Selective Substitution of 31/42–OH in Rapamycin Guided by an in Situ IR Technique

Abstract
:1. Introduction


2. Results and Discussion
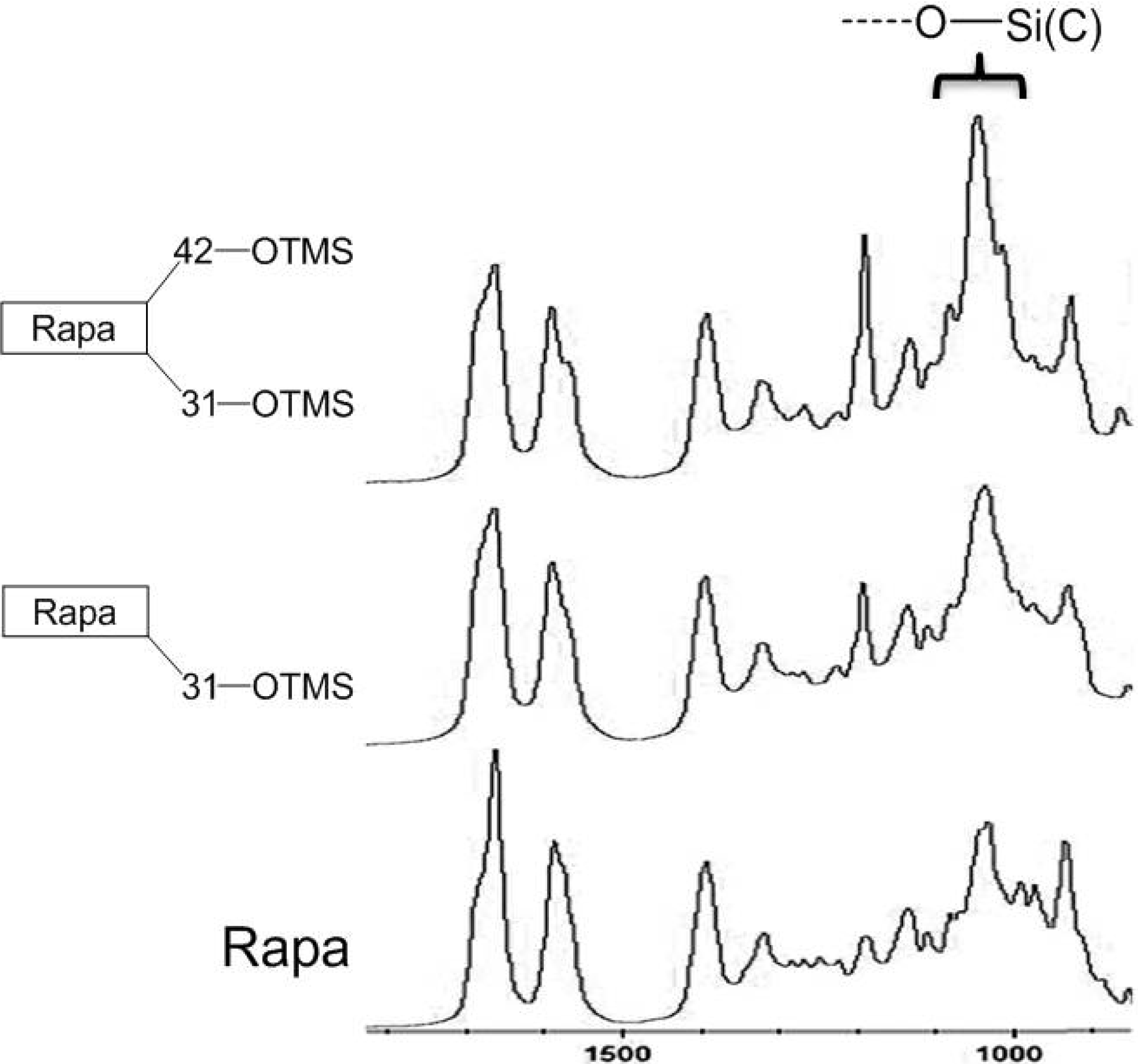
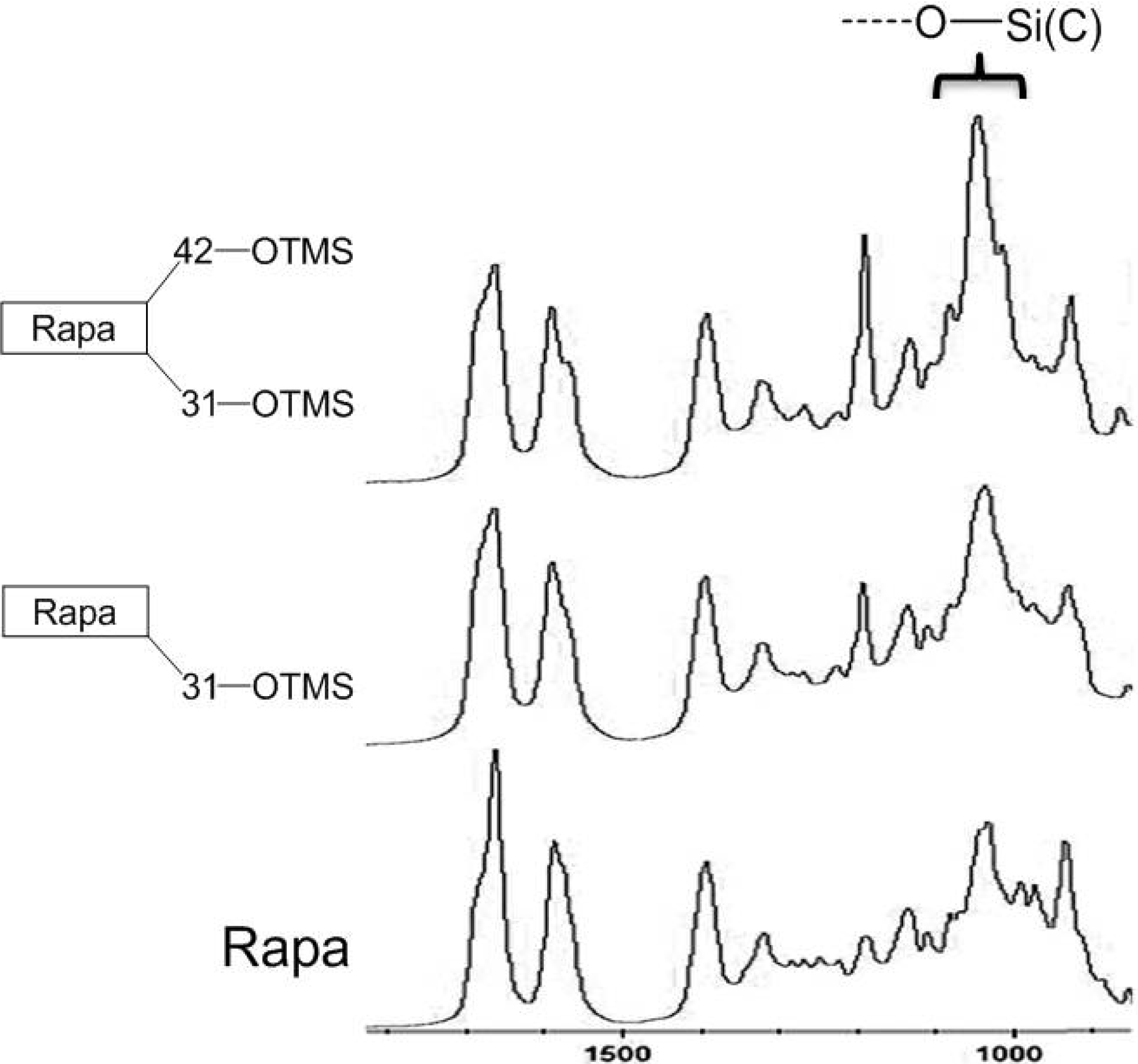
2.1. In Situ IR


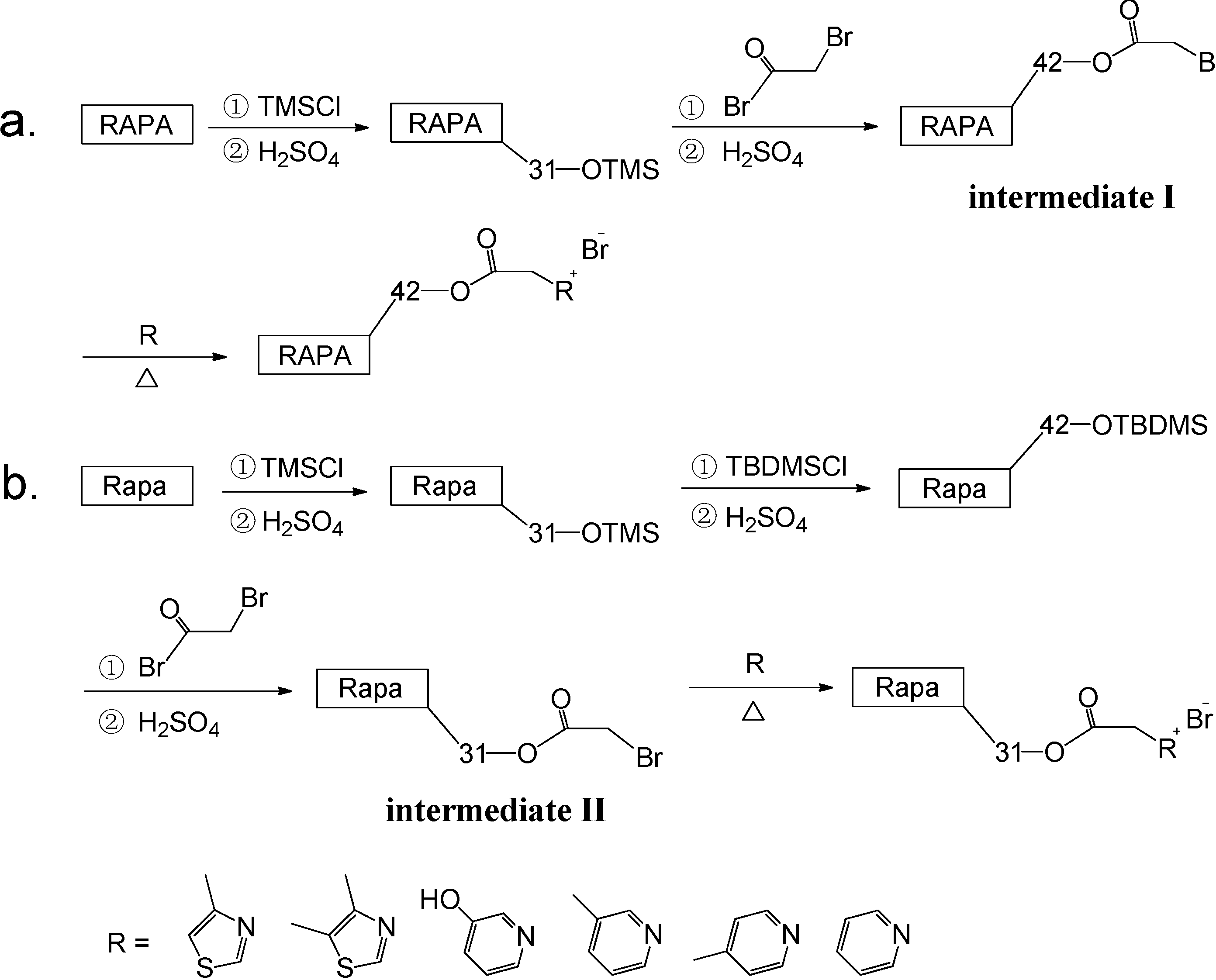
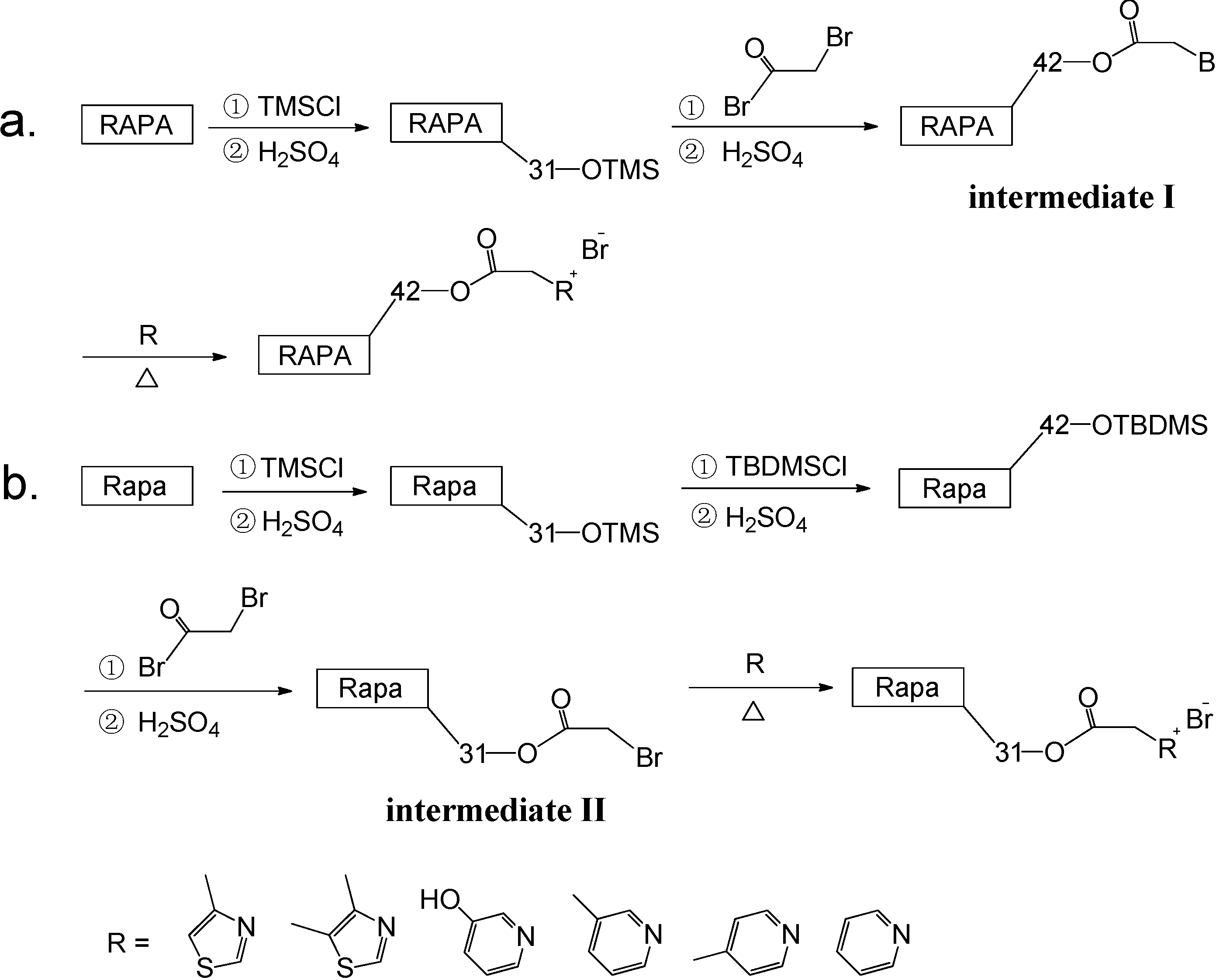
2.2. Synthesis
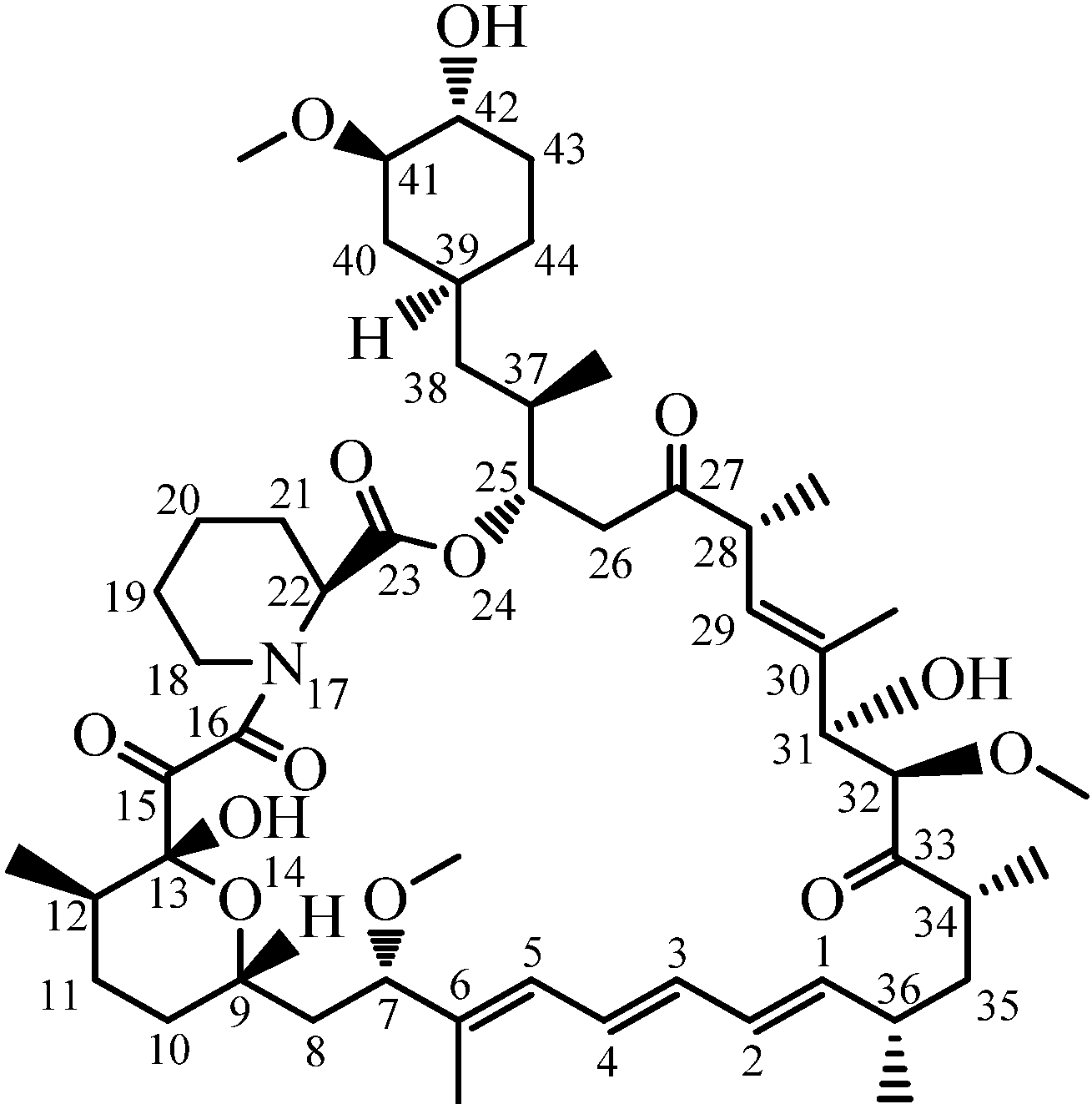
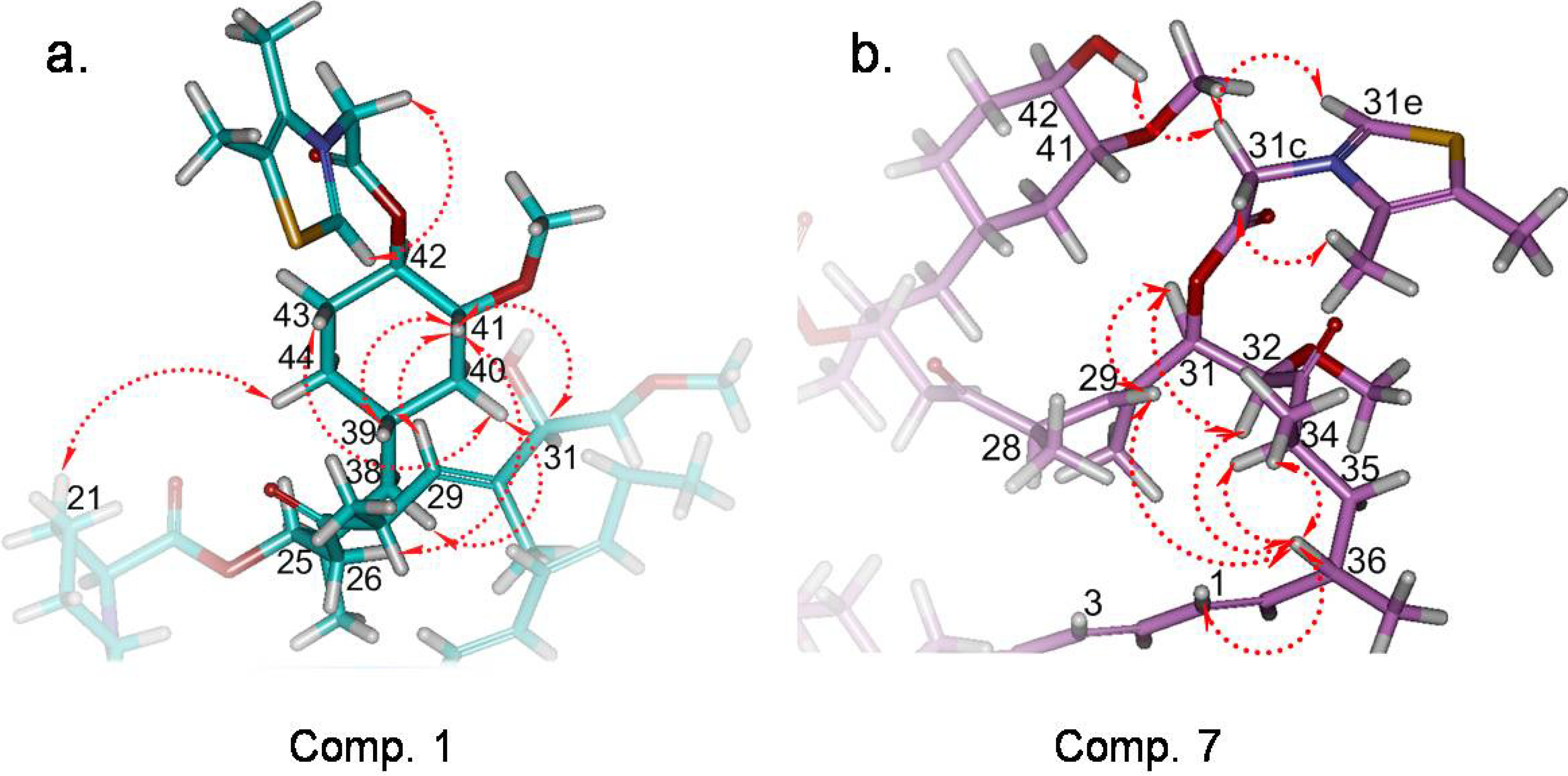
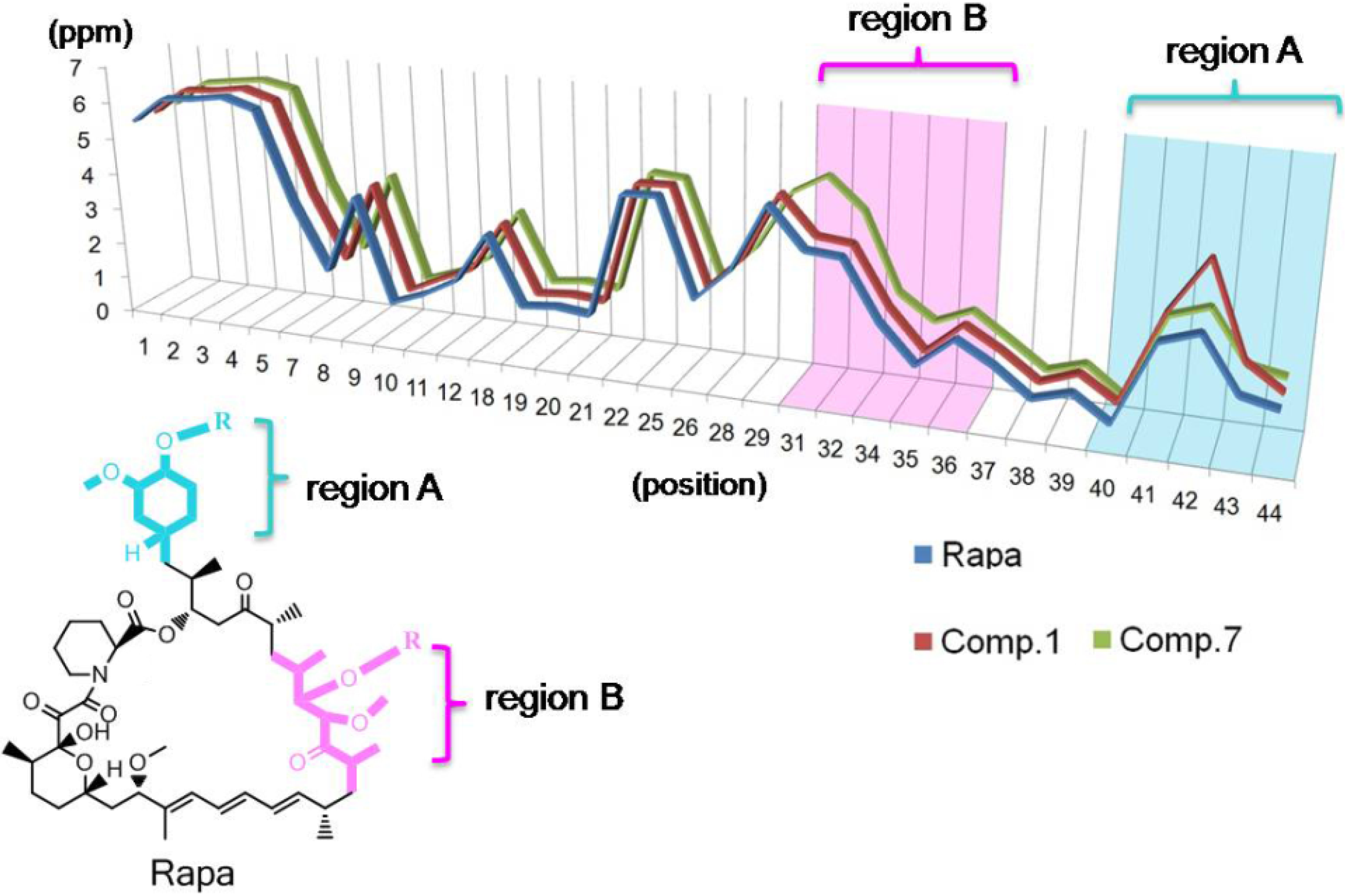
2.3. Structure

{kind=link}
{kind=link}
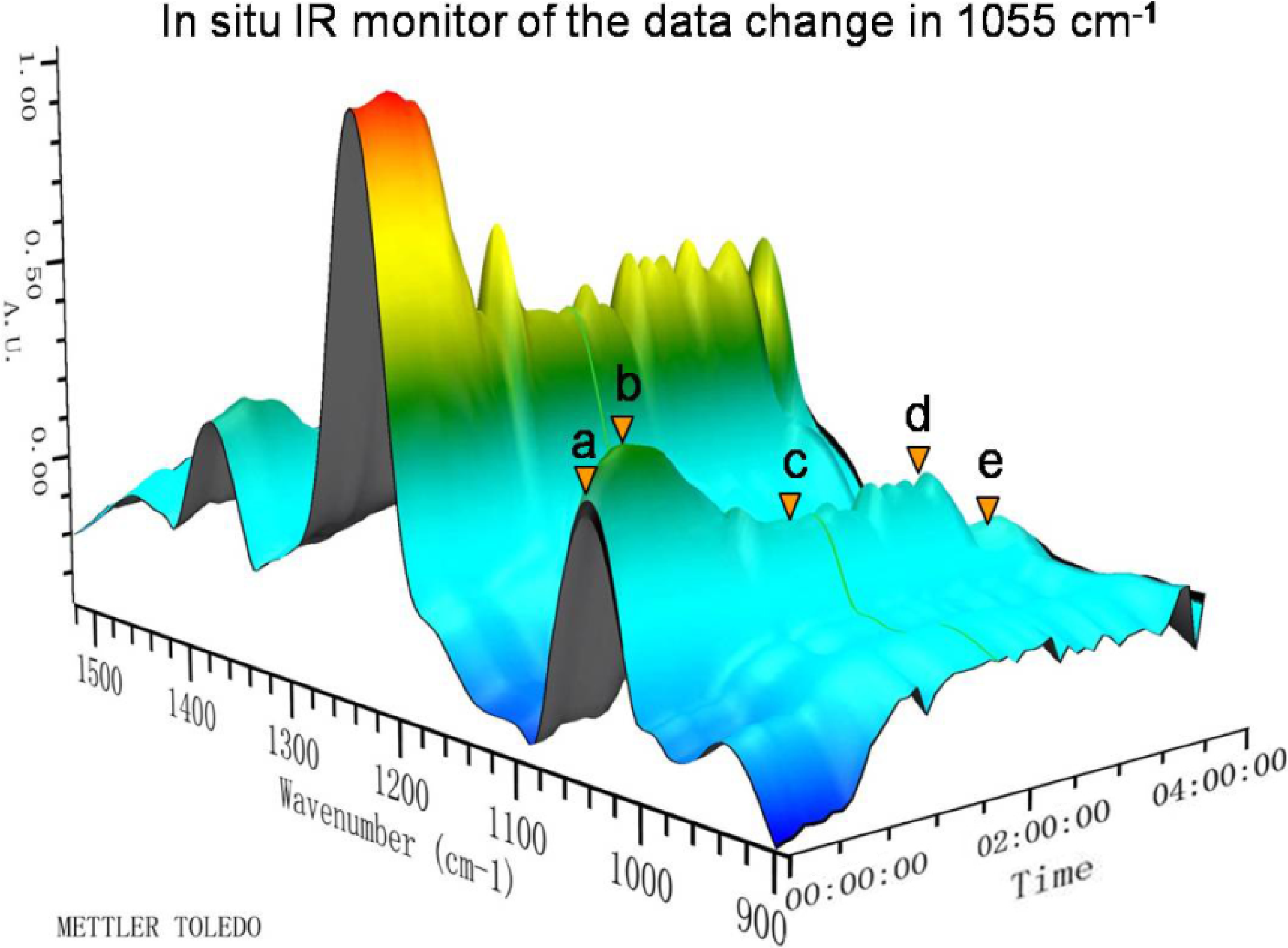{kind=link}
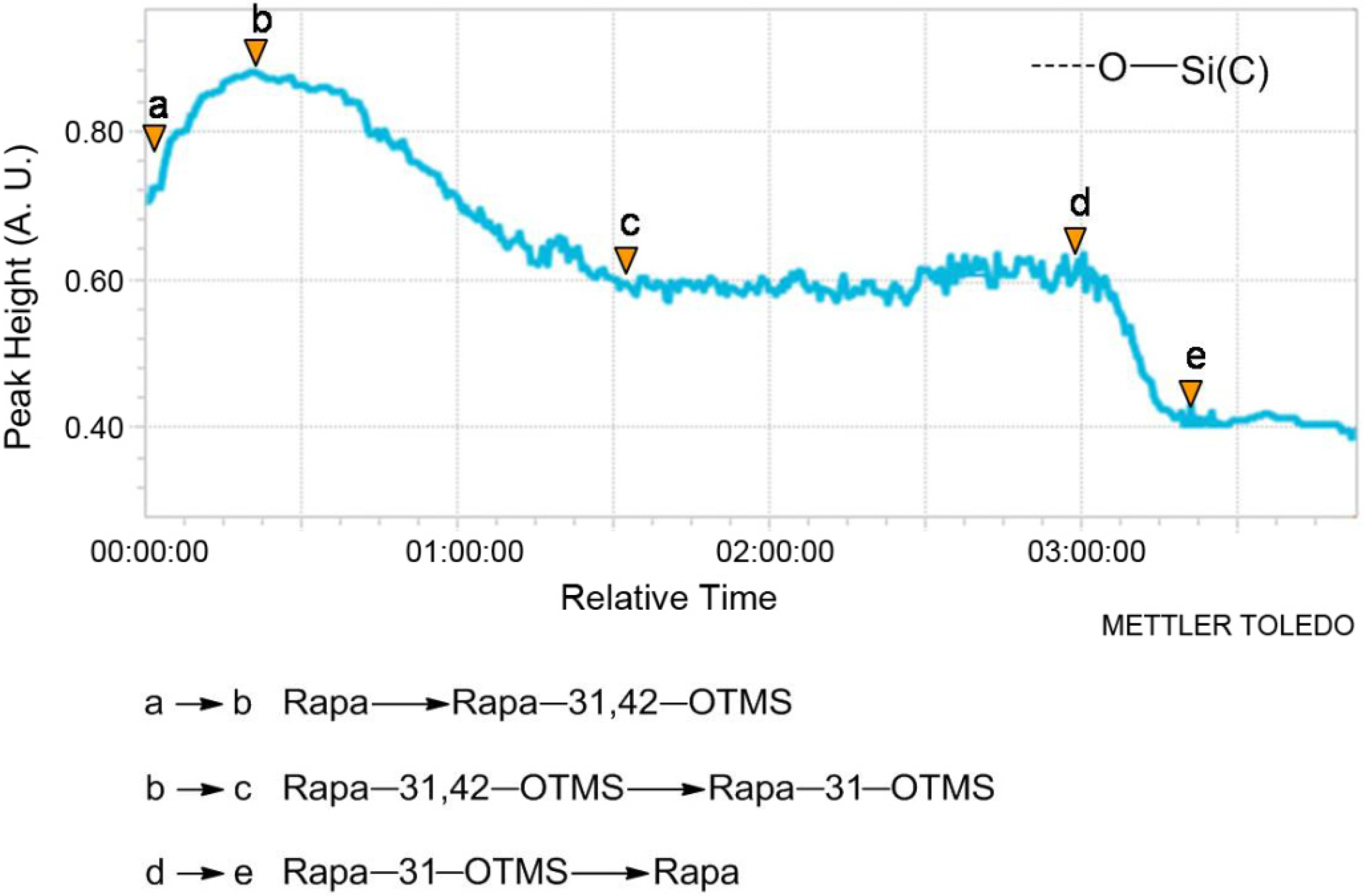{kind=link}
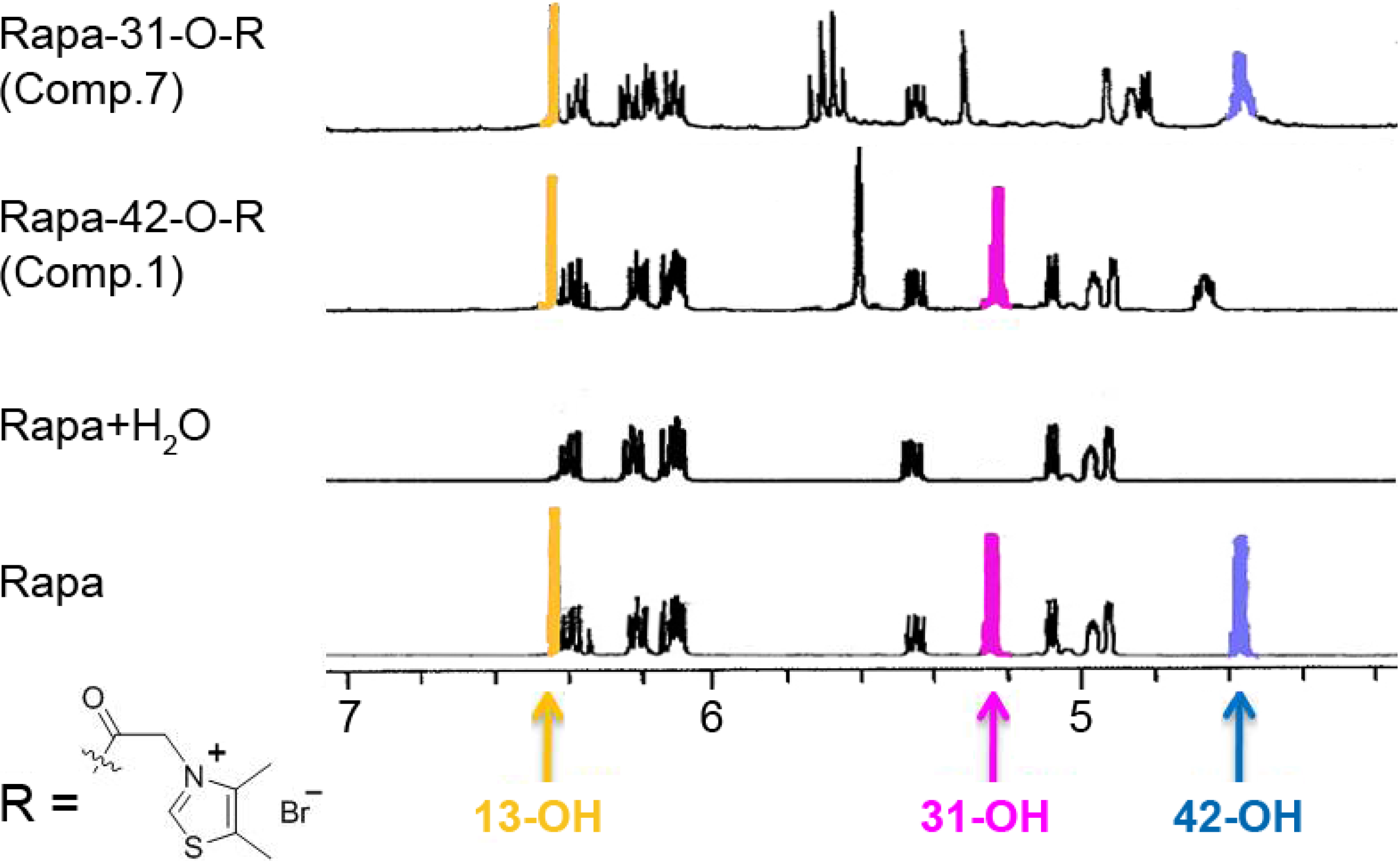{kind=link}
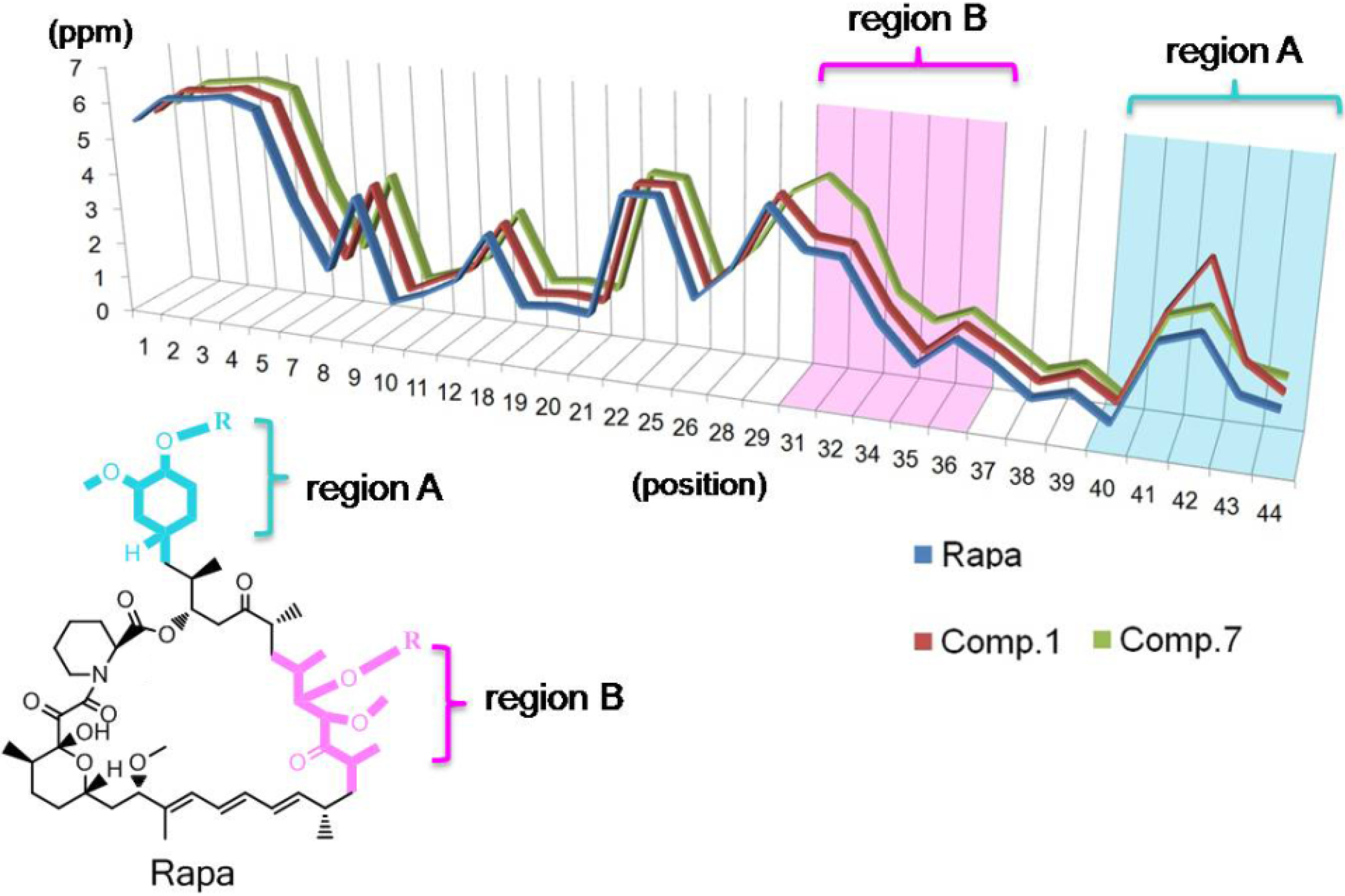{kind=link}
{kind=link}
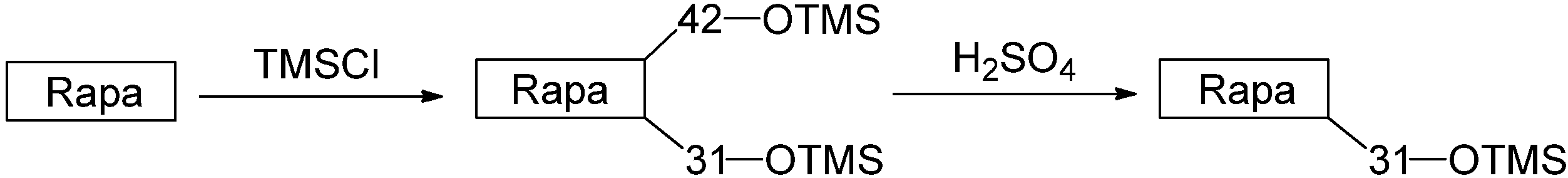{kind=link}
{kind=link}
| Comp. |  | Aqueous Solubility (mg/mL) | Times Increased of Aqueous Solubility (Comp. vs. Rapa) | |
|---|---|---|---|---|
| R1 | R2 | |||
| Rapa | H | H | 0.0026 | 1 |
| 1 | 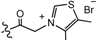 | H | 4.2 | 1615 |
