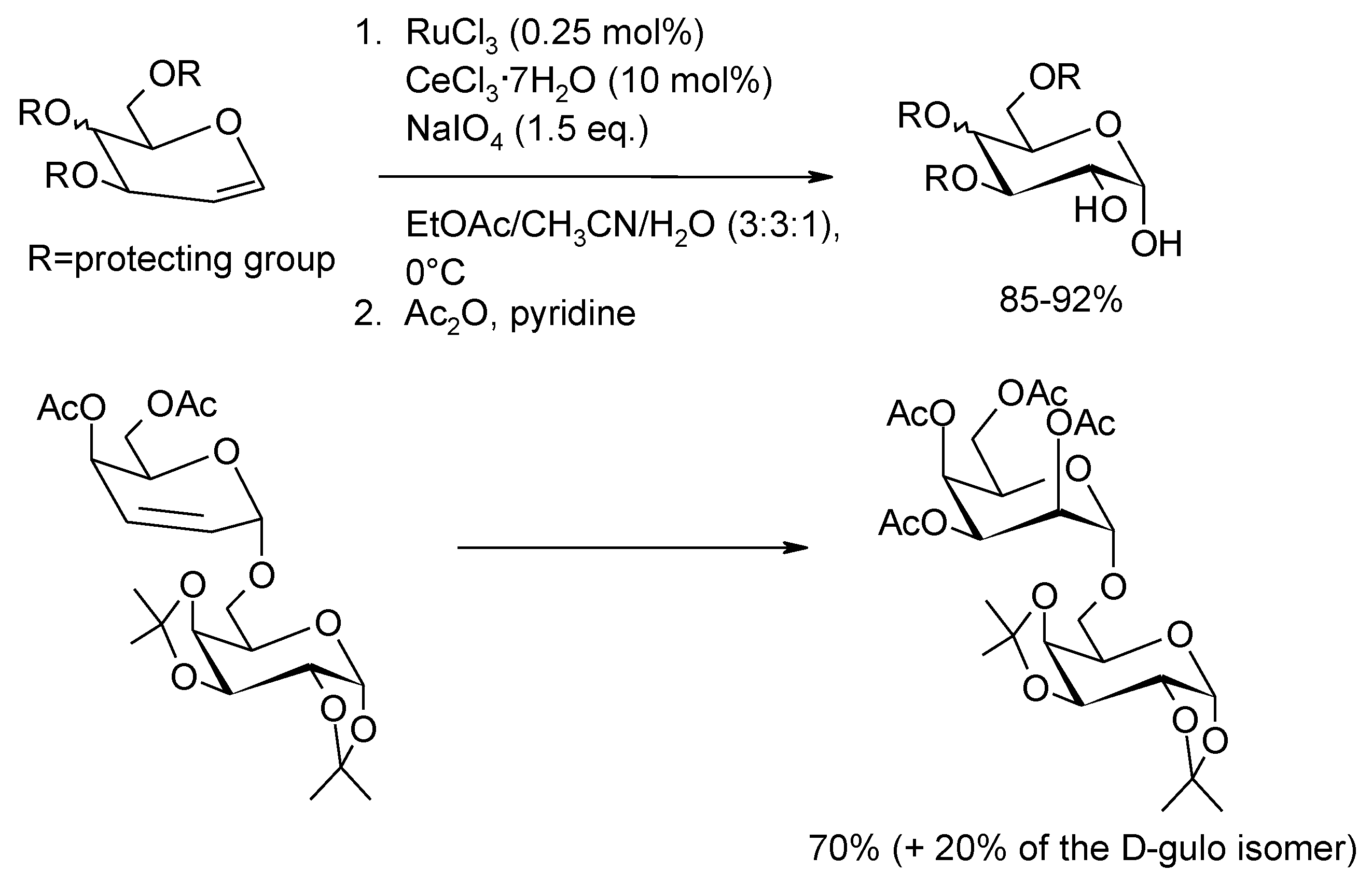
2.6. Oxidation of Ethers





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
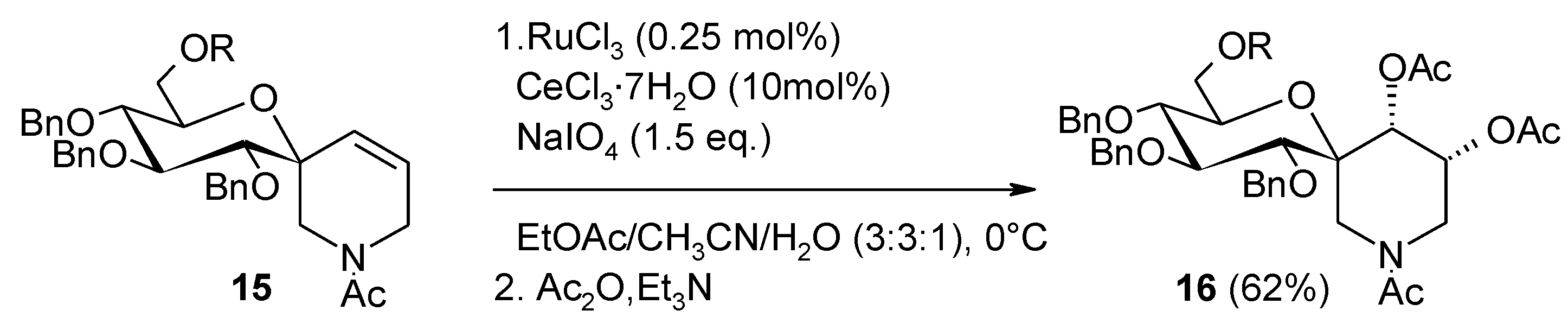{kind=link}
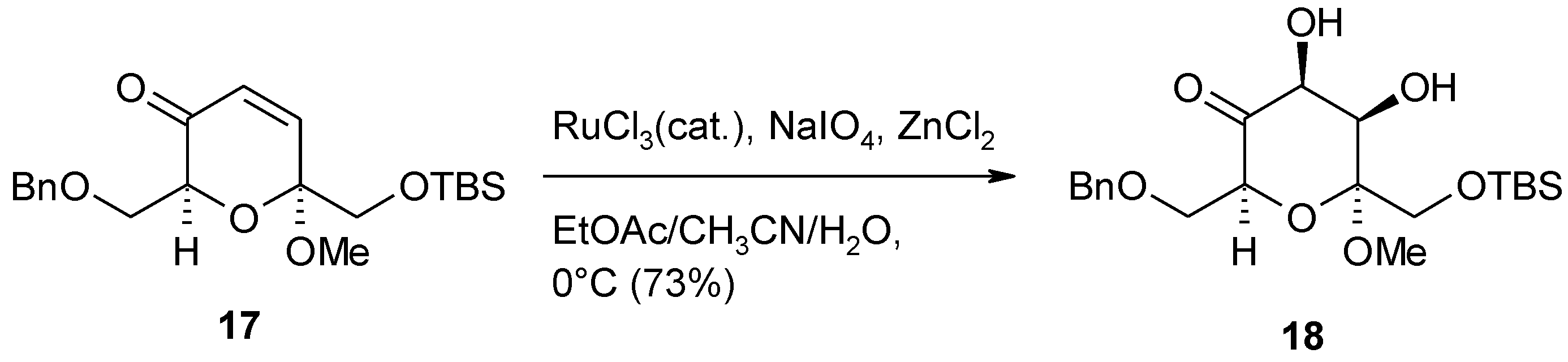{kind=link}
{kind=link}
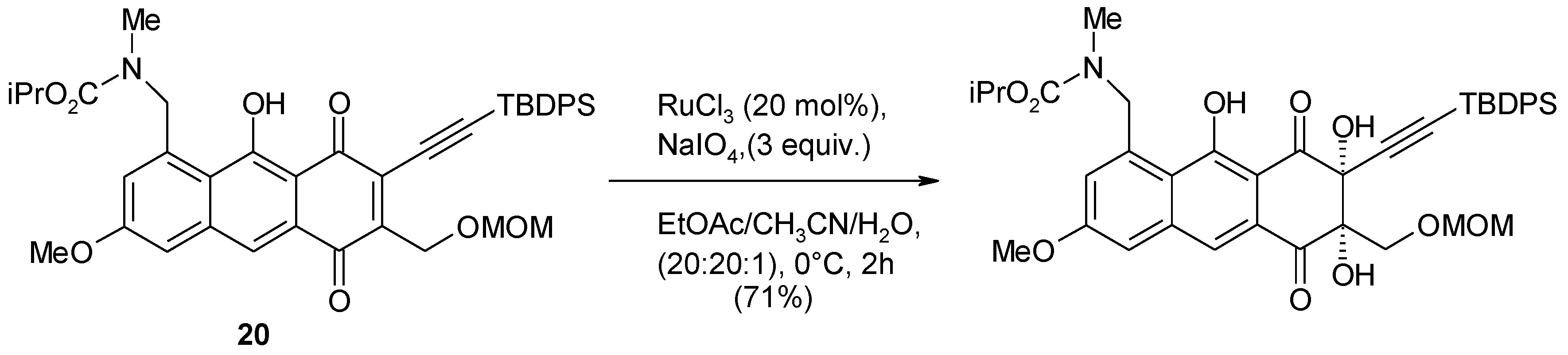{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
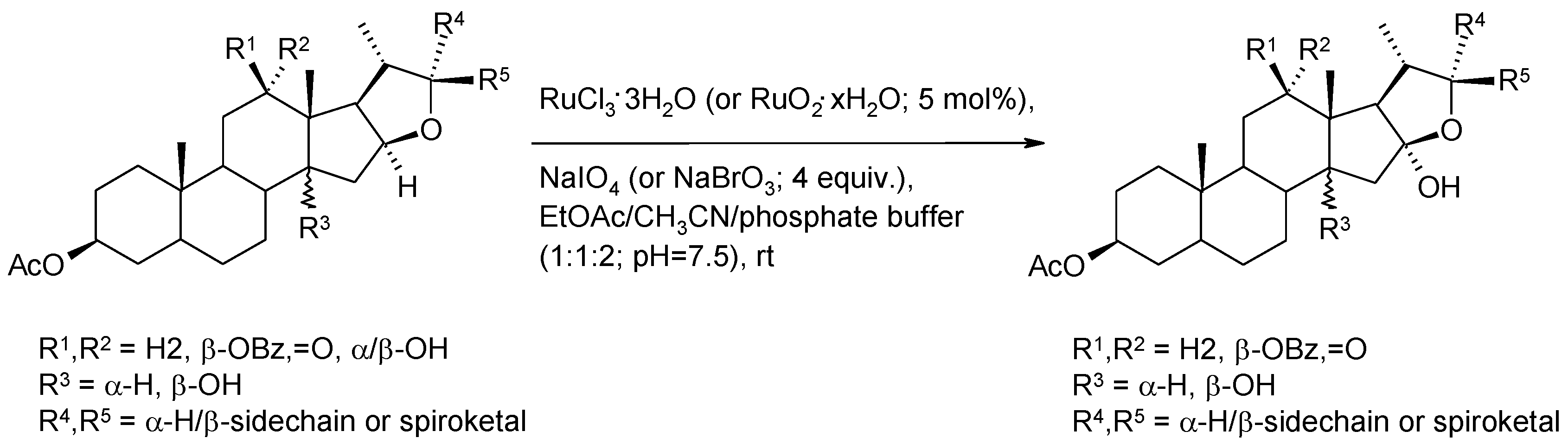{kind=link}
{kind=link}
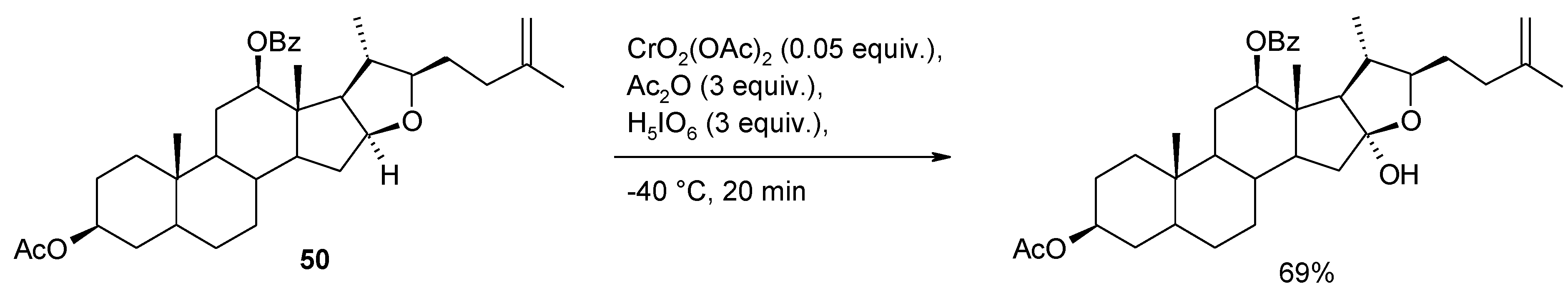{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
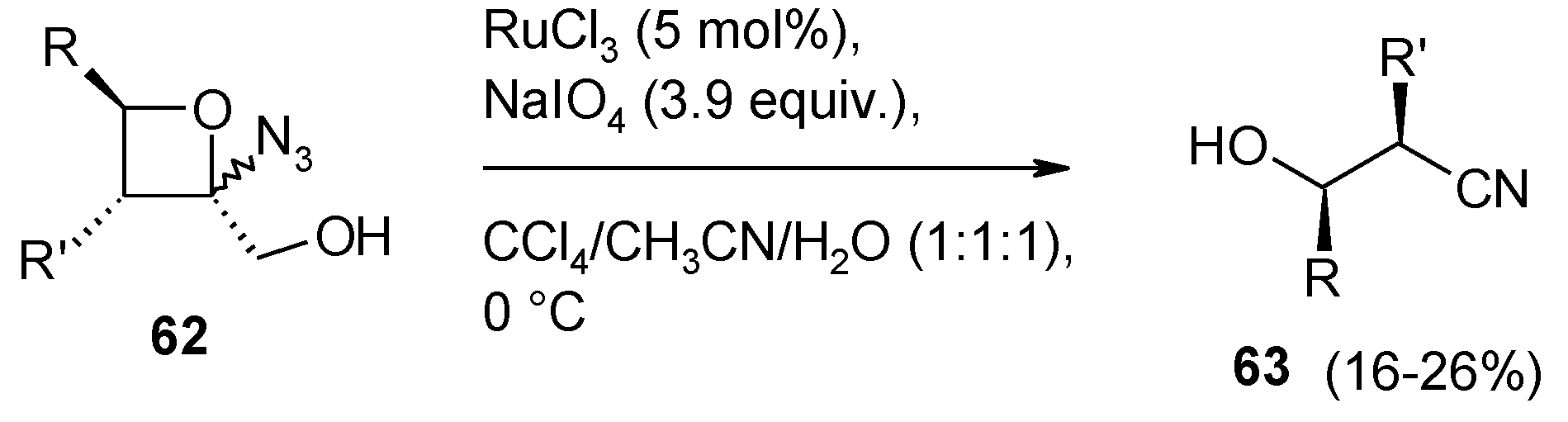{kind=link}
{kind=link}
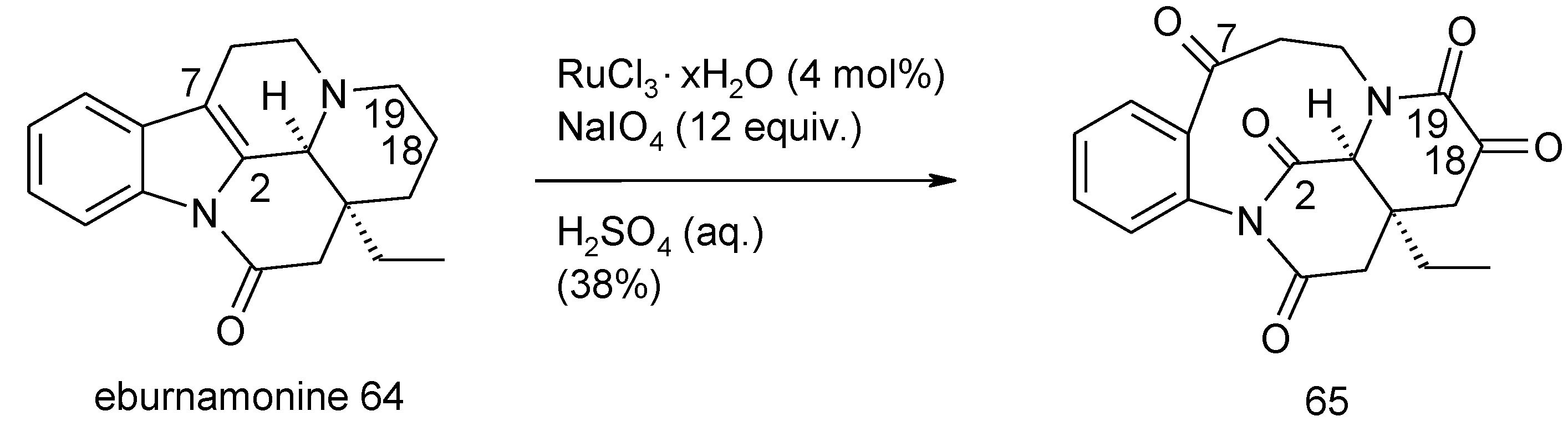{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
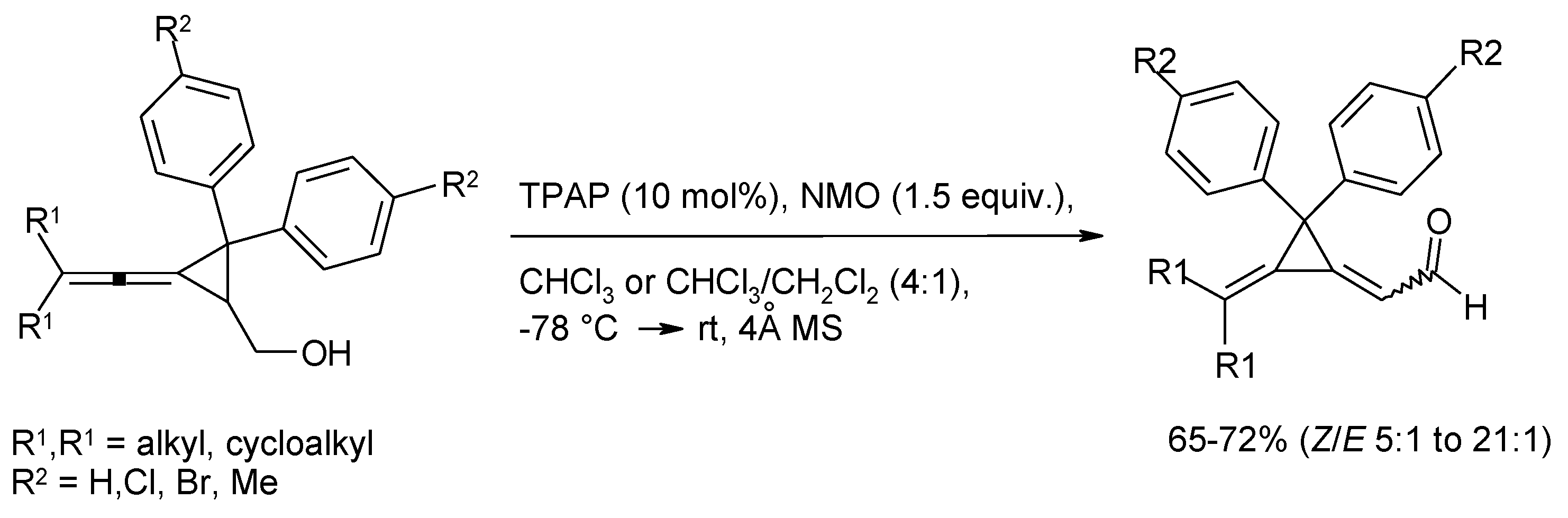{kind=link}
{kind=link}


2.6.1. Oxidative Spiroketalization




2.7. Some Unexpected Results




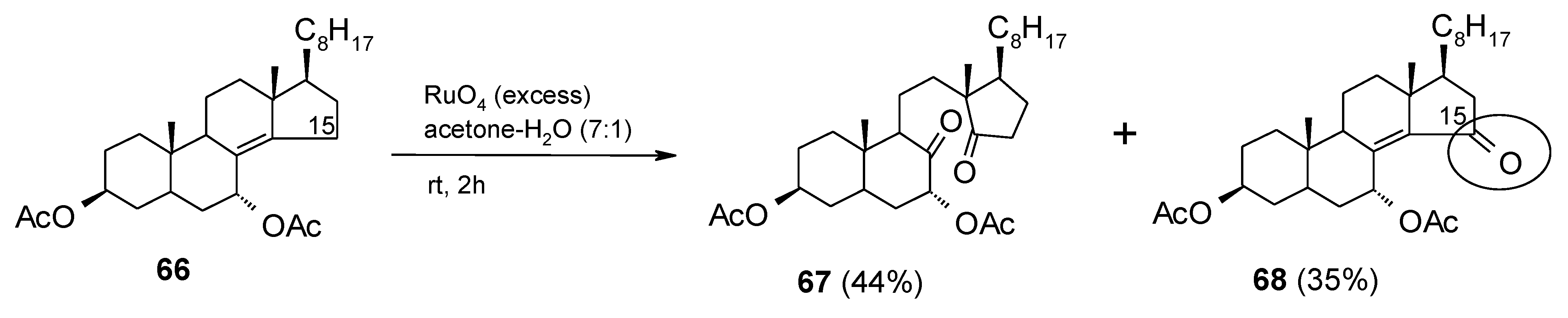
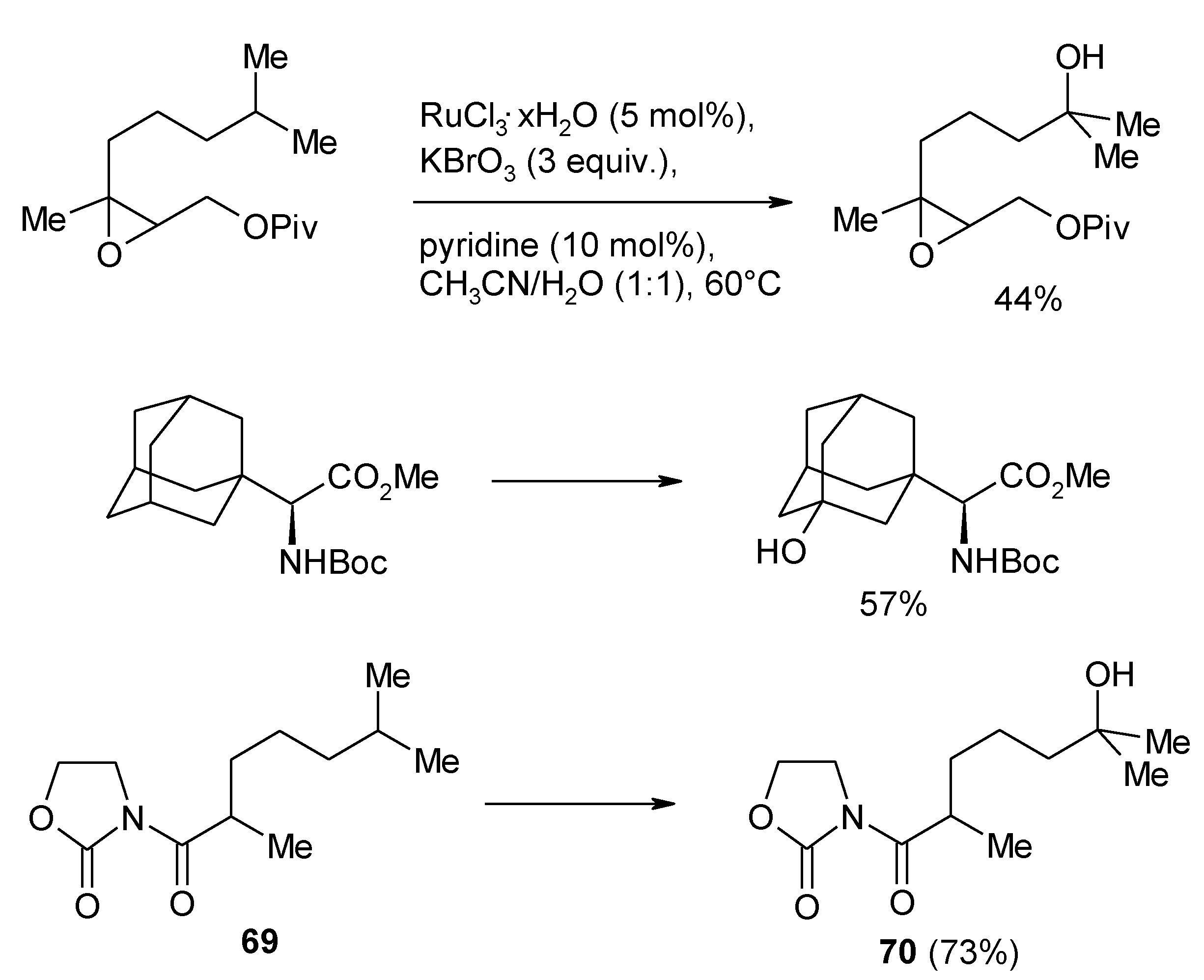
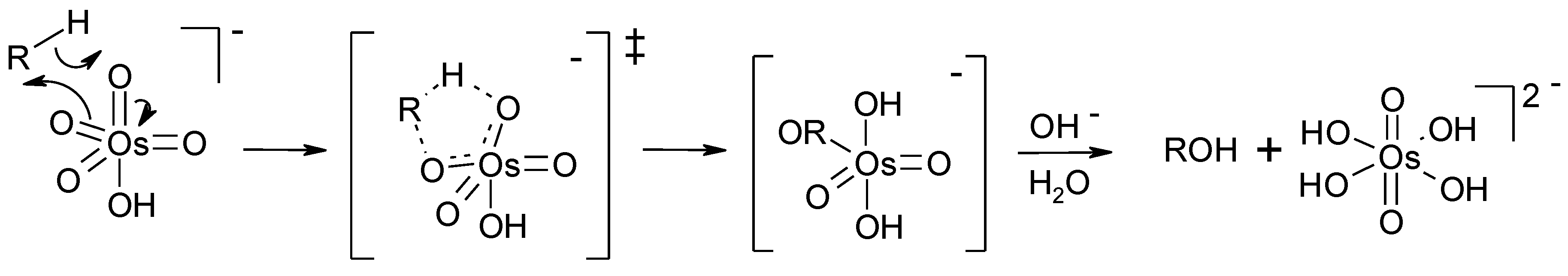
2.8. Oxidation of Alkanes


3. Perruthenate Chemistry
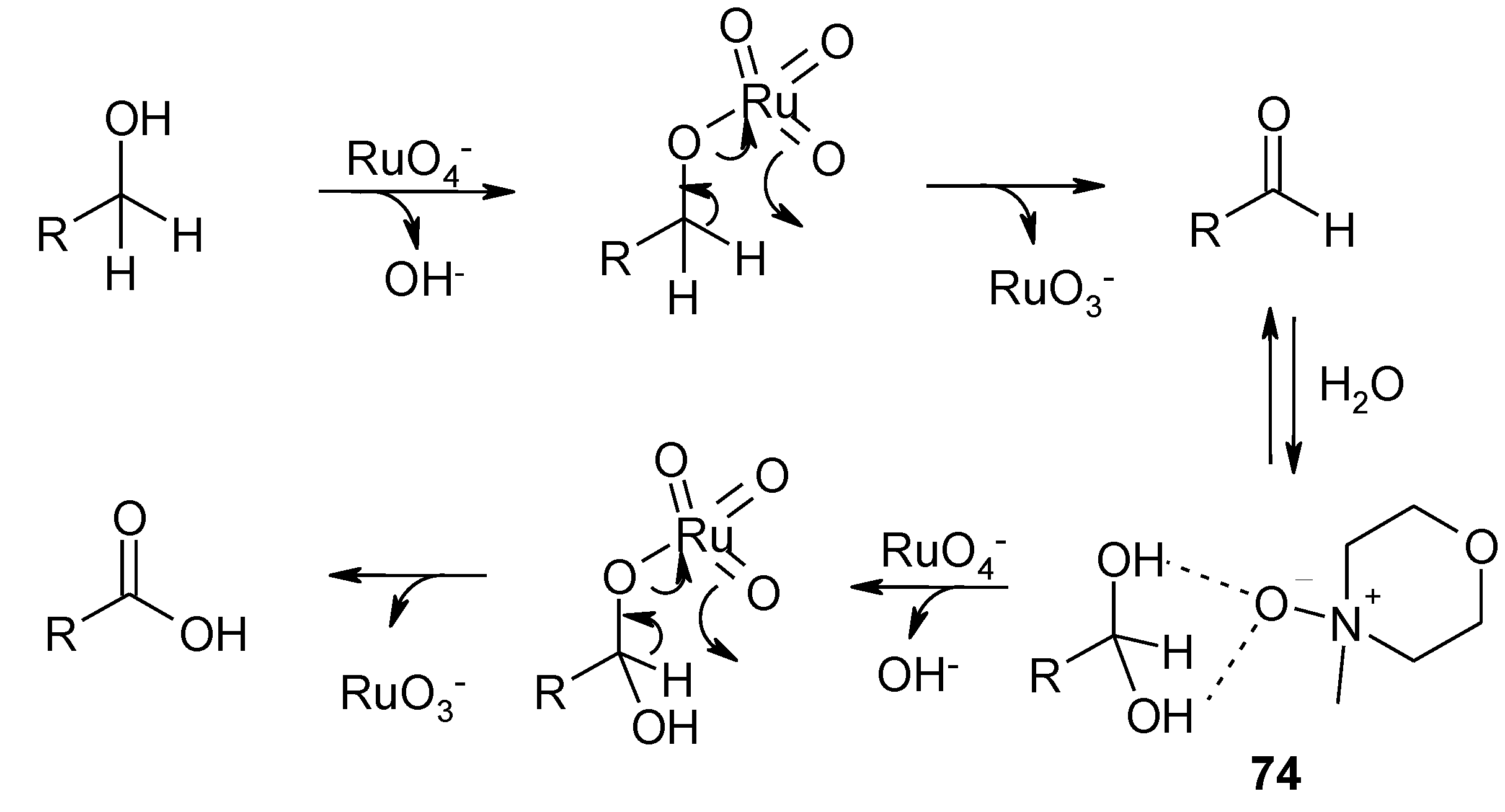
3.1. An Overview of the Previous Work
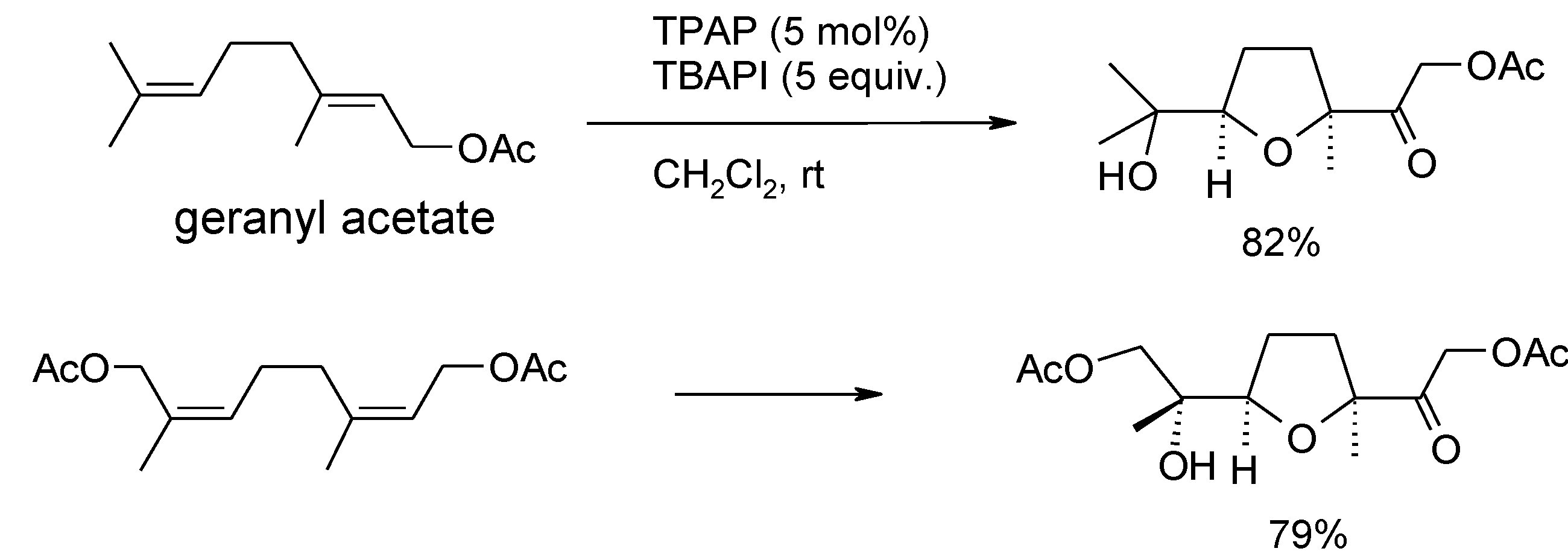
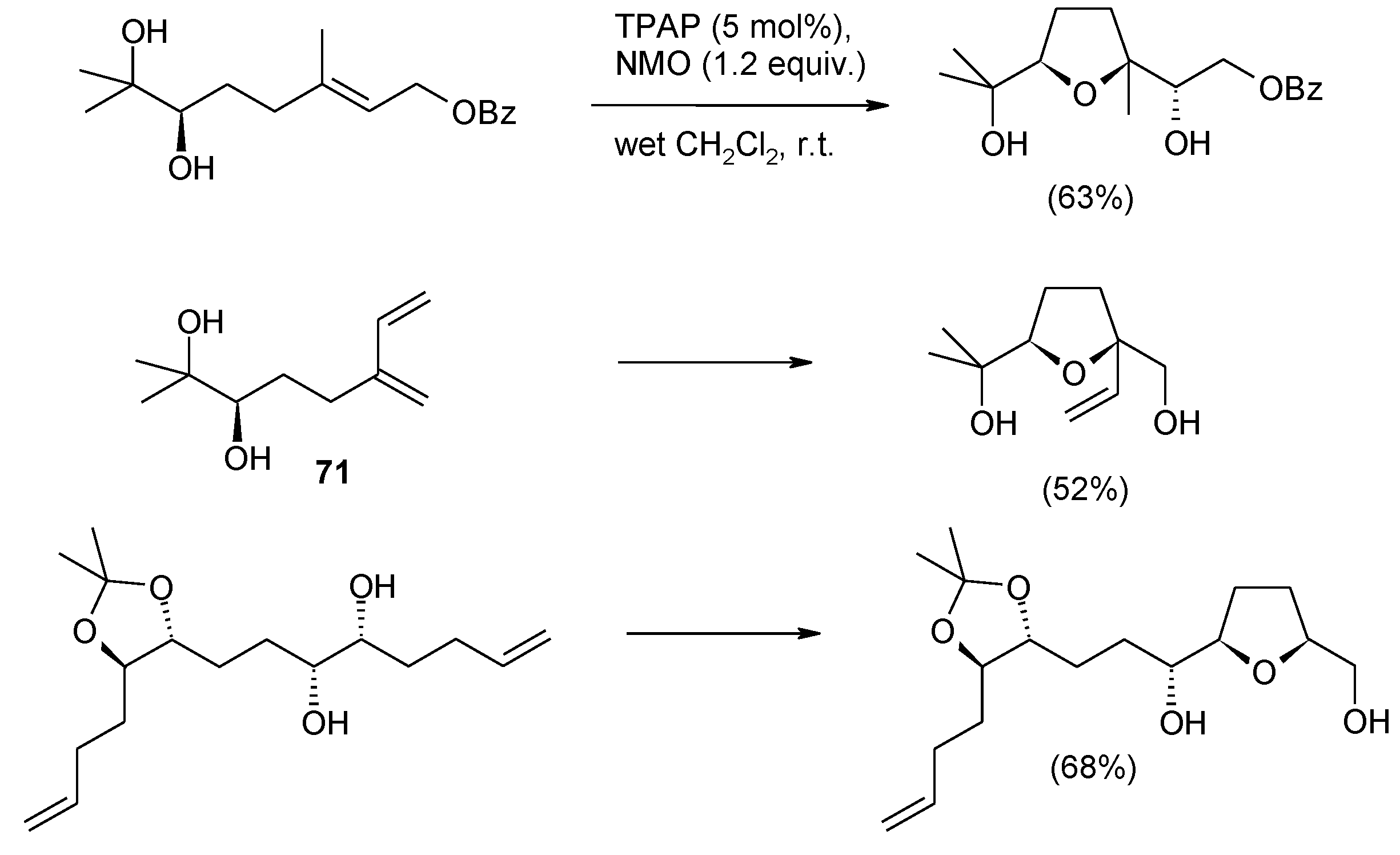
3.2. Formation of THF-Diols from 5,6-Dihydroxyalkenes




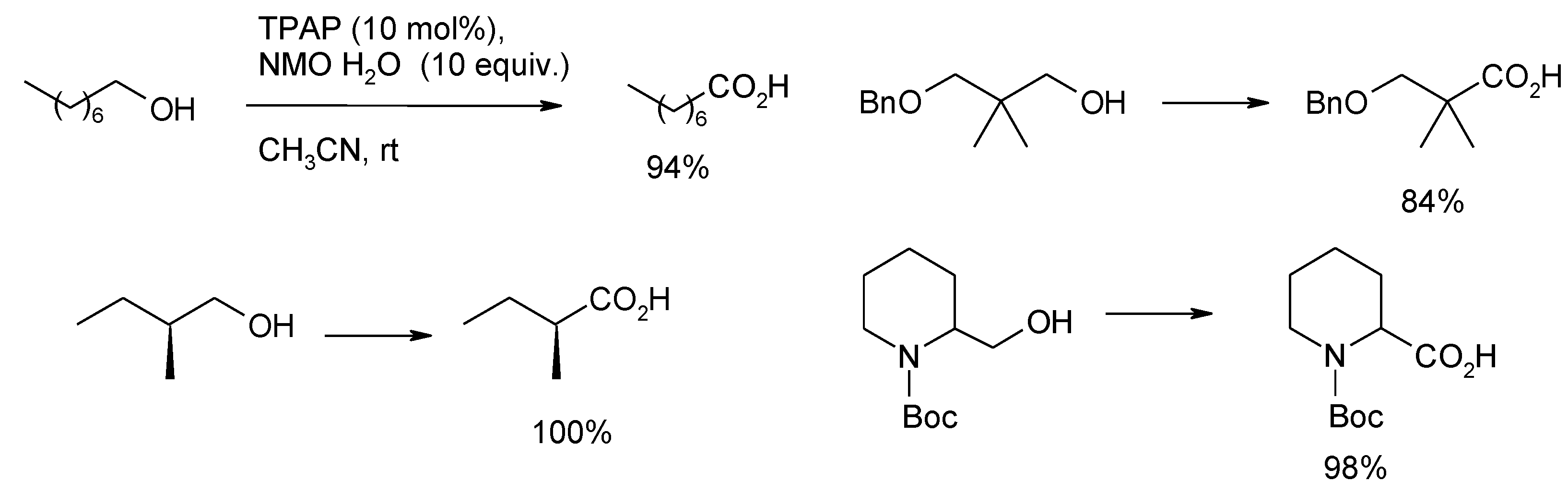
3.3. Other TPAP-Catalyzed Oxidations





4. Conclusions
Conflicts of Interest
References and Notes
- Schroder, M. Osmium tetraoxide cis hydroxylation of unsaturated substrates. Chem. Rev. 1980, 80, 187–213. [Google Scholar] [CrossRef]
- Muñiz, K. Imido-osmium(VIII) compounds in organic synthesis: Aminohydroxylation and diamination reactions. Chem. Soc. Rev. 2004, 33, 166–174. [Google Scholar] [CrossRef]
- Dash, S.; Patel, S.; Mishra, B.K. Oxidation by permanganate: synthetic and mechanistic aspects. Tetrahedron 2009, 65, 707–739. [Google Scholar] [CrossRef]
- Singh, N.; Lee, D.G. Permanganate: A green and versatile industrial oxidant. Org. Process Res. Dev. 2001, 5, 599–603. [Google Scholar] [CrossRef]
- Fatiadi, A.J. The classical permanganate ion: still a novel oxidant in organic chemistry. Synthesis 1987, 85–127. [Google Scholar] [CrossRef]
- Kühn, F.E.; Scherbaum, A.; Herrmann, W.A. Methyltrioxorhenium and its applications in olefin oxidation, metathesis and aldehyde olefination. J. Organomet. Chem. 2004, 689, 4149–4164. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Rost, A.M.J.; Mitterpleininger, J.K.M.; Szesni, N.; Sturm, S.; Fischer, R.W.; Kühn, F.E. A cheap, efficient, and environmentally benign synthesis of the versatile catalyst methyltrioxorhenium (MTO). Angew. Chem. Int. Ed. 2007, 46, 7301–7303. [Google Scholar] [CrossRef]
- Piancatelli, G.; Scettri, A.; D’Auria, M. Pyridinium chlorochromate: A versatile oxidant in organic synthesis. Synthesis 1982, 245–258. [Google Scholar]
- Mijs, W.J.; De Jonge, C.R.H.I. (Eds.) Organic Syntheses by Oxidation with Metal Compounds; Plenum Press: New York, NY, USA, 1986.
- Baeckvall, J.-E. (Ed.) Modern Oxidation Methods, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2010.
- Lee, D.G.; van den Hengh, M. The oxidation of organic compounds by RuO4. In Oxidation in Organic Chemistry; Trahanovsky, W.S., Ed.; Academic Press: New York, NY, USA, 1973; Volume 5, pp. 177–227. [Google Scholar]
- Haines, A.H. (Ed.) Methods for the Oxidation of Organic Compounds: alkanes, alkenes, alkynes and arenes; Academic Press: London, UK, 1985.
- Haines, A.H. (Ed.) Methods for the Oxidation of Organic Compounds: alcohols, alcohol derivatives, alkyl halides, nitroalkanes, alkyl azides, carbonyl compounds, hydroxyarenes and aminoarenes; Academic Press: London, UK, 1988.
- Courtney, J.L. Ruthenium tetroxide oxidations. In Organic Syntheses by Oxidation with Metal Compounds; Mijs, W.J., De Jonge, C.R.H.I., Eds.; Plenum Press: New York, NY, USA, 1986; pp. 445–467. [Google Scholar]
- Murahashi, S.-I.; Komiya, N. Ruthenium-catalyzed oxidation for organic synthesis. In Modern Oxidation Methods, 2nd ed.; Baeckvall, J.-E., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 241–275. [Google Scholar]
- Djerassi, C.; Engle, R.R. Oxidations with ruthenium tetroxide. J. Am. Chem. Soc. 1953, 75, 3838–3840. [Google Scholar] [CrossRef]
- Berkowitz, L.M.; Rylander, P.N. Use of ruthenium tetroxide as a multi-purpose oxidant. J. Am. Chem. Soc. 1958, 80, 6682–6684. [Google Scholar] [CrossRef]
- Wolfe, S.; Hasan, S.K.; Campbell, J.R. Ruthenium trichloride-catalyzed hypochlorite oxidation of organic compounds. Chem. Commun. 1970, 1420–1421, and references therein. [Google Scholar] [CrossRef]
- Charlsen, P.H.J.; Katsuki, T.; Martin, V.S.; Sharpless, K.B. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds. J. Org. Chem. 1981, 46, 3936–3938. [Google Scholar] [CrossRef]