| 2 | 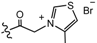 | H | 0.028 | 11 |
| 3 | 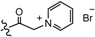 | H | 0.025 | 10 |
| 4 | 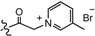 | H | 6.7 | 2577 |
| 5 | 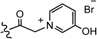 | H | 0.01 | 4 |
| 6 | 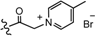 | H | 6 | 2308 |
| 7 | H | 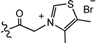 | 23.3 | 8962 |
| 8 | H |  | 10 | 3846 |
| 9 | H |  | 26.7 | 10269 |
| 10 | H | 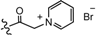 | 52.6 | 20231 |
| 11 | H | 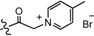 | 62.5 | 24038 |
| C | Rapa | Comp. 1 | Comp. 7 | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 1 | 139.26 | 5.46 | 139.28 | 5.46 | 138.73 | 5.46 |
| 2 | 130.38 | 6.15 | 130.37 | 6.15 | 130.64 | 6.12 |
| 3 | 132.28 | 6.22 | 132.33 | 6.22 | 132.23 | 6.25 |
| 4 | 126.99 | 6.37 | 126.94 | 6.37 | 127.18 | 6.37 |
| 5 | 126.94 | 6.11 | 126.87 | 6.11 | 126.94 | 6.20 |
| 6 | 137.81 | — | 137.83 | — | 138.03 | — |
| 7 | 82.22 | 3.61 | 82.19 | 3.61 | 81.91 | 3.63 |
| 8 | 39.91 | 1.24,1.83 | 39.92 | 1.21,1.83 | 40.03 | 1.08,1.84 |
| 9 | 66.18 | 4.01 | 66.17 | 4.06 | 66.00 | 4.03 |
| 10 | 29.60 | 1.80,1.16 | 29.65 | 1.80,1.17 | 29.54 | 1.16,1.88 |
| 11 | 31.08 | 0.83,1.51 | 30.27 | 0.97,1.61 | 31.02 | 0.82,1.49 |
| 12 | 34.76 | 2.01 | 34.77 | 2.01 | 34.57 | 1.99 |
| 13 | 98.97 | — | 98.97 | — | 98.91 | — |
| 13-OH | — | 6.44 | — | 6.49 | — | 6.45 |
| 15 | 198.84 | — | 198.76 | — | 198.21 | — |
| 16 | 166.95 | — | 166.97 | — | 167.00 | — |
| 18 | 43.45 | 3.41,3.15 | 43.46 | 3.42,3.15 | 43.67 | 2.99,3.43 |
| 19 | 24.42 | 1.55,1.26 | 24.33 | 1.28,1.56 | 24.41 | 1.28,1.57 |
| 20 | 20.32 | 1.65,1.38 | 20.31 | 1.65,1.38 | 20.40 | 1.43,1.69 |
| 21 | 26.39 | 2.08,1.56 | 26.40 | 1.59,2.08 | 26.27 | 1.60,2.20 |
| 22 | 50.72 | 4.93 | 50.81 | 4.93 | 51.25 | 4.93 |
| 23 | 169.15 | — | 169.24 | — | 169.46 | — |
| 25 | 73.55 | 4.98 | 73.57 | 4.97 | 73.56 | 4.87 |