- Rossiter, B.E.; Katsuki, T.; Sharpless, K.B. Asymmetric epoxidation provides shortest routes to four chiral epoxy alcohols which are key intermediates in syntheses of methymycin, erythromycin, leukotriene C-1, and disparlure. J. Am. Chem. Soc. 1981, 103, 464–465. [Google Scholar] [CrossRef]
- Kasai, M.; Ziffer, H. On the absolute stereochemistries of (−)-benzocyclohepten-3-ol, (−)-2,3,4,5-tetrahydro-l-benzoxepin-5-ol, and (−)-benzocycloocten-3-ol. J. Org. Chem. 1983, 48, 712–715. [Google Scholar] [CrossRef]
- Kasai, M.; Ziffer, H. Ruthenium tetroxide catalyzed oxidations of aromatic and heteroaromatic rings. J. Org. Chem. 1983, 48, 2346–2349. [Google Scholar] [CrossRef]
- Schuda, P.F.; Cichowicz, M.B.; Heimann, M.R. A facile method for the oxidative removal of benzyl ethers: the oxidation of benzyl ethers to benzoates by ruthenium tetraoxide. Tetrahedron Lett. 1983, 24, 3829–3830. [Google Scholar] [CrossRef]
- Chong, J.M.; Sharpless, K.B. Nucleophilic openings of 2,3-epoxy acids and amides mediated by Ti(O-i-Pr)4. Reliable C-3 selectivity. J. Org. Chem. 1985, 50, 1560–1563. [Google Scholar] [CrossRef]
- Webster, F.X.; Rivas-Enterrios, J.; Silverstein, R.M. Synthesis of diacids and keto acids by ruthenium tetraoxide catalyzed oxidation of cyclic allylic alcohols and α,β-unsaturated ketones. J. Org. Chem. 1987, 52, 689–691. [Google Scholar] [CrossRef]
- Adam, G.; Zibuck, R.; Seebach, D. Total synthesis of (+)-gloeosporone: assignment of absolute configuration. J. Am. Chem. Soc. 1987, 109, 6176–6177. [Google Scholar] [CrossRef]
- Martin, V.S.; Nuñez, M.T.; Tonn, C.E. Easy and general method to synthesize chiral 2-hydroxyacid benzoates. Tetrahedron Lett. 1988, 29, 2701–2702. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kusakabe, M.; Kitano, Y.; Sato, F. Preparation of optically active 2-furylcarbinols by kinetic resolution using the Sharpless reagent. J. Org. Chem. 1988, 53, 1586–1587. [Google Scholar] [CrossRef]
- Zibuck, R.; Seebach, D. The preparation of 1,2-diketones from acetylenes. Helv. Chim. Acta 1988, 71, 237–240. [Google Scholar] [CrossRef]
- Gao, Y.; Sharpless, K.B. Vicinal diol cyclic sulfates: like epoxides only more reactive. J. Am. Chem. Soc. 1988, 110, 7538–7538. [Google Scholar] [CrossRef]
- Caron, M.; Carlier, P.R.; Sharpless, K.B. Regioselective azide opening of 2,3-epoxy alcohols by [Ti(O-i-Pr)2(N3)2]: synthesis of α-amino acids. J. Org. Chem. 1988, 53, 5185–5187. [Google Scholar] [CrossRef]
- Nuñez, M.T.; Martin, V.S. Efficient oxidation of phenyl groups to carboxylic acids with ruthenium tetraoxide. A simple synthesis of (R)-γ-caprolactone, the pheromone of Trogoderma granarium. J. Org. Chem. 1990, 55, 1928–1932. [Google Scholar] [CrossRef]
- Baumer, U.-S.; Schäfer, H.J. Cleavage of olefinic double bonds by mediated anodic oxidation. Electrochim. Acta 2003, 48, 489–495. [Google Scholar] [CrossRef]
- Cornely, J.; Su Ham, L.M.; Meade, D.E.; Dragojlovic, V. Dimethyl carbonate-water: An environmentally friendly solvent system for ruthenium tetraoxide oxidations. Green Chem. 2003, 5, 34–37. [Google Scholar]
- Zimmermann, F.; Meux, E.; Mieloszynski, J.-L.; Lecuire, J.-M.; Oget, N. Ruthenium catalysed oxidation without CCl4 of oleic acid, other monoenic fatty acids and alkenes. Tetrahedron Lett. 2005, 46, 3201–3203. [Google Scholar] [CrossRef]
- Rup, S.; Zimmermann, F.; Meux, E.; Schneider, M.; Sindt, M.; Oget, N. The ultrasound-assisted oxidative scission of monoenic fatty acids by ruthenium tetroxide catalysis: Influence of the mixture of solvents. Ultrason. Sonochem. 2009, 16, 266–272. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, C. Ruthenium-catalyzed oxidative cleavage of olefins to aldehydes. J. Org. Chem. 2001, 66, 4814–4818. [Google Scholar] [CrossRef]
- Gopal, H.; Gordon, A.J. Ruthenium tetroxide oxidation of alkynes. A new one step synthesis of α-diketones. Tetrahedron Lett. 1971, 12, 2941–2944. [Google Scholar] [CrossRef]
- Mukai, C.; Miyakawa, M.; Hanaoka, M. A highly stereoselective synthesis of the N-terminal aminoacid analogue of nikkomycin B and Bx. Synlett 1994, 165–166. [Google Scholar] [CrossRef]
- Griffith, W.P.; Shoair, A.G.; Suriaatmaja, M. Ruthenium-catalysed cleavage of alkenes and alkynes to carboxylic acids. Synth. Commun. 2000, 30, 3091–3095. [Google Scholar] [CrossRef]
- Yang, D.; Chen, F.; Dong, Z.-M.; Zhang, D.-W. Ruthenium-catalyzed oxidative cleavage of alkynes to carboxylic acids. J. Org. Chem. 2004, 69, 2221–2223. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Akashi, K. Osmium-catalyzed vicinal hydroxylation of olefins by tert-butyl hydroperoxide under alkaline conditions. J. Am. Chem. Soc. 1976, 98, 1986–1987. [Google Scholar] [CrossRef]
- Piccialli, V.; Smaldone, D.M.A.; Sica, D. Studies towards the synthesis of polyoxygenated steroids. Reaction of some tri- and tetrasubstituted monoene steroids with RuO4. Tetrahedron 1993, 49, 4211–4228. [Google Scholar] [CrossRef]
- Albarella, L.; Piccialli, V.; Smaldone, S.; Sica, D. Ruthenium tetroxide oxidation of alkenes. Part 7: A more complete picture. J. Chem. Res. (S) 1996, 400–401. [Google Scholar]
- Notaro, G.; Piccialli, V.; Sica, D.; Smaldone, D. Studies towards the synthesis of polyoxygenated steroids. Reaction of 7,9(11)-diene steroids with RuO4. Tetrahedron 1994, 50, 4835–4852. [Google Scholar] [CrossRef]
- Piccialli, V.; Sica, D.; Smaldone, D. Reaction of 7-dehydrocholesteryl acetate with RuO4. First isolation of a cyclic ruthenium (VI) diester. Tetrahedron Lett. 1994, 35, 7093–7096. [Google Scholar] [CrossRef]
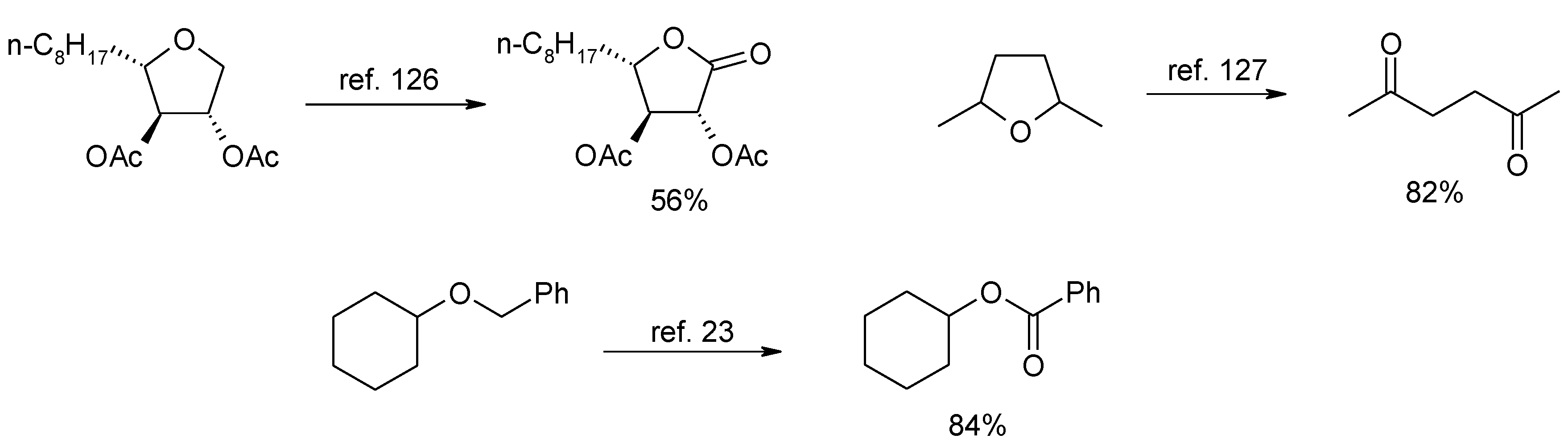
- Shing, T.K.M.; Tai, V.W.-F.; Tam, E.K.W. Practical and rapid vicinal hydroxylation of alkenes by catalytic ruthenium tetroxide. Angew. Chem. Int. Ed. 1994, 33, 2312–2313. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Tam, E.K.W.; Tai, V.W.-F.; Chung, I.H.F.; Jiang, Q. Ruthenium-catalyzed cis-dihydroxylation of alkenes: scope and limitations. Chem. Eur. J. 1996, 2, 50–57, These authors erroneously reported (see Scheme 2 in this publication) that the best yields of diols obtained in our investigation (see Piccialli et al., ref. 43) was 32% and was relevant to the oxidation of cholesteryl acetate. Actually, we obtained yields of the diol products in the range 55%–80% for the tested steroids embodying trisubstituted double bonds by conducting the process in carbon tetrachloride. In most of the substrates we tested, the yields are in line with those later reported by these authors for the dihydroxylation of cyclohexene derivatives embodying trisubstituted bouble bonds. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Tam, E.K.W. Solvent effect on ruthenium catalyzed dihydroxylation. Tetrahedron Lett. 1999, 40, 2179–2180. [Google Scholar] [CrossRef]
- Plietker, B.; Niggemann, M. An improved protocol for the RuO4-catalyzed dihydroxylation of olefins. Org. Lett. 2003, 5, 3353–3356. [Google Scholar] [CrossRef]
- Plietker, B.; Niggemann, M. An improved protocol for the RuCl3/CeCl3/NaIO4: A new bimetallic oxidation system for the mild and efficient dihydroxylation of unreactive olefins. J. Org. Chem. 2005, 70, 2402–2405. [Google Scholar] [CrossRef]
- Piccialli, V.; Cavallo, N. Improved RuO4-catalyzed oxidative cyclization of geraniol-type 1,5-dienes to cis-2,5-bis(hydroxymethyl)-tetrahydrofuranyldiols. Tetrahedron Lett. 2001, 42, 4695–4699. [Google Scholar] [CrossRef]
- Albarella, L.; Musumeci, D.; Sica, D. Reaction of RuO4 with carbon-carbon double bonds, part 9. Reaction of 1,5-dienes with ruthenium tetroxide: Stereoselective synthesis of tetrahydrofurandiols. Eur. J. Org. Chem. 2001, 997–1003. [Google Scholar] [CrossRef]
- Piccialli, V. RuO4-catalysed oxidative cyclization of 1,6-dienes to trans-2,6-bis(hydroxymethyl)-tetrahydropyrans. A novel stereoselective process. Tetrahedron Lett. 2000, 48, 3731–3733. [Google Scholar] [CrossRef]
- Piccialli, V.; Borbone, N.; Oliviero, G. Ruthenium-catalyzed oxidative cyclization of 1,7-dienes. A novel diasteroselective synthesis of 2,7-disubstitueted trans-oxepane diols. Tetrahedron Lett. 2007, 48, 5131–5135. [Google Scholar] [CrossRef]
- Centore, R.; Piccialli, V.; Tuzi, A. Rac-2,7-bis(2-hydroxy-2-propyl)-trans-oxepane. Acta Cryst. 2007, E63, o2907–o2908. [Google Scholar]
- Bifulco, G.; Caserta, T.; Gomez-Paloma, G.; Piccialli, V. RuO4-promoted syn-oxidative polycyclization of isoprenoid polyenes: a new stereoselective cascade process. Tetrahedron Lett. 2002, 43, 9265–9269. [Google Scholar] [CrossRef]
- Bifulco, G.; Caserta, T.; Gomez-Paloma, L.; Piccialli, V. RuO4-promoted oxidative polycyclization of isoprenoid polyenes. A further insight into the stereochemistry of the process. Tetrahedron Lett. 2003, 44, 5499–5503. [Google Scholar] [CrossRef]
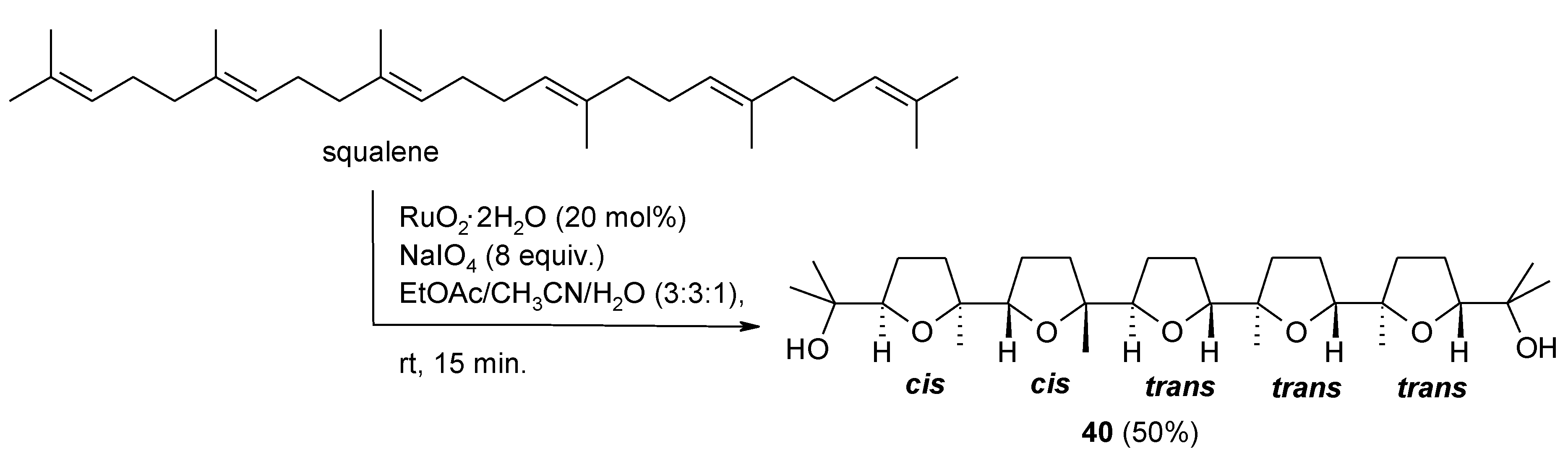
- Caserta, T.; Piccialli, V.; Gomez-Paloma, L.; Bifulco, G. RuO4-catalyzed oxidative cyclization of squalene. Determination of the configuration of the penta-tetrahydrofuranyl diol product. Tetrahedron 2005, 61, 927–939. [Google Scholar] [CrossRef]
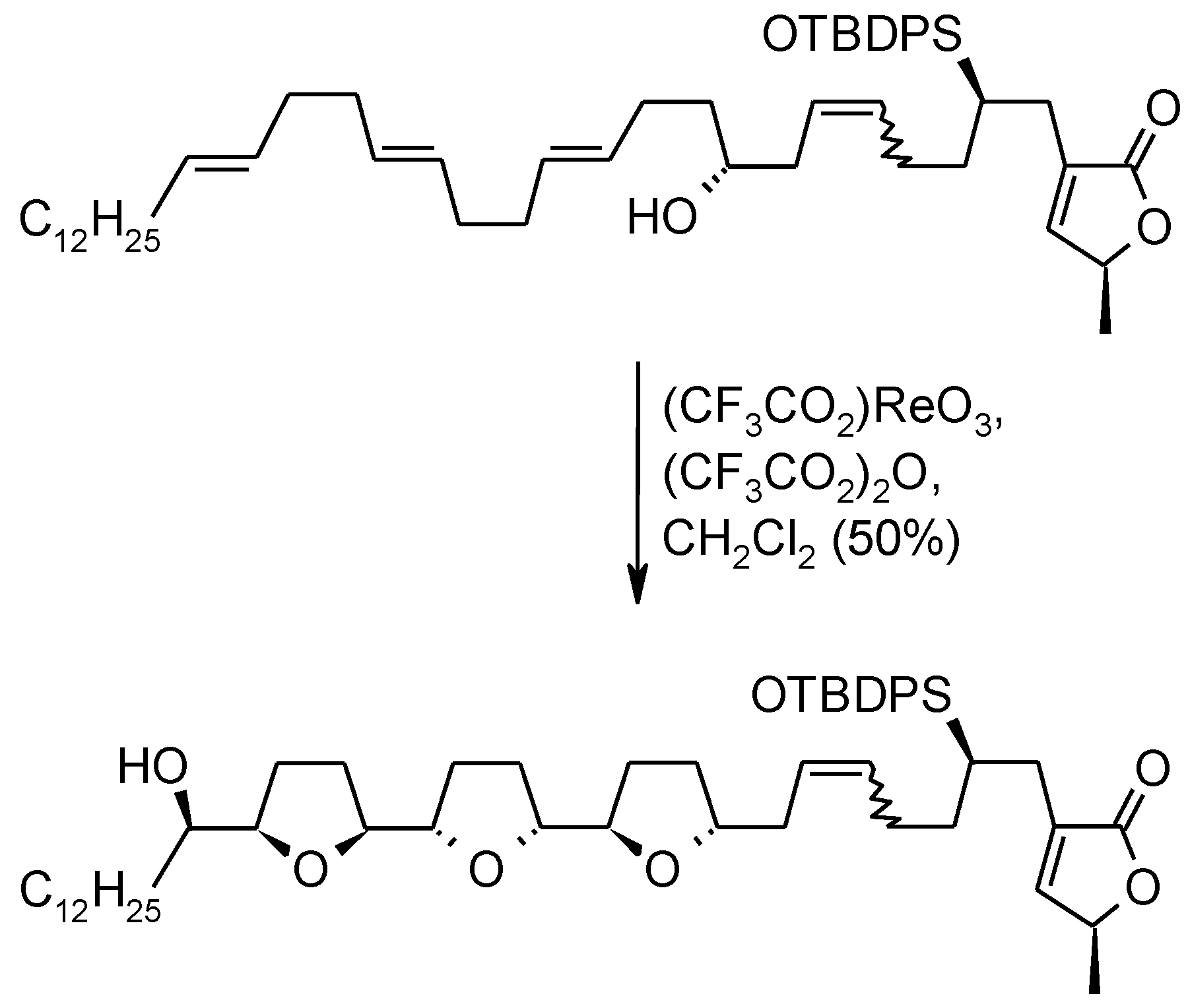