| 26 | 39.50 | 2.72,2.37 | 39.50 | 2.79,2.38 | 40.03 | 2.34,2.66 |
| 27 | 207.48 | — | 207.47 | — | 207.03 | — |
| 28 | 45.16 | 3.25 | 45.18 | 3.24 | 45.07 | 3.20 |
| 29 | 124.93 | 5.08 | 124.73 | 5.08 | 124.30 | 4.81 |
| 30 | 137.09 | — | 137.06 | — | 138.03 | — |
| 31-OH | — | 5.25 | — | 5.23 | — | — |
| 31 | 75.70 | 4.01 | 75.77 | 4.06 | 78.51 | 5.35 |
| 32 | 85.52 | 3.94 | 85.41 | 3.97 | 80.89 | 4.54 |
| 33 | 210.47 | — | 210.31 | — | 208.09 | — |
| 34 | 39.37 | 2.39 | 39.36 | 2.43 | 39.92 | 2.48 |
| 35 | 39.22 | 1.39,1.02 | 39.22 | 1.39,1.02 | 40.02 | 1.07,1.83 |
| 36 | 35.15 | 2.20 | 35.11 | 2.22 | 35.05 | 2.24 |
| 37 | 33.34 | 1.66 | 33.27 | 1.67 | 33.57 | 1.59 |
| 38 | 38.36 | 1.03,0.94 | 37.98 | 0.99,1.06 | 38.24 | 0.92,0.99 |
| 39 | 32.50 | 1.23 | 31.81 | 1.35 | 32.42 | 1.22 |
| 40 | 35.41 | 1.88,0.58 | 34.93 | 2.05,0.73 | 35.26 | 0.51,1.87 |
| 41 | 83.71 | 2.81 | 79.85 | 3.15 | 83.68 | 2.76 |
| 42 | 73.55 | 3.15 | 78.14 | 4.67 | 73.15 | 3.10 |