- Piccialli, V.; Caserta, C.; Caruso, L.; Gomez-Paloma, L.; Bifulco, G. RuO4-mediated oxidative polycyclization of linear polyenes. A new approach to the synthesis of the bis-THF diol core of antitumour cis-cis adjacent bis-THF annonaceous acetogenins. Tetrahedron 2006, 62, 10989–11007. [Google Scholar] [CrossRef]
- Roth, S.; Göhler, S.; Cheng, H.; Stark, C.B.W. A highly efficient procedure for ruthenium tetroxide catalyzed oxidative cyclzation of 1,5-dienes. Eur. J. Org. Chem. 2005, 4109–4118. [Google Scholar]
- Göhler, S.; Roth, S.; Cheng, H.; Göksel, H.; Rupp, A.; Haustedt, L.O.; Stark, C.B.W. Multigram synthesis of diastereomerically pure tetrahydrofuran-diols. Synthesis 2007, 17, 2751–2754. [Google Scholar]
- Roth, S.; Stark, C.B.W. Efficient oxidative cyclization of 1,6-dienes: A highly diastereoselective entry to substituted tetrahydropyrans. Angew. Chem. Int. Ed. 2006, 45, 6218–6221. [Google Scholar] [CrossRef]
- Plietker, B. RuO4-catalyzed ketohydroxylation of olefins. J. Org. Chem. 2003, 68, 7123–7125. [Google Scholar] [CrossRef]
- Plietker, B. RuO4-catalyzed ketohydroxylation. Part 1. Development, scope, and limitation. J. Org. Chem. 2004, 69, 8287–8296. [Google Scholar] [CrossRef]
- Plietker, B. The RuO4-catalyzed ketohydroxylation, Part II: A regio-, chemo- and stereoselectivity study. Eur. J. Org. Chem. 2005, 1919–1929. [Google Scholar] [CrossRef]
- Plietker, B. New oxidative pathways for the synthesis of α-hydroxyketones, the α-hydroxylation and ketohydroxylation. Tetrahedron 2005, 16, 3453–3459. [Google Scholar] [CrossRef]
- Plietker, B. Alkenes as ketol surrogates-A new approach toward enantiopure acyloins. Org. Lett. 2003, 6, 289–291. [Google Scholar] [CrossRef]
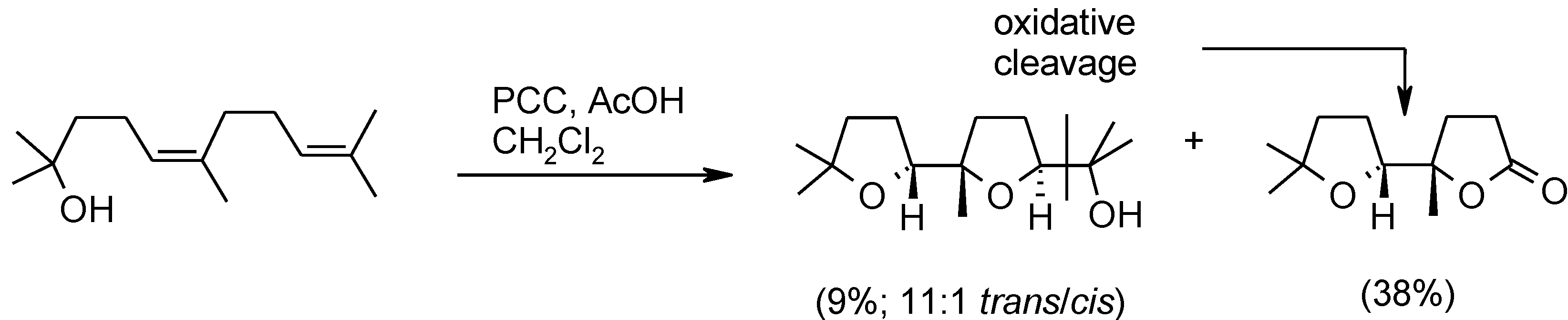
- Albarella, L.; Giordano, F.; Lasalvia, M.; Piccialli, V.; Sica, D. Evidence for the existence of a cyclic ruthenium(VI) diester as an intermediate in the oxidative scission of (−)-α-pinene with RuO4. Tetrahedron Lett. 1995, 36, 5267–5270. [Google Scholar]
- Plietker, B.; Niggemann, M. The RuO4-catalysed dihydroxylation, ketohydroxylation and mono-oxidation- novel oxidation reactions for the synthesis of diols and α-hydroxy ketones. Org. Biomol. Chem. 2004, 2, 2403–2407. [Google Scholar] [CrossRef]
- Plietker, B. Selectivity versus reactivity. Recent advances in RuO4-catalyzed oxidations. Synthesis 2005, 15, 2453–2472. [Google Scholar] [CrossRef]
- Piccialli, V. Oxidative cyclization of dienes and polyenes mediated by transition-metal-oxo species. Synthesis 2007, 17, 2585–2607. [Google Scholar] [CrossRef]
- Lee, A.W.M.; Chan, W.H.; Yuen, W.H.; Xia, P.F.; Wong, W.Y. Ruthenium catalyzed asymmetric dihydroxylation with sultams as chiral auxiliaries. Tetrahedron: Asymmetry 1999, 10, 1421–1424. [Google Scholar] [CrossRef]
- Neisius, N.M.; Plietker, B. Diastereoselcetive Ru-catalyzed cross-methatesis-dihydroxylation sequence. An efficient approach toward enantiomerically enriched syn-diols. J. Org. Chem. 2008, 73, 3218–3227. [Google Scholar] [CrossRef]
- Pagliaro, M.; Campestrini, S.; Criminna, R. Ru-based oxidation catalysis. Chem. Soc. Rev. 2005, 34, 837–845. [Google Scholar] [CrossRef]
- Zhou, M.; Crabtree, R.H. C-H oxidation by platinum group metal oxo or peroxo species. Chem. Soc. Rev. 2011, 40, 1875–1884. [Google Scholar] [CrossRef]
- Bataille, J.R.; Donohoe, T.J. Osmium-free direct syn-dihydroxylation of alkenes. Chem. Soc. Rev. 2011, 40, 114–128. [Google Scholar] [CrossRef]
- Sperry, S. The oxidation of amides to imides: A powerful synthetic transformation. Synthesis 2011, 22, 3569–3580. [Google Scholar] [CrossRef]
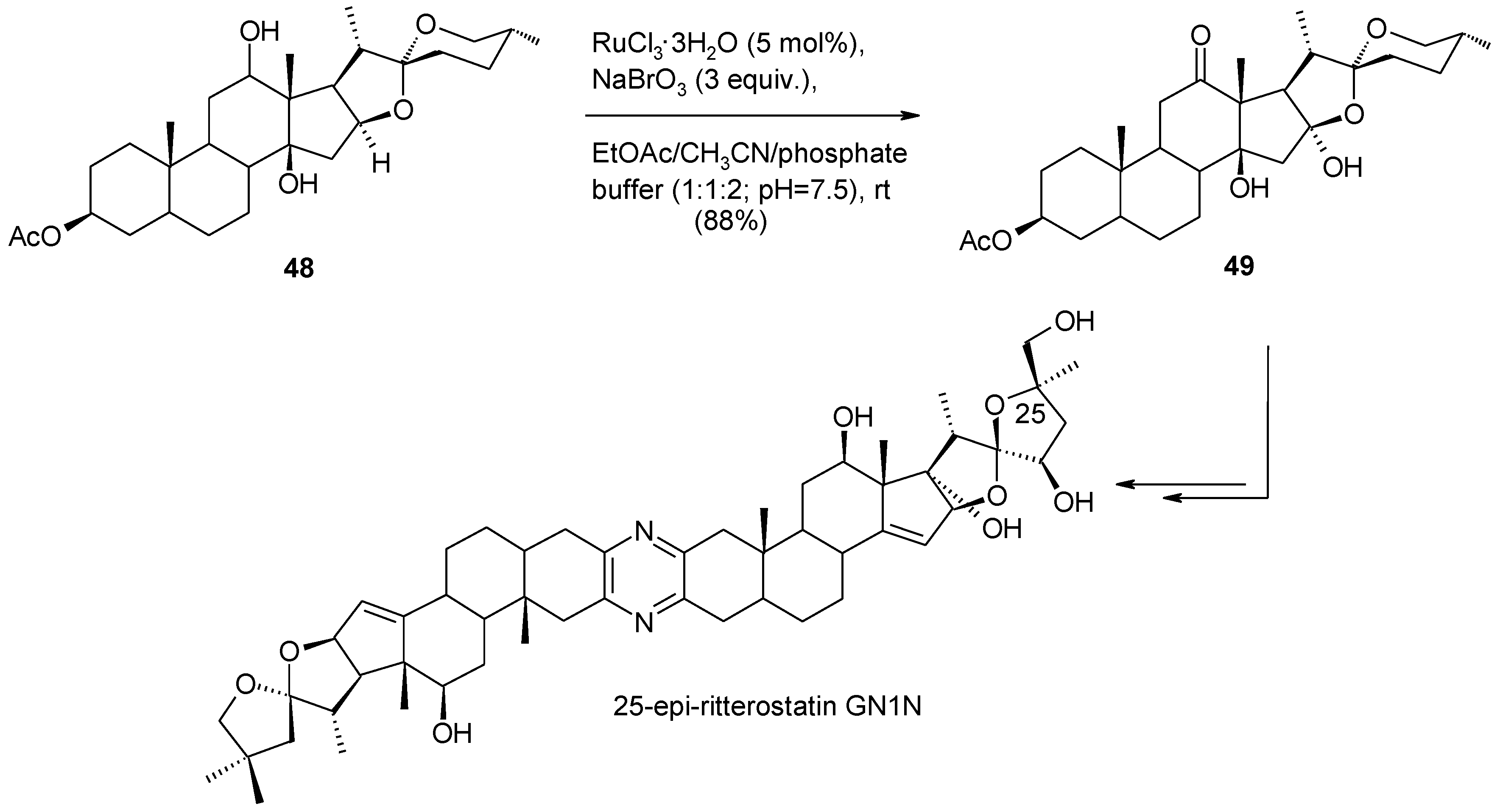
- Piccialli, V.; Zaccaria, S.; Borbone, N.; Oliviero, G.; D’Errico, S.; Hemminki, A.; Cerullo, V.; Romano, V.; Tuzi, A.; Centore, R. Discovery of a novel one-step RuO4-catalysed tandem oxidative polycyclization/double spiroketalization process. Access to a new type of polyether bis-spiroketal compound displaying antitumour activity. Tetrahedron 2010, 66, 9370–9378. [Google Scholar]
- Beligny, S.; Eibauer, S.; Maechling, S.; Blechert, S. Sequential catalysis: A metathesis/dihydroxylation sequence. Angew. Chem. Int. Ed. 2006, 45, 1900–1903. [Google Scholar] [CrossRef]
- Sholte, A.A.; An, M.H.; Snapper, M.L. Ruthenium-catalyzed tandem olefin methatesis-oxidations. Org. Lett. 2006, 8, 4759–4762. [Google Scholar] [CrossRef]
- Malik, M.; Witkowsky, G.; Ceborska, M.; Jarosz, S. Synthesis of polyhydroxylated quinolizidines and azaspiro[4.5]decanes from D-xylose. Org. Lett. 2013, 15, 6214–6217. [Google Scholar] [CrossRef]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
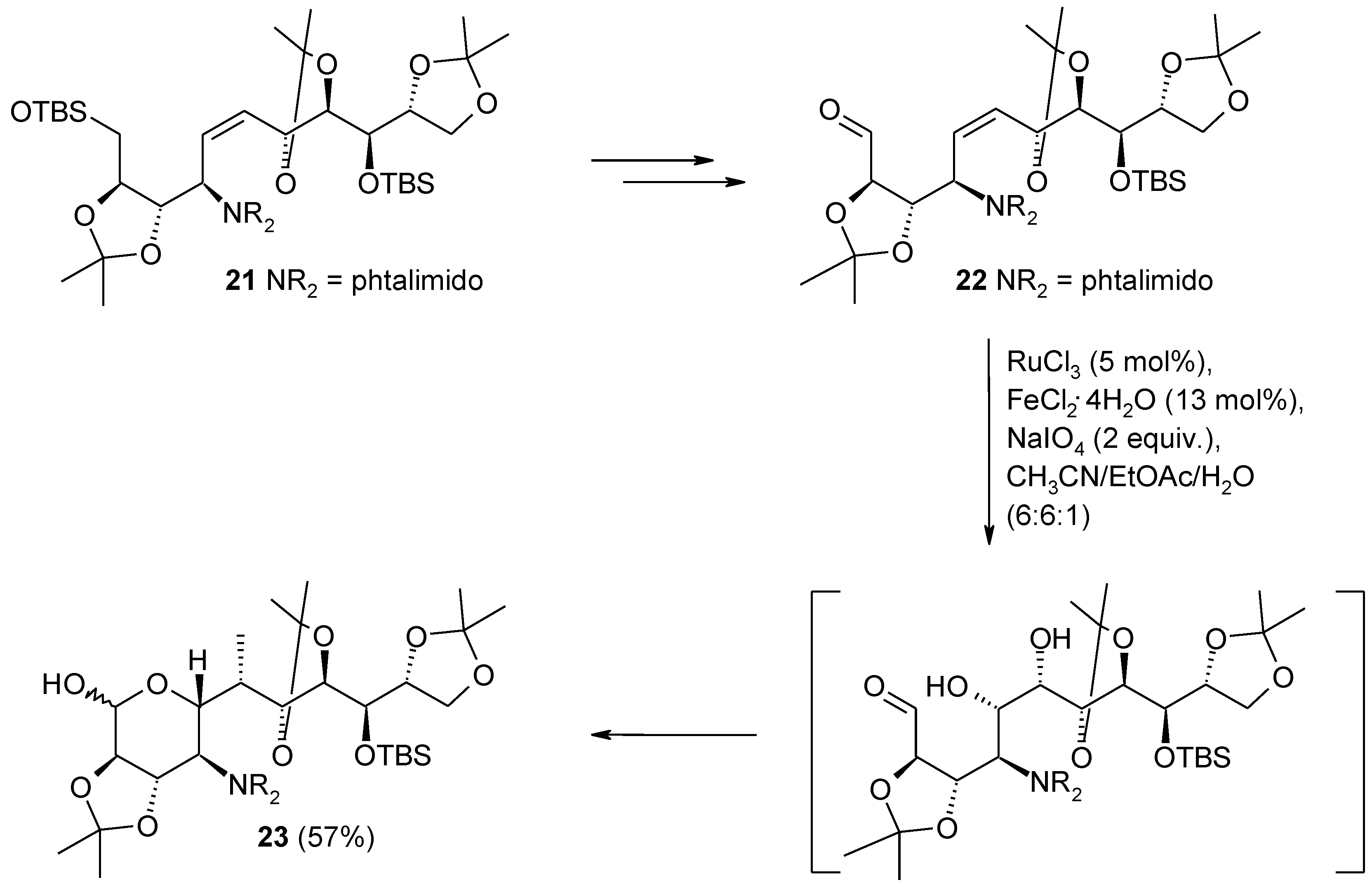
- Niggemann, M.; Jelonek, A.; Biber, N.; Wuchrer, M.; Plietker, B. A general, iterative, and modular approach toward carbohydrate libraries based on ruthenium-catalyzed oxidative cyclization. J. Org. Chem. 2008, 73, 7028–7036. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Mizuno, N. Scope, kinetics, and mechanistic aspects of aerobic oxidations catalyzed by ruthenium supported on alumina. Chem. Eur. J. 2003, 9, 4353–4361. [Google Scholar]
- Tiwari, P.; Misra, A.K. An efficient stereoselective dihydroxylation of glicals using a bimetallic system, RuCl3/CeCl3/NaIO4. J. Org. Chem. 2006, 71, 2911–2913. [Google Scholar] [CrossRef]
- John Pal, A.P.; Gupta, P.; Reddy, Y.S.; Vankar, Y.D. Synthesis of fused oxa-aza spiro sugars from D-glucose-derived δ-lactone as glycosidase inhibitors. Eur. J. Org. Chem. 2010, 6957–6966. [Google Scholar]
- Takamura, H.; Tsuda, K.; Kawakubo, Y.; Kadota, I.; Uemura, D. Stereoselective synthesis of the C94-C104 fragment of symbiodinolide. Tetrahedron Lett. 2012, 53, 4317–4319. [Google Scholar] [CrossRef]
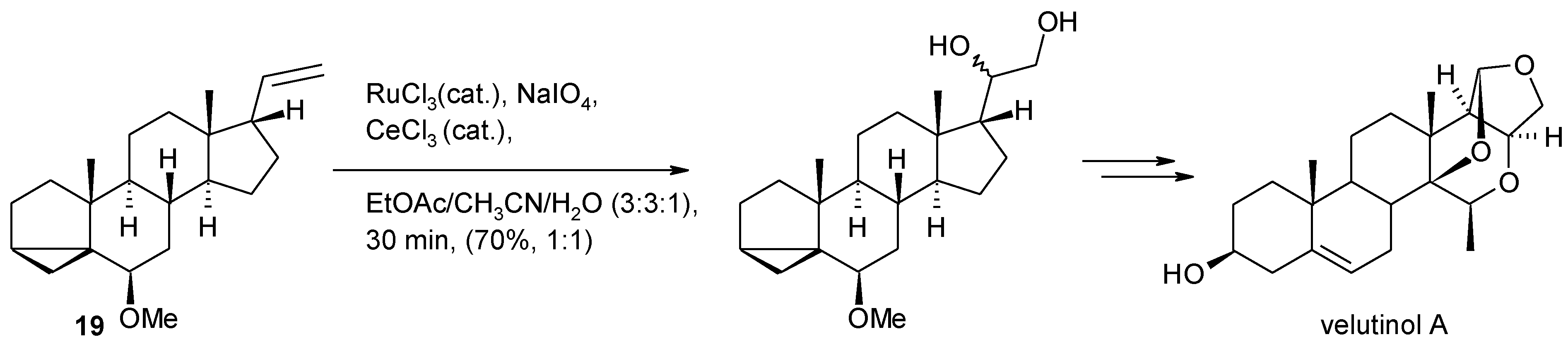
- Isaka, N.; Tamyia, M.; Hasegawa, A.; Ishiguro, M. A concise total synthesis of the non-peptide bradykidin B1 receptor antagonist velutinol A. Eur. J. Org. Chem. 2012, 665–668. [Google Scholar]
- Adachi, S.; Watanabe, K.; Iwata, Y.; Kameda, S.; Miyaoka, Y.; Onozuka, M.; Mitsui, R.; Saikawa, Y.; Nakata, M. Total syntheses of lactonamycin and lactonamycin Z with late-stage A-ring formation and glycosylation. Angew. Chem. Int. Ed. 2013, 52, 2087–2091. [Google Scholar] [CrossRef]
- Furstner, A.; Wuchrer, M. Concise approach to the “higher sugar” core of the nucleoside antibiotic hikizimycin. Chem. Eur. J. Chem. 2006, 12, 76–89. [Google Scholar] [CrossRef]
- Tao, Z.L.; Zhang, W.-Q.; Chen, D.-F.; Adele, A.; Gong, L.-Z. Pd-catalyzed asymmetric allylic alkylation of pyrazol-5-ones with allylic alcohols: the role of the chiral phosphoric acid in C-O bond cleavage and stereocontrol. J. Am. Chem. Soc. 2013, 135, 9255–9258. [Google Scholar] [CrossRef]
- Kawamoto, H.; Ohmori, Y.; Maekawa, M.; Shimada, M.; Mano, N.; Iida, T. An efficient synthesis of 4α- and 4β-hydroxy-7-dehydrocholesterol, biomarkers for patients with and animal models of the Smith-Lemli-Opitz syndrome. Chem. Phys. Lipids 2013, 175–176, 73–78. [Google Scholar] [CrossRef]
- Lee, D.G.; Spitzer, U.A. The Oxidation of methyl cinnamate by ruthenium tetroxide. J. Org. Chem. 1976, 41, 3644. [Google Scholar]
- Albarella, L.; Lasalvia, M.; Piccialli, V.; Sica, D. Reaction of RuO4 with carbon-carbon double bonds. Part 8. Reaction of 7-dehydrocholesteryl acetate and cholesteryl acetate with RuO4 and OsO4. A comparative view. J. Chem. Soc. Perkin Trans. 1998, 2, 737–743. [Google Scholar]
- Strassner, T.; Drees, M. Rutheniumtetraoxide oxidation of alkenes—A density functional theory study. J. Mol. Struct. (Theochem) 2004, 197–204. [Google Scholar] [CrossRef]
- Frunzke, J.F.; Loschen, C.; Frenking, G. Why are olefins oxidized by RuO4 under cleavage of the carbon-carbon bond whereas oxidation by OsO4 yields cis-diols. J. Am. Chem. Soc. 2004, 126, 3642–3652. [Google Scholar] [CrossRef]
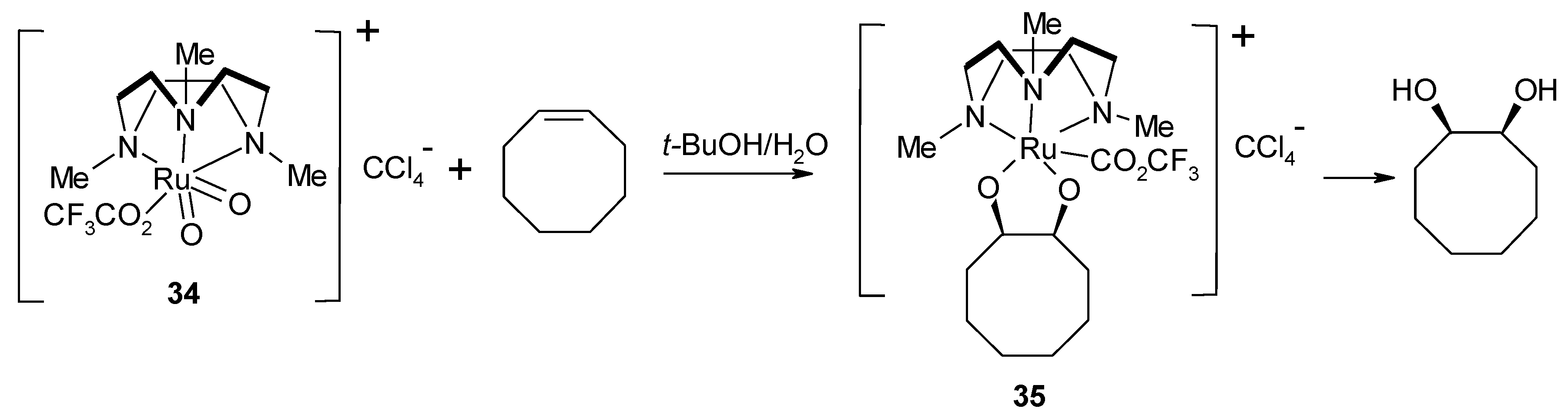
- Yip, W.-P.; Yu, W.-Y.; Zhu, N.; Che, C.-M. Alkene cis-dihydroxylation by [(Me3tacn)(CF3CO2)RuVIO2]ClO4 (Me3tacn=1,4,7-trimethyl-1,4,7-triazacyclononane): Structural characterization of [3 + 2] cycloadducts and kinetic studies. J. Am. Chem. Soc. 2005, 127, 14239–14249. [Google Scholar] [CrossRef]
- Cecil, A.R.L.; Brown, R.C.D. Stereoselective synthesis of cis-2,6-bis-hydroxyalkyl-tetrahydropyrans by permanganate promoted cyclisation of 1,6-dienes. Tetrahedron Lett. 2004, 45, 7269–7271. [Google Scholar] [CrossRef]
- Pilgrim, B.S.; Donohoe, T.J. Osmium-catalyzed oxidative cyclization of dienes and their derivatives. J. Org. Chem. 2013, 78, 2149–2167. [Google Scholar] [CrossRef]
- Di Dio, P.J.; Zahn, S.; Stark, C.B.W.; Kirchner, B. Understanding selectivities in ligand-free oxidative cyclizations of 1,5- and 1,6-dienes with RuO4 from density functional theory. Z. Naturforsch. 2010, 65B, 367–375. [Google Scholar]
- Göhler, S.; Stark, C.B.W. Catalytic diastereo- and positionselective mono-cyclization of 1,5,9-trienes and polyenes. Org. Biomol. Chem. 2007, 5, 1605–1614. [Google Scholar] [CrossRef]
- An efficient stirring and the complete dissolution of periodate are of prime importance to obtain high and reproducible yields in this process. We were able to obtain a 50% yield (5.5 g of the penta-THF 40) by performing the process on 21 mmol of squalene under mechanical stirring, in 15 min at rt (unpublished results).