| 42-OH | — | 4.57 | — | — | — | 4.56 |
| 43 | 32.86 | 1.73,1.15 | 35.11 | 2.01,2.21 | 32.75 | 1.16,1.74 |
| 44 | 31.08 | 1.52 | 26.40 | 1.55 | 31.02 | 1.56 |


2.4. Aqueous Solubility
3. Experimental
3.1. Physical Measurements
3.2. Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gomez-Pinillos, A.; Ferrari, A.C. mTOR signaling pathway and mTOR inhibitors in Cancer therapy. Hematol. Oncol. Clin. N. 2012, 26, 483–505. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Huang, S.L. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin. J. Cancer 2012, 31, 8–18. [Google Scholar]
- Lv, X.; Ma, X.; Hu, Y. Furthering the design and the discovery of small molecule ATP-competitive mTOR inhibitors as an effective cancer treatment. Expert Opin. Drug Discov. 2013, 8, 991–1012. [Google Scholar]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Simamora, P.; Alvarez, J.M.; Yalkowsky, S.H. Solubilization of rapamycin. Int. J. Pharm. 2001, 213, 25–29. [Google Scholar] [CrossRef]
- Crowe, A.; Bruelisauer, A.; Duerr, L. Absorption and intestinal metabolism of SDZ-RAD and rapamycin in rats. Drug Metab. Dispos. 1999, 27, 627–632. [Google Scholar]
- Gu, J.X.; Ruppen, M.E. Proline CCI-779, Production of and Uses Therefor, and Two-step Enzymatic Synthesis of Proline CCI-779 and CCI-779. US Patent 20050234086A1, 12 April 2005. [Google Scholar]
- Shaw, C.C. Treating Rapamycin with Chlorotrimethylsilane in an Inert Solvent in the Presence of a Suitable Base to Provide Rapamycin 31,42-bis-Trimethylsilyl ether; Treating with Dilute Acid to prOvide rapamycin 31-Trimethylsilyl Ether. US Patent 6277983, 21 August 2001. [Google Scholar]
- Niznansky, D.; Rehspringer, J.L. Infrared study of SiO 2 sol to gel evolution and gel aging. J. Non-Cryst. Solids 1995, 180, 191–196. [Google Scholar] [CrossRef]
- Yokogawa, H.; Yokogawa, M. Hydrophobic silica aerogels. J. Non-Cryst. Solids 1995, 186, 23–29. [Google Scholar]