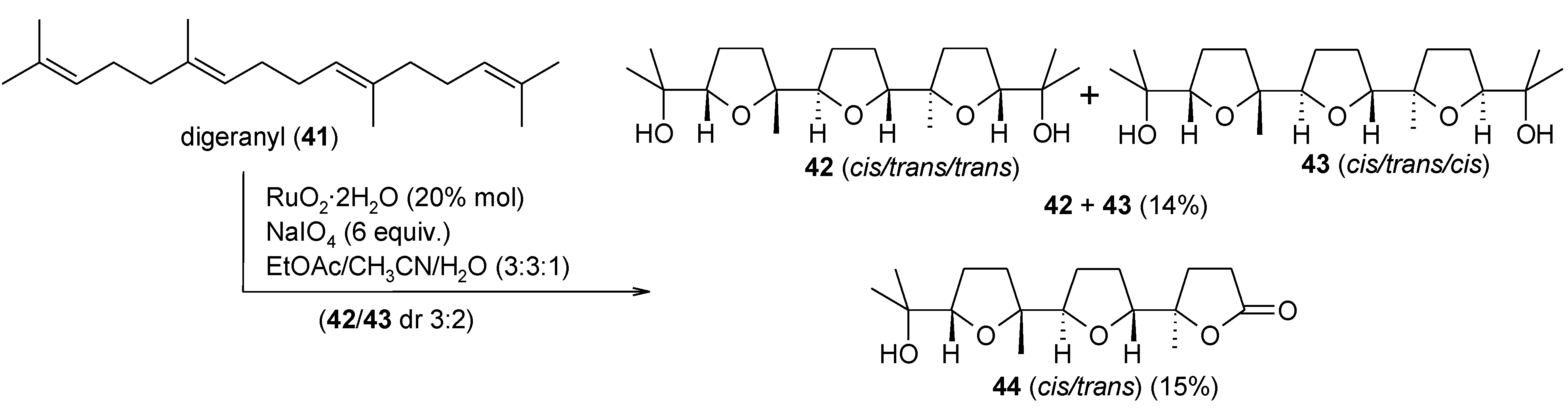
- Piccialli, V.; Borbone, N.; Oliviero, G. RuO4-catalyzed oxidative polycyclization of the CS-symmetric isoprenoid polyene digeranyl. An unexpected stereochemical outcome. Tetrahedron 2008, 64, 11185–11192. [Google Scholar] [CrossRef]
- Klein, E.; Rojahn, W. Die permanganatoxydation von 1,5-dienverbindungen. Tetrahedron 1965, 21, 2353–2358. [Google Scholar] [CrossRef]
- Poethig, A.; Strassner, T. The mechanism of the permanganate-promoted oxidative cyclization of 1,5-dienes—A DFT study. Collect. Czech. Chem. Commun. 2007, 72, 715–727. [Google Scholar] [CrossRef]
- de Champdoré, M.; Lasalvia, M.; Piccialli, V. OsO4-catalyzed oxidative cyclization of geranyl and neryl acetate to cis-2,5-bis(hydroxymethyl)tetrahydrofurans. Tetrahedron Lett. 1998, 39, 9781–9784. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Winter, J.J.G.; Helliwell, M.; Stemp, G. Hydrogen bonding control in the oxidative cyclisation of 1,5-dienes. Tetrahedron Lett. 2001, 42, 971–974. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Butterworth, S. A general oxidative cyclization of 1,5-dienes using catalytic osmium tetroxide. Angew. Chem. Int. Ed. 2003, 42, 948–951. [Google Scholar] [CrossRef]
- Poethig, A.; Strassner, T. Stereoselective OsO4-catalyzed oxidative cyclization of 1,5-dienes. J. Org. Chem. 2010, 75, 1967–1973. [Google Scholar] [CrossRef]
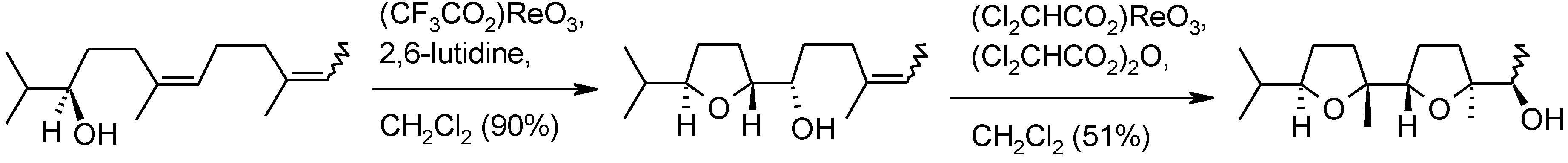
- Piccialli, V.; Caserta, T. Perruthenate ion. Another metal oxo species able to promote the oxidative cyclization of 1,5-dienes to 2,5-disubstituted cis-tetrahydrofurans. Tetrahedron Lett. 2004, 45, 303–308. [Google Scholar] [CrossRef]
- Towne, T.B.; McDonald, F.E. syn-Oxidative polycyclizations of hydroxypolyenes: highly stereoselective and potentially biomimetic syntheses of all-trans-polytetrahydrofurans. J. Am. Chem. Soc. 1997, 119, 6022–6028. [Google Scholar] [CrossRef]
- McDonald, F.E.; Towne, T.B.; Schultz, C.C. Metal-oxo induced syn-oxidative polycyclizations of hydroxypolyenes: Biomimetic synthesis of polycyclic ether natural products. Pure Appl. Chem. 1998, 70, 355–358. [Google Scholar]
- Keinan, E.; Sinha, S.C. Oxidative polycyclizations with rhenium(VII) oxides. Pure Appl. Chem. 2002, 74, 93–105. [Google Scholar]
- McDonald, F.E.; Towne, T.B. Acylperrhenate-induced syn-oxidative cyclizations of hydroxydienes. J. Org. Chem. 1995, 60, 5750–5751. [Google Scholar] [CrossRef]
- Morimoto, Y.; Iwai, T. Highly diastereoselective cyclizations of bishomoallylic tertiary alcohols promoted by rhenium(VII) oxide. Critical steric versus chelation effects in alkoxyrhenium intermediates. J. Am. Chem. Soc. 1998, 120, 1633–1634. [Google Scholar] [CrossRef]
- Morimoto, Y.; Kinoshita, T.; Iwai, T. Asymmetric total synthesis of highly symmetric squalene-derived cytotoxic polyethers. Chirality 2002, 14, 578–586. [Google Scholar] [CrossRef]
- Sinha, S.C.; Sinha-Bagchi, A.; Keinan, E. Combined osmium-rhenium approach to synthesis of naturally occurring polyethers. J. Am. Chem. Soc. 1995, 117, 1447–1448. [Google Scholar] [CrossRef]
- Sinha, S.C.; Keinan, E.; Sinha, S.C. Rules of stereoselectivity in tandem oxidative polycyclization reaction with rhenium(VII) oxides. J. Am. Chem. Soc. 1998, 120, 9076–9077. [Google Scholar] [CrossRef]
- Das, S.; Li, L.-S.; Abraham, S.; Chen, Z.; Sinha, S.C. A bidirectional approach to the synthesis of complete library of adjacent-bis-THF annonaceous acetogenins. J. Org. Chem. 2005, 70, 5922–5931. [Google Scholar] [CrossRef]
- Sinha, S.C.; Sinha, A.; Sinha, S.C.; Keinan, E. Tandem oxidative cyclization with rhenium oxide. Total synthesis of 17,18-bisepi-goniocin. J. Am. Chem. Soc. 1997, 119, 12014–12015. [Google Scholar] [CrossRef]
- Kennedy, R.M.; Tang, S. Directed oxidative cyclization of 5-hydroxyalkenes with rhenium oxide. Tetrahedron Lett. 1992, 33, 3729–3732. [Google Scholar] [CrossRef]
- McDonald, F.E.; Towne, T.B. Syn-oxidative polycyclization of hydroxyl polyenes: A new approach to polyether synthesis. J. Am. Chem. Soc. 1994, 116, 7921–7922. [Google Scholar] [CrossRef]
- Lee, D.G.; van den Hengh, M. The oxidation of tetrahydrofuran by ruthenium tetroxide. Can. J. Chem. 1972, 50, 3129–3134. [Google Scholar] [CrossRef]
- Bakke, J.M.; Frøhaug, A.E. The mechanism of RuO4-mediated oxidations of ethers: Isotope effects, solvent effects and substituent effects. Acta Chem. Scand. 1995, 49, 615–622. [Google Scholar] [CrossRef]
- Diaz, D.D.; Ramirez, M.A.; Cenal, J.P.; Saad, J.R.; Tonn, C.E.; Martin, V.S. Stereocontrolled synthesis of 1-acetylen-2,3-di-O-benzyl-tetrahydrofurans, 1,4-anhydro-arabinitol, and α,β-dihydroxy-γ-alkyl-butyrolactones. Chirality 2003, 15, 148–155. [Google Scholar] [CrossRef]
- Smith, A.B.; Scarborough, R.M., Jr. Ruthenium tetroxide oxidation of simple ethers: A systematic study. Synth. Commun. 1980, 10, 205–211. [Google Scholar] [CrossRef]
- Miranda, L.S.M.; Vasconcellos, L.A.A. Chemoselective RuO4 oxidation of phenyl or p-methoxyphenyl groups to carboxylic acid functions in the presence of tetrahydropyran ring. Synthesis 2004, 11, 1767–1770. [Google Scholar] [CrossRef]
- Piccialli, V.; D’Errico, S.; Borbone, N.; Oliviero, G.; Centore, R.; Zaccaria, S. A general synthesis of bis-α-acyloxy-1,4- and -1,5-diketones through catalytic oxidative opening of acylated THF and THP diols. Eur. J. Org. Chem. 2013, 1781–1789. [Google Scholar]
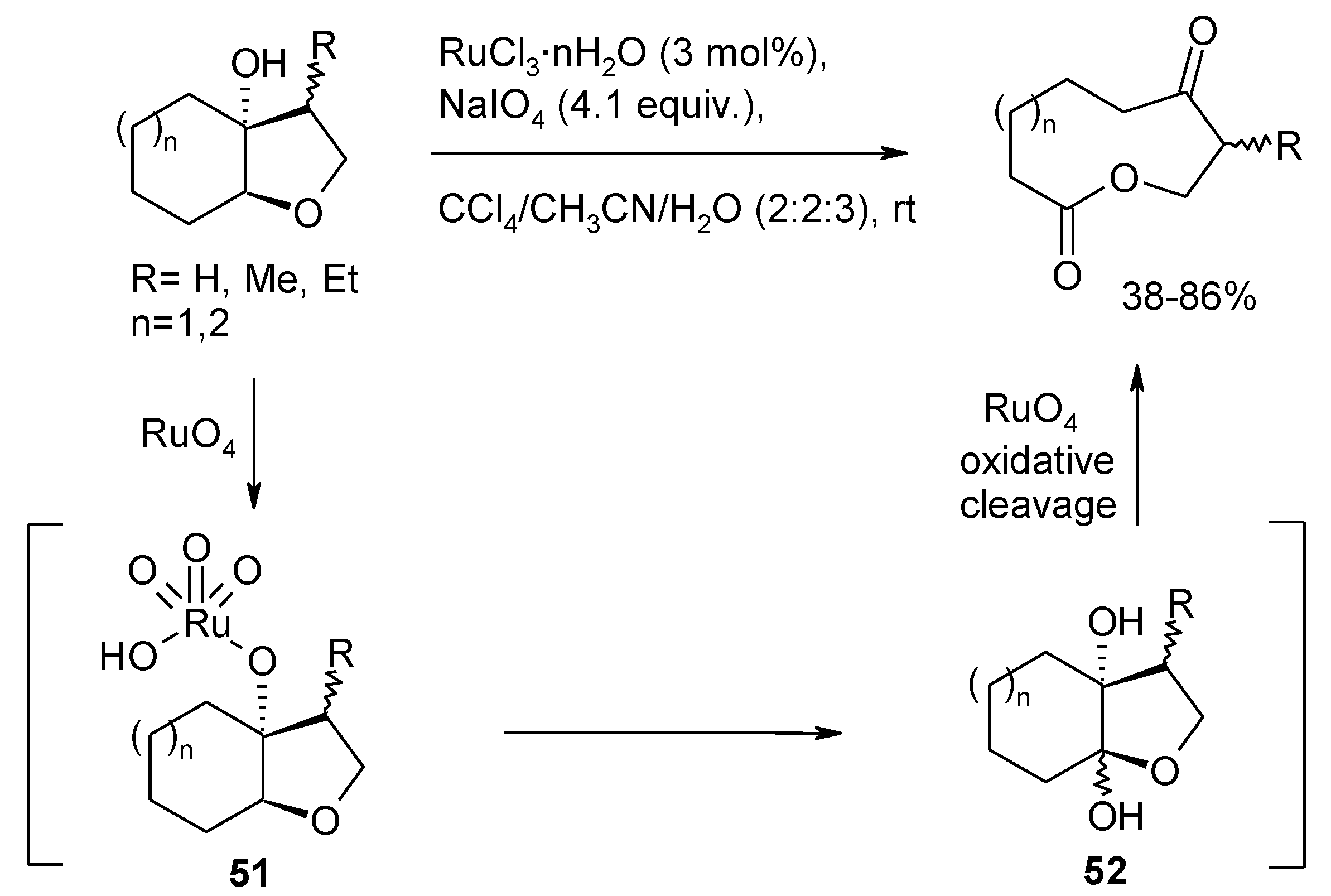
- Lee, J.S.; Cao, H.; Fuchs, P.L. Ruthenium-catalyzed mild C-H oxyfunctionalization of cyclic steroidal ethers. J. Org. Chem. 2007, 72, 5820–5823. [Google Scholar] [CrossRef]
- Kanduluru, A.K.; Banerjee, P.; Beutler, J.A.; Fuchs, P.L. A convergent total synthesis of the potent cephalostatin/ritterazine hybrid-25-epi ritterostatin GN1N. J. Org. Chem. 2013, 78, 9085–9092. [Google Scholar] [CrossRef]
- Lee, S.; Fuchs, P.L. Chemospecific chromium(VI) catalyzed oxidation of C-H bonds at -40 °C. J. Am. Chem. Soc. 2002, 124, 13978–13979. [Google Scholar] [CrossRef]
- Ferraz, H.M.; Longo, L.S., Jr. Efficient entry into medium-ring keto-lactones. Ruthenium tetraoxide-promoted oxidative cleavage of β-hydroxyethers. Org. Lett. 2003, 5, 1337–1339. [Google Scholar] [CrossRef]
- Ferraz, H.M.; Longo, L.S., Jr. Bicyclic β-hydroxytetrahydrofurans as precursors of medium ring keto-lactones. J. Org. Chem. 2007, 72, 2945–2950. [Google Scholar] [CrossRef]
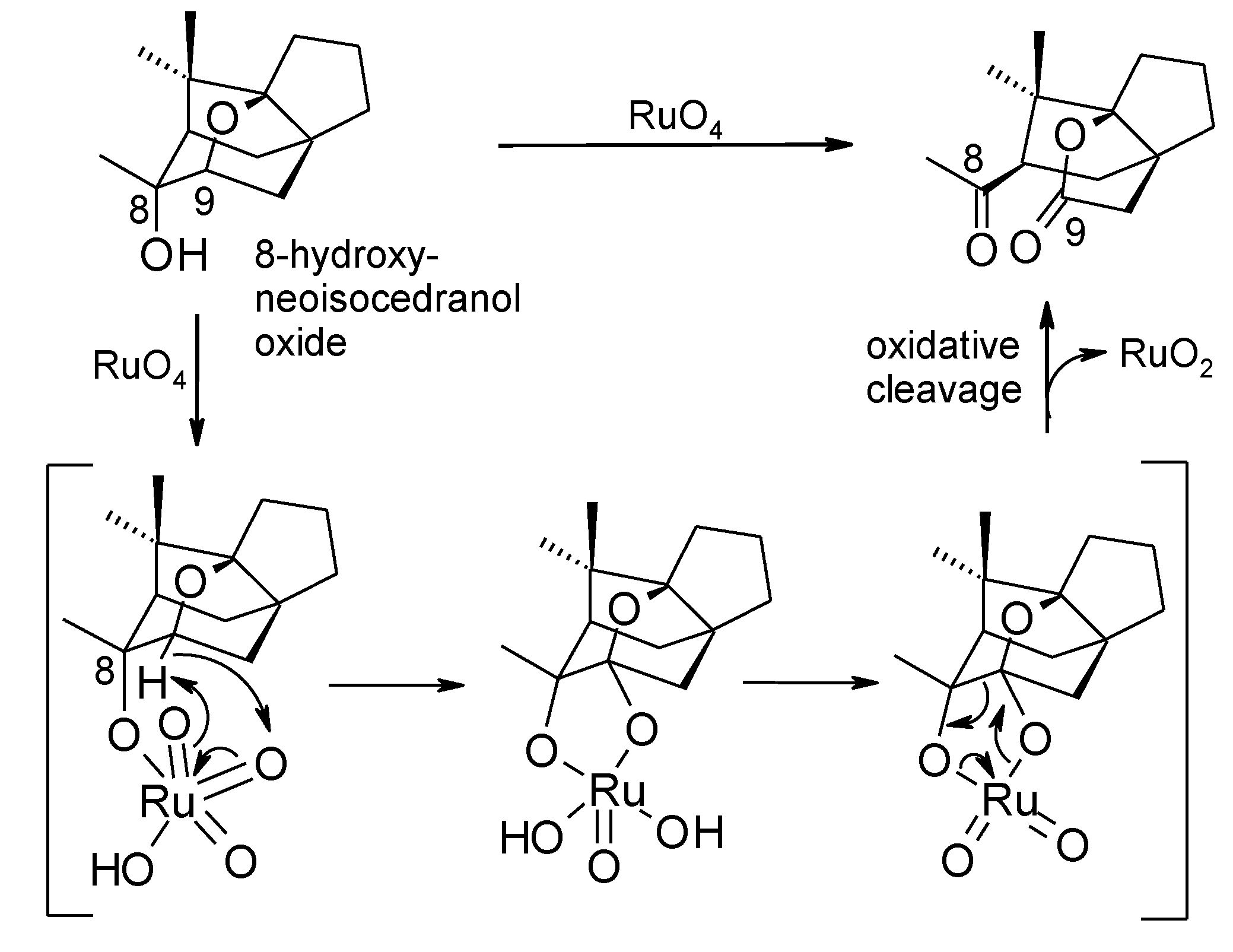
- Tenaglia, A.; Terranova, E.; Waegell, B. Ruthenium-catalyzed carbon-hydrogen bond activation. Oxyfunctionalization of nonactivated carbon-hydrogen bonds in the cedrane series with ruthenium tetraoxide generated in situ. J. Org. Chem. 1992, 57, 5523–5528. [Google Scholar] [CrossRef]
- Baskaran, S.; Chandrasekaran, S. Oxidation of tetrahydrofuran methanol derivatives with pyridinium chlorochromate: a facile synthesis of γ-butyrolactones. Tetrahedron Lett. 1990, 31, 2775–2778. [Google Scholar] [CrossRef]
- Ali, S.M.; Ramesh, K.; Borchardt, R.T. Efficient enantioselective synthesis of carbocyclic nucleoside and prostaglandin synthons. Tetrahedron Lett. 1990, 31, 1509–1512. [Google Scholar] [CrossRef]
- Edmunds, A.J.F.; Trueb, W.; Oppolzer, W.; Cowley, P. Herboxidiene: Determination of absolute configuration by degradation and synthetic studies. Tetrahedron 1997, 53, 2785–2802. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, M.S.; Prasad, A.R. A convergent route to β-hydroxy δ-lactones through Prins cyclization as the key step: Synthesis of (+)-prelactones B, C, and V. Tetrahedron Lett. 2005, 46, 2133–2136. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, M.S.; Prasad, A.R. Stereoselective syntheses of (-)-tetrahydrolipstatin via Prins cyclisations. Tetrahedron Lett. 2006, 47, 4995–4998. [Google Scholar] [CrossRef]