- Latthe, S.S.; Nadargi, D.Y. TMOS based water repellent silica thin films by co-precursor method using TMES as a hydrophobic agent. Appl. Surf. Sci. 2009, 225, 3600–3604. [Google Scholar] [CrossRef]
- Orcel, O.; Hench, L.L. Effect of formamide additive on the chemistry of silica sol-gels II. gel structure. J. Non-Cryst. Solids 1988, 105, 223–231. [Google Scholar] [CrossRef]
- Cao, S.; Zhou, X.B.; Zhang, H. Novel cross-link breaker based on zwitterion structure: Synthesis, structure and druggability studies. Eur. J. Med. Chem. 2013, 68, 89–95. [Google Scholar] [CrossRef]
- Gupta, N. Process for the Preparation of Temsirolimus and Its Intermediates. US Patent 20110184167A1, 28 July 2011. [Google Scholar]
- Lee, K.C. Process for Preparation of Temsirolimus. US Patent 20100249415A1, 30 September 2010. [Google Scholar]
- Swindells, D.C.; White, P.S.; Findlay, J.A. The X-ray crystal structure of rapamycin, C51H79NO13. Can. J. Chem. 1978, 56, 2491–2492. [Google Scholar] [CrossRef]
- Xie, L.; Huang, J.; Zuo, J. 41-Azido-41-de-oxy-rapamycin. Acta Crystallogr. E 2012, 68, (Pt. 4). o1206–o1207. [Google Scholar] [CrossRef]
- Xie, L.; Zuo, J.; Yang, G. 40-De-oxy-40(S)-iodo-rapamycin. Acta Crystallogr. E 2012, 68, (Pt. 8). o2331. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–11 are available from the authors.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cao, S.; Zhou, X.; Yang, Y.; Zhong, W.; Sun, T. Selective Substitution of 31/42–OH in Rapamycin Guided by an in Situ IR Technique. Molecules 2014, 19, 7770-7784. https://doi.org/10.3390/molecules19067770
Cao S, Zhou X, Yang Y, Zhong W, Sun T. Selective Substitution of 31/42–OH in Rapamycin Guided by an in Situ IR Technique. Molecules. 2014; 19(6):7770-7784. https://doi.org/10.3390/molecules19067770
Chicago/Turabian StyleCao, Shuang, Xinbo Zhou, Yuanshuai Yang, Wu Zhong, and Tiemin Sun. 2014. "Selective Substitution of 31/42–OH in Rapamycin Guided by an in Situ IR Technique" Molecules 19, no. 6: 7770-7784. https://doi.org/10.3390/molecules19067770