- Reddy, M.S.; Narender, M.; Rao, K.R. Stereoselective synthesis of simplalactone B via Prins cyclization. Tetrahedron 2007, 63, 11011–11015. [Google Scholar] [CrossRef]
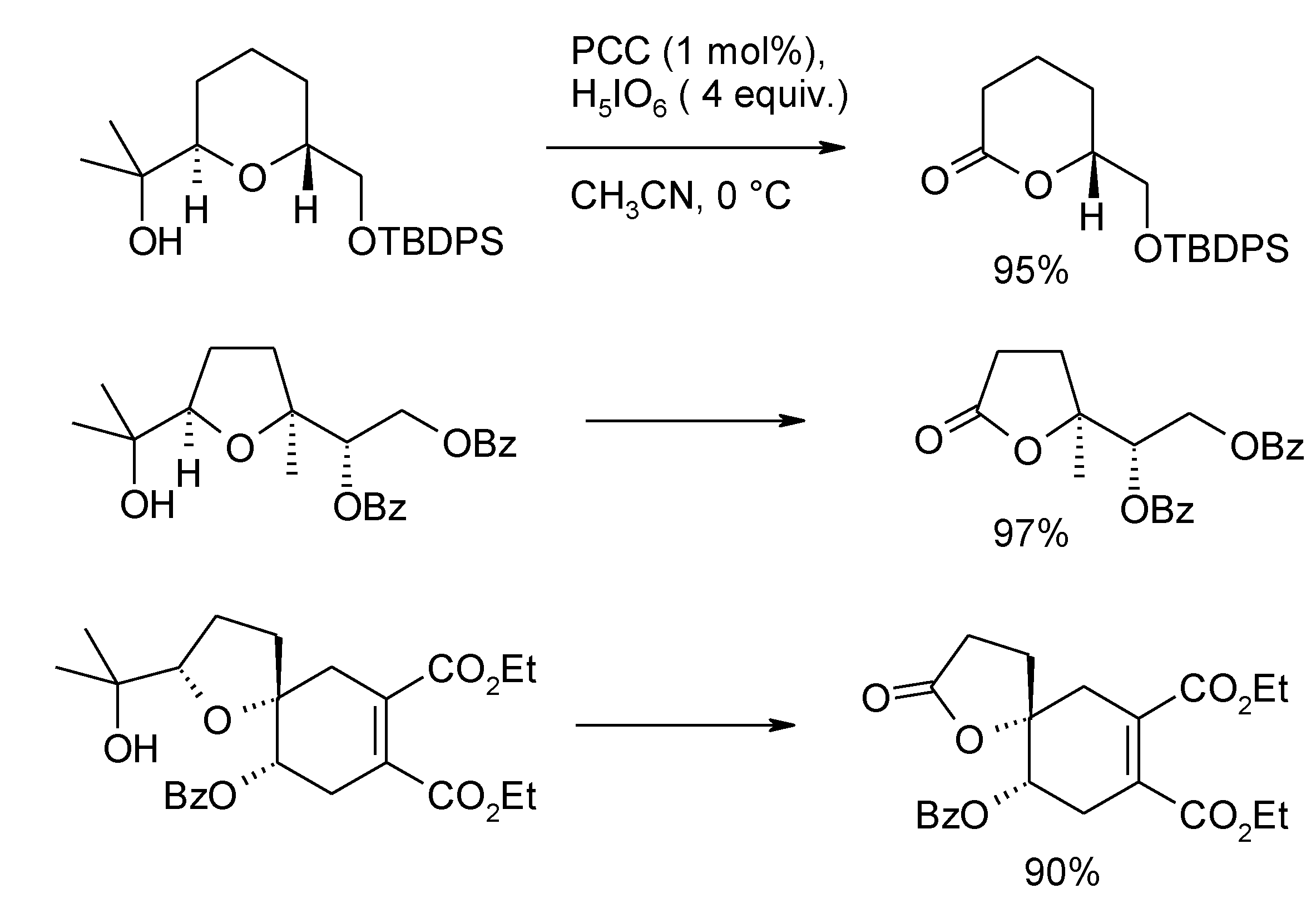
- Roth, S.; Stark, C.B.W. A new catalytic oxidative cleavage reaction to furnish lactones. J. Chem. Soc. Chem. Commun. 2008, 6411–6413. [Google Scholar] [CrossRef]
- Okamura, A.; Kitani, M.; Murata, M. Kinetic studies of the catalytic oxygen exchange of chromate ions with water by periodate ions. Bull. Chem. Soc. Jpn. 1994, 67, 1522–1530. [Google Scholar] [CrossRef]
- Piccialli, V.; Oliviero, G.; Borbone, N.; Tuzi, A.; Centore, R.; Hemminki, A.; Ugolini, M.; Cerullo, V. Discovery of a new PCC-mediated stereoselective oxidative spiroketalization process. An access to a new type of poly-THF spiroketal compound displaying anticancer activity. Org. Biomol. Chem. 2009, 7, 3036–3039. [Google Scholar] [CrossRef]
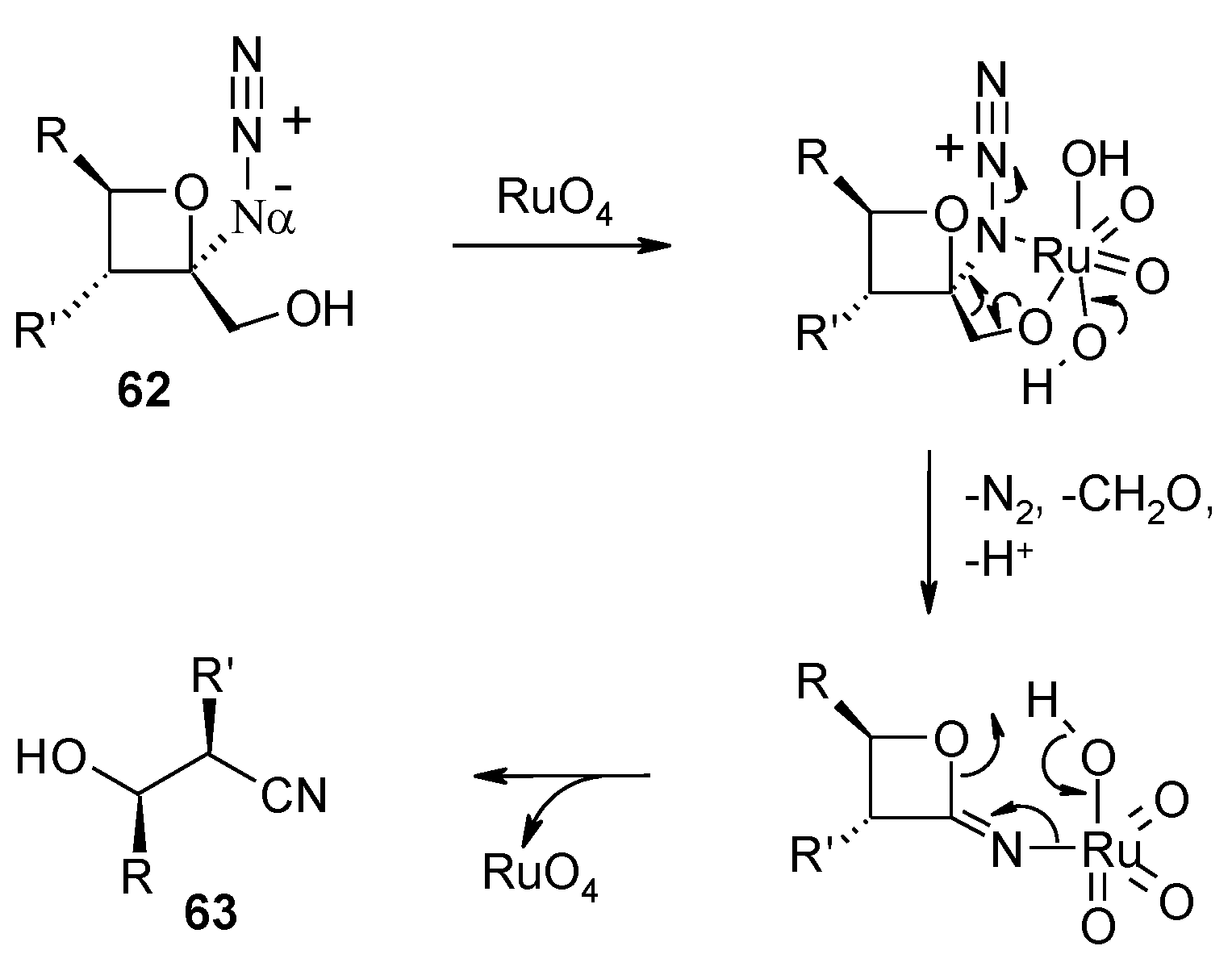
- Faber, E.; Herget, J.; Gascon, J.A.; Howell, A.R. Unexpected cleavage of 2-azido-2-(hydroxymethyl)oxetanes: conformation determines reaction pathway? J. Org. Chem. 2010, 75, 7565–7572. [Google Scholar] [CrossRef]
- Cenini, S.; Gallo, M.; Caselli, A.; Ragaini, F.; Fantauzzi, S.; Piangiolino, C. Coordination chemistry of organic azides and amination reactions catalyzed by transition metal complexes. Coord. Chem. Rev. 2006, 250, 1234–1253. [Google Scholar] [CrossRef]
- Chiba, S.; Xu, Y.-J.; Wang, Y.-F. A Pd(II)-catalyzed ring-expansion reaction of cyclic 2-azidoalcohol derivatives: synthesis of azaheterocycles. J. Am. Chem. Soc. 2009, 12886–12887. [Google Scholar] [CrossRef]
- Lancefield, C.S.; Zhou, L.; Slawin, A.M.Z.; Westwood, N.J. The synthesis of melohenine B and a related natural product. Org. Lett. 2012, 14, 6166–6169. [Google Scholar] [CrossRef]
- Rodewal, W.J.; Jagodzinski, J.J. C(14a)-homo-B-norlanostanes. Part I. Transformation of lanosterol into derivatives of C(14a)-homo-B-norlanost-8(14a)-ene. Pol. J. Chem. 1979, 53, 1203–1210. [Google Scholar]
- Rodewal, W.J.; Jagodzinski, J.J. C(14a)-homo-B-norlanostanes. Part III. Synthesis of some C(14a)-homo-B-norlanostanes modified in the side chain. Pol. J. Chem. 1981, 55, 1751–1757. [Google Scholar]
- Weidmann, V.; Maison, W. Allylic oxidations of olefins to enones. Synthesis 2013, 45, 2201–2221. [Google Scholar] [CrossRef]
- Company, A.; Lloret, J.; Gómez, L.; Costas, M. Alkane C-H oxygenation catalyzed by transition metal complexes. In Alkane C-H Activation by Single-Site Metal Catalysis; Pérez, P.J., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 143–228. [Google Scholar]
- Bakke, J.M.; Lundquist, M. The RuO4 oxidation of cyclic saturated hydrocarbons. Formation of alcohols. Acta Chem. Scand. 1986, B40, 430–433. [Google Scholar] [CrossRef]
- Bakke, J.M.; Brænden, J.E. Derivatization of saturated hydrocarbons. The mechanism of RuO4 oxidation. Acta Chem. Scand. 1991, 45, 418–423. [Google Scholar] [CrossRef]
- Bakke, J.M.; Bethell, D. The mechanism of RuO4-mediated oxidations of saturated hydrocarbons. Reactivity, kinetic isotope effect and activation parameters. Acta Chem. Scand. 1992, 46, 644–649. [Google Scholar] [CrossRef]
- Bakke, J.M.; Fröhaug, A.E. The mechanism of RuO4-mediated oxidations of saturated hydrocarbons. Solvent effect and substituent effect. Acta Chem. Scand. 1994, 48, 160–164. [Google Scholar] [CrossRef]
- Bakke, J.M.; Fröhaug, A.E. Mechanism of RuO4-mediated oxidations of saturated hydrocarbons. Isotope effect, solvent effects and substituent effects. J. Phys. Org. Chem. 1996, 9, 507–513. [Google Scholar] [CrossRef]
- Bakke, J.M.; Fröhaug, A.E. Ruthenium tetroxide mediated reactions: The mechanism of oxidations of hydrocarbons and ethers. J. Phys. Org. Chem. 1996, 9, 310–318. [Google Scholar] [CrossRef]
- Tenaglia, A.; Terranova, E.; Waegell, B. Ruthenium-catalyzed C-H bond activation oxidation of bridged bicyclic and ticyclic alkanes. Tetrahedron Lett. 1989, 30, 5271–5274. [Google Scholar] [CrossRef]
- Tenaglia, A.; Terranova, E.; Waegell, B. Ruthenium-catalysed C-H bond activation. Evidence for a concerted mechanism in oxyfunctinalization of cyclic saturated hydrocarbons. J. Chem Soc.Chem. Commun. 1990, 1344–1345. [Google Scholar] [CrossRef]
- Coudret, J.L.; Waegell, B. Oxidation of cis- and trans-pinane with RuO4 generated in situ. Inorg. Chim. Acta 1994, 222, 115–122. [Google Scholar] [CrossRef]
- Coudret, J.L.; Zöllner, S.; Ravoo, B.J.; Malara, L.; Hanisch, C.; Dörre, K.; de Meijere, A.B. Waegell Role of cyclopropanes as activating groups during oxidation reactions with RuO4 generated in situ. Tetrahedron Lett. 1996, 37, 2425–2428. [Google Scholar] [CrossRef]
- Drees, M.; Strassner, T. Ruthenium tetroxide oxidations of alkanes: DFT calculations of barrier heights and kinetic isotope effects. J. Org. Chem. 2006, 71, 1755–1760. [Google Scholar] [CrossRef]
- McNeill, E.; Du Bois, J. Ruthenium-catalyzed hydroxylation of unactivated tertiary C-H bonds. J. Am. Chem. Soc. 2010, 132, 10202–10204. [Google Scholar] [CrossRef]
- Bales, B.C.; Brown, P.; Dehestani, A.; Mayer, J.M. Alkane oxidation by osmium tetroxide. J. Am. Chem. Soc. 2005, 127, 2832–2833. [Google Scholar] [CrossRef]
- Mayer, J.M.; Mader, E.A.; Roth, J.P.; Bryant, J.R.; Matsuo, T.; Dehestani, A.; Bales, B.C.; Watson, E.J.; Osako, T.; Valliant-Saunder, K.; et al. Stoichiometric oxidations of σ-bonds: Radical and possible non-radical pathways. J. Mol. Cat. A: Chem. 2006, 251, 24–33. [Google Scholar] [CrossRef]
- Dehestani, A.; Lam, W.H.; Hrovat, D.A.; Davidson, E.R.; Borden, W.T.; Mayer, J.M. Ligand-assisted reduction of osmium tetroxide with molecular hydrogen via s [3+2] mechanism. J. Am. Chem. Soc. 2005, 127, 3423–3432. [Google Scholar] [CrossRef]
- Gardner, K.A.; Kuehnert, L.L.; Mayer, J.M. Hydrogen atom abstraction by permanganate: Oxidations of arylalkanes in organic solvents. Inorg. Chem. 1997, 36, 2069–2078. [Google Scholar] [CrossRef]
- Wiberg, K.B. Oxidation by chromic acid and chromyl compounds. In Oxidation in Organic Chemistry; Wiberg, K.B., Ed.; Academic Press: New York, NY, USA, 1965; pp. 69–184. [Google Scholar]
- Freeman, F. Oxidation by oxochromium compounds. In Organic Syntheses by Oxidation with Metal Compounds; Mijs, W.J., De Jonge, C.R.H.I., Eds.; Plenum Press: New York, NY, USA, 1986; pp. 41–118. [Google Scholar]
- Mayer, J.M. Hydrogen atom abstraction by metal-oxo complexes: Understanding the analogy with organic radical reactions. Acc. Chem. Res. 1998, 31, 441–450. [Google Scholar] [CrossRef]
- Tkachenko, B.A.; Shubina, T.E.; Gusev, D.V.; Gunchenko, P.A.; Yurchenko, A.G.; Schreiner, P.R.; Fokin, A.A. Mechanism of C-H activation in alkanes by chromium-oxo reagents. Theor. Exp. Chem. 2003, 39, 90–95. [Google Scholar] [CrossRef]
- Griffith, W.P.; Ley, S.V.; Whitcombe, G.P.; White, A.D. Preparation and use of tetra-n-butylammonium per-ruthenate (TBAP reagent) and tetra-n-propylammonium per-ruthenate (TPAP reagent) as new catalytic oxidants for alcohols. Chem. Commun. 1987, 1625–1627. [Google Scholar]
- Griffith, W.P.; Ley, S.V. TPAP: Tetra-n-propylammonium perruthenate, a mild and convenient oxidant for alcohols. Aldrichim. Acta 1990, 23, 13–19. [Google Scholar]
- Ley, S.V.; Norman, J.; Griffith, W.P.; Marsden, S.P. Tetrapropylammonium perruthenate, Pr4N+ RuO4-, TPAP: A catalytic oxidant for organic synthesis. Synthesis 1994, 639–666. [Google Scholar]
- Langer, P. Tetra-n-propyl ammonium perruthenate (TPAP). An efficient and selective reagent for oxidation reactions in solution and on the solid phase. J. Prakt. Chem. 2000, 342, 728–730. [Google Scholar] [CrossRef]
- Gonsalvi, L.; Arends, I.W.C.E.; Sheldon, R.A. Selective ruthenium-catalyzed oxidation of 1,2:4,5-di-O-isopropylidene-β-D-fructopyranose and other alcohols with NaOCl. Org. Lett. 2002, 4, 1659–1661. [Google Scholar] [CrossRef]
- Gonsalvi, L.; Arends, I.W.C.E.; Moilanen, P.; Sheldon, R.A. The effect of pH control on the selective ruthenium-catalyzed oxidation of ethers and alcohols with sodium hypochlorite. Adv. Synth. Catal. 2003, 345, 1321–1328. [Google Scholar] [CrossRef]
- Lenz, R.; Ley, S.V. Tetra-n-propylammonium perruthenate (TPAP)-catalysed oxidations of alcohols using molecular oxygen as a co-oxidant. J. Chem. Soc. Perkin Trans. 1 1997, 3291–3292. [Google Scholar] [CrossRef]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Chellé-Regnaut, I.; Urch, C.J.; Brown, S.M. Efficient, aerobic, ruthenium-catalyzed oxidation of alcohols into aldehydes and ketones. J. Am. Chem. Soc. 1997, 119, 12661–12662. [Google Scholar] [CrossRef]
- Arends, I.W.C.E.; Sheldon, R.A. Modern oxidation of alcohols using environmentally benign oxidants. In Modern Oxidation Methods, 2nd ed.; Baeckvall, J.-E., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 147–185. [Google Scholar]
- Hinzen, B.; Ley, S.V. Polymer supported perruthenate (PSP): A new oxidant for clean organic synthesis. J. Chem. Soc. Perkin Trans. 1 1997, 1907–1908. [Google Scholar] [CrossRef]
- Hinzen, B.; Lenz, R.; Ley, S.V. Polymer supported perruthenate (PSP): Clean oxidation of primary alcohols to carbonyl compounds using oxygen as cooxidant. Synthesis 1998, 977–979. [Google Scholar] [CrossRef]
- Bleloch, A.; Johnson, B.F.G.; Steven, V.; Ley, S.V.; Price, A.J.; Shephard, D.S.; Thomas, A.W. Modified mesoporous silicate MCM-41 materials: Immobilised perruthenate-a new highly active heterogeneous oxidation catalyst for clean organic synthesis using molecular oxygen. Chem. Commun. 1999, 1907–1908. [Google Scholar]
- Pagliaro, M.; Ciriminna, R. New recyclable catalysts for aerobic alcohols oxidation: Sol-gel ormosils doped with TPAP. Tetrahedron Lett. 2001, 42, 4511–4514. [Google Scholar] [CrossRef]
- Campestrini, M.; Carraro, M.; Franco, L.; Ciriminna, R.; Pagliaro, M. Stabilization of catalytic-sol-gel entrapped perruthenate. Tetrahedron Lett. 2007, 49, 419–423. [Google Scholar]
- Campestrini, M.; Carraro, M.; Ciriminna, R.; Pagliaro, M.; Tonellato, U. Alcohols oxidation with hydrogen peroxide promoted by TPAP-doped ormosils. Tetrahedron Lett. 2004, 45, 7283–7286. [Google Scholar] [CrossRef]
- Ciriminna, R.; Hesemann, P.; Moreau, J.J.E.; Carraro, M.; Campestrini, M.; Pagliaro, M. Aerobic oxidation of alcohols in carbon dioxide with silica-supported ionic liquids doped with perruthenate. Chem. Eur. J. 2006, 12, 5220–5224. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, Z.; Hu, S.; Song, J.; Li, W.; Han, B. Aerobic oxidation of benzyl alcohol in supercritical CO2 catalyzed by perruthenate immobilized on polymer supported ionic liquid. Green Chem. 2008, 10, 278–282. [Google Scholar] [CrossRef]
- Ciriminna, R.; Carraro, M.; Campestrini, M.; Pagliaro, M. Sol-gel entrapped TPAP: An off-the-shelf catalyst set for the clean oxidation of alcohols. Curr. Org. Chem. 2008, 12, 257–261. [Google Scholar]
- Goti, A.; Romani, M. Catalytic oxidation of secondary amines with tetra-n-propylammonium perruthenate. Tetrahedron 1994, 35, 6567–6570. [Google Scholar] [CrossRef]
- Goti, A.; de Sarlo, F.; Romani, M. Highly efficient and mild synthesis of nitrones by catalytic oxidation of hydroxylamines with tetra-n-propylammonium perruthenate. Tetrahedron 1994, 35, 6571–6574. [Google Scholar] [CrossRef]
- Murugan, R.; Ramamoorthy, S.; Sundarrajan, S.; Ramakrishna, S. Simple and efficient synthesis of 2,6-dialkyl-3,5-dialkoxycarbonyl-4-(3-aryl-1-phenyl-pyrazol-4-yl)pyridines using TPAP/NMO as a catalyst under mild conditions. Tetrahedron 2011, 67, 2998–3002. [Google Scholar] [CrossRef]
- Pulvannam, K.; Hale, R.; Sedehi, D.; Chen, T. Cyclization of 1,2,4-triazenes using oxidizing reagents- NaOCl, Ca(ClO)2, Dess-Martin periodinane and Ley’s TPAP/NMO. Tetrahedron 2001, 57, 9677–9682. [Google Scholar] [CrossRef]
- Virlouvet, M.; Podlech, J. lactam-containing peptidomimetics. Tetrahedron 2010, 66, 6174–6180. [Google Scholar] [CrossRef]
- Yates, M.H. One-pot conversion of olefins to carbonyl compounds by hydroboration/TPAP-NMO oxidation. Tetrahedron Lett. 1997, 38, 2813–2816. [Google Scholar] [CrossRef]
- Markó, I.E.; Gautier, A.; Tsukazaki, M.; Llobet, A.; Plantalech-Mir, E.; Urch, C.J.; Brown, S.M. Novel and efficient isomerization of allylic alcohols promoted by a tetrapropylammonium perruthenate catalyst. Angew. Chem. Int. Ed. 1999, 38, 1960–1962. [Google Scholar] [CrossRef]
- Cheng, H.; Stark, C.B.W. A double donor-activated ruthenium(VII) catalyst: Synthesis of enantiomerically pure THF-diols. Angew. Chem. Int. Ed. 2010, 49, 1587–1590. [Google Scholar]
- Donohoe, T.J.; Butterworth, S. xidative cyclization of diols derived from 1,5-dienes: formation of enantiopure cis-tetrahydrofurans by using catalytic osmium tetroxide; formal synthesis of (+)-cis-solamin. Angew. Chem. Int. Ed. 2005, 44, 4766–4768. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Wheelhouse, K.M.P.; Lindsay-Scott, P.J.; Glossop, P.A.; Nash, I.A.; Parker, J.S. Pyridine-N-oxide as a mild reoxidant which transforms osmium-catalyzed oxidative cyclization. Angew. Chem. Int. Ed. 2008, 47, 2872–2875. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Winship, P.C.M.; Walter, D.S.A. Lewis acid promoted oxidative cyclization. J. Org. Chem. 2009, 74, 6394–6397. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Lipński, R.M. Interplay of cascade oxidative cyclization and hydride shifts in the synthesis of the ABC spiroketal ring system of pectenotoxin-4. Angew. Chem. Int. Ed. 2013, 52, 1–5. [Google Scholar] [CrossRef]
- Hammock, B.D.; Gill, S.S.; Casida, J.E. Synthesis and morphogenetic activity of derivatives and analogs of aryl geranyl ether juvenoids. J. Agric. Food Chem. 1974, 22, 379–385. [Google Scholar] [CrossRef]
- Walba, D.M.; Stoudt, G.S. Oxidative cyclization of 5,6-dihydroxyalkenes promoted by chromium (VI) oxo species: A novel cis-2,5-disubstituted tertahydrofuran synthesis. Tetrahedron Lett. 1982, 23, 727–730. [Google Scholar] [CrossRef]
- Corey, E.J.; Ha, D.C. Total synthesis of venustatriol. Tetrahedron Lett. 1988, 29, 3171–3174. [Google Scholar] [CrossRef]
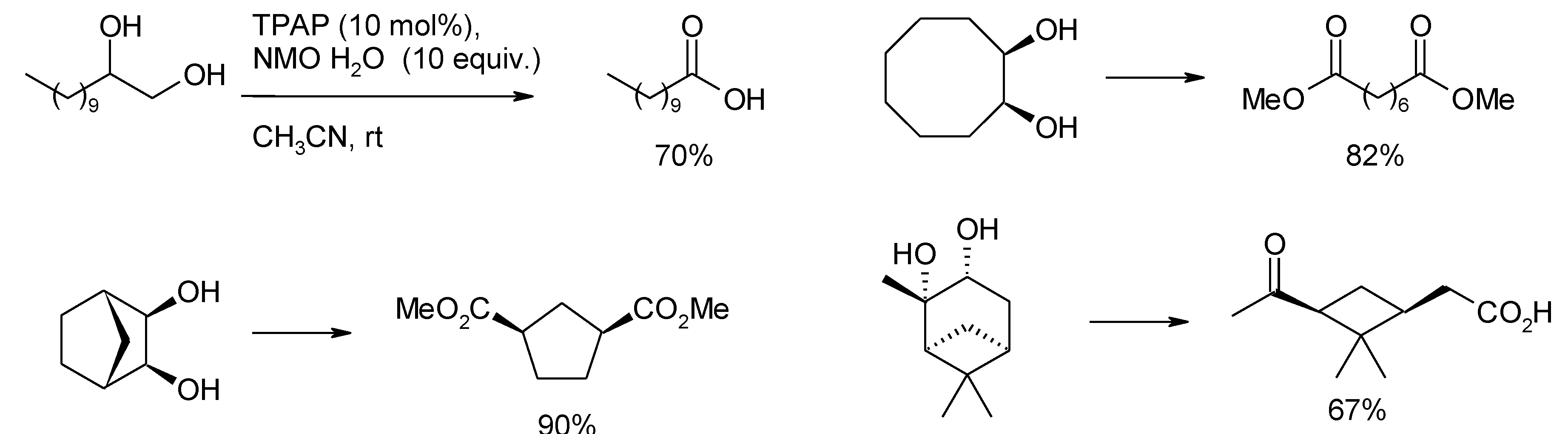
- Schmidt, A.-K.; Stark, C.B.W. TPAP-catalyzed direct oxidation of primary alcohols to carboxylic acids through stabilized aldehyde hydrates. Org. Lett. 2011, 13, 4164–4167. [Google Scholar] [CrossRef]
- Schmidt, A.-K.; Stark, C.B.W. Tetrapropylammonim prruthenate catalyzed glycol cleavage to carboxylic acids. Org. Lett. 2011, 13, 5788–5791. [Google Scholar] [CrossRef]
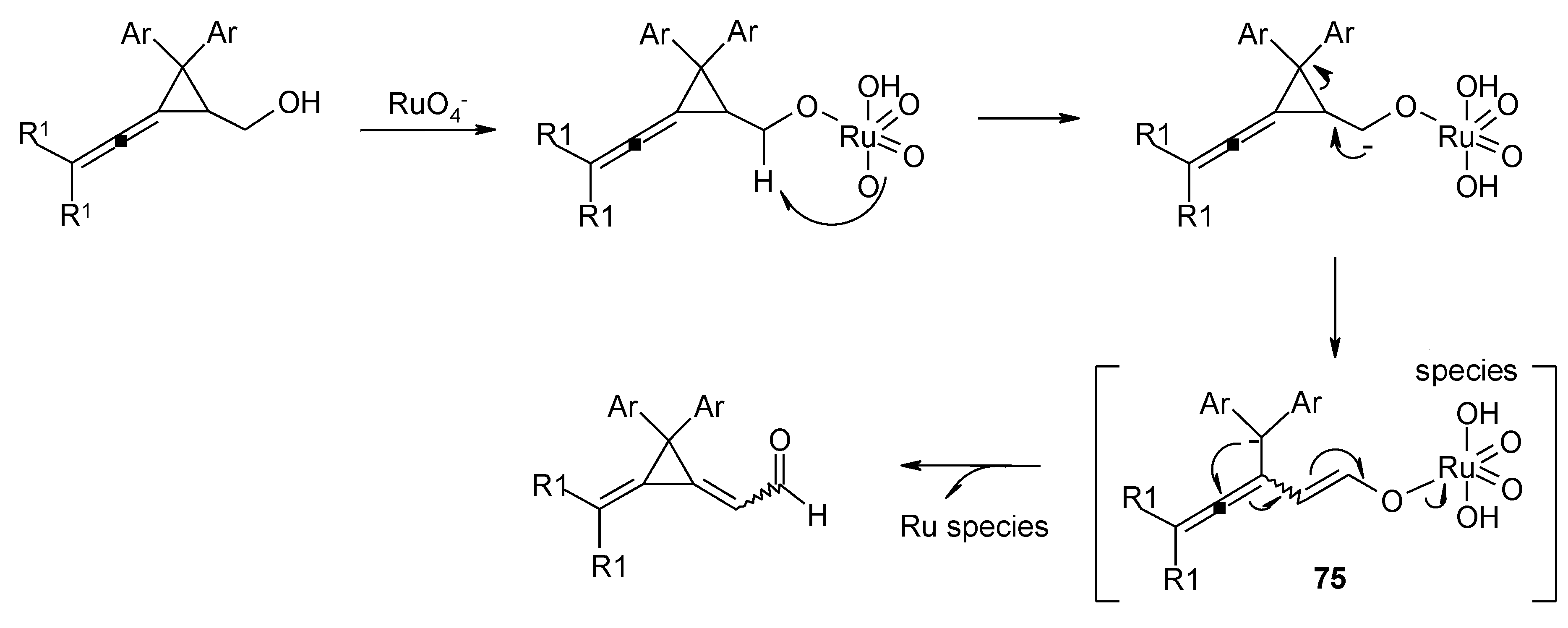
- Wiberg, K.B.; Snoonian, J.R. Synthesis, reactions, and structural studies of two-carbon bridged spiropentanes. J. Org. Chem. 1998, 63, 1402–1407. [Google Scholar] [CrossRef]
- Lu, B.-L.; Shi, M. Oxidative isomerization of vinylidenecyclopropanes to dimethylenecyclopropanes and Brønsted acid-catalyzed further transformation. Eur. J. Org. Chem. 2011, 243–248. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piccialli, V. Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-species. Molecules 2014, 19, 6534-6582. https://doi.org/10.3390/molecules19056534
Piccialli V. Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-species. Molecules. 2014; 19(5):6534-6582. https://doi.org/10.3390/molecules19056534
Chicago/Turabian StylePiccialli, Vincenzo. 2014. "Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-species" Molecules 19, no. 5: 6534-6582. https://doi.org/10.3390/molecules19056534
APA StylePiccialli, V. (2014). Ruthenium Tetroxide and Perruthenate Chemistry. Recent Advances and Related Transformations Mediated by Other Transition Metal Oxo-species. Molecules, 19(5), 6534-6582. https://doi.org/10.3390/molecules19056534