Elucidation of the Relationships between H-Bonding Patterns and Excited State Dynamics in Cyclovalone


Abstract
:1. Introduction

2. Results and Discussion
2.1. IR Spectroscopy and H-Bonding Patterns

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ν(O-H) cm−1 | |||
|---|---|---|---|
| Free | Intra H-Bond | Inter H-Bond | |
| KBr disk | 3370 | ||
| Nujol | 3375 | ||
| Thin film | 3378 | ||
| Carbon tetrachloride | 3553 | ||
| Dichloromethane | 3530 | ||
| Acetonitrile | 3636 | 3542 | 3392 |
| Acetone | 3620 * | 3540 * | 3350 |
| Dimethylsulfoxide | 3450 * | 3080 | |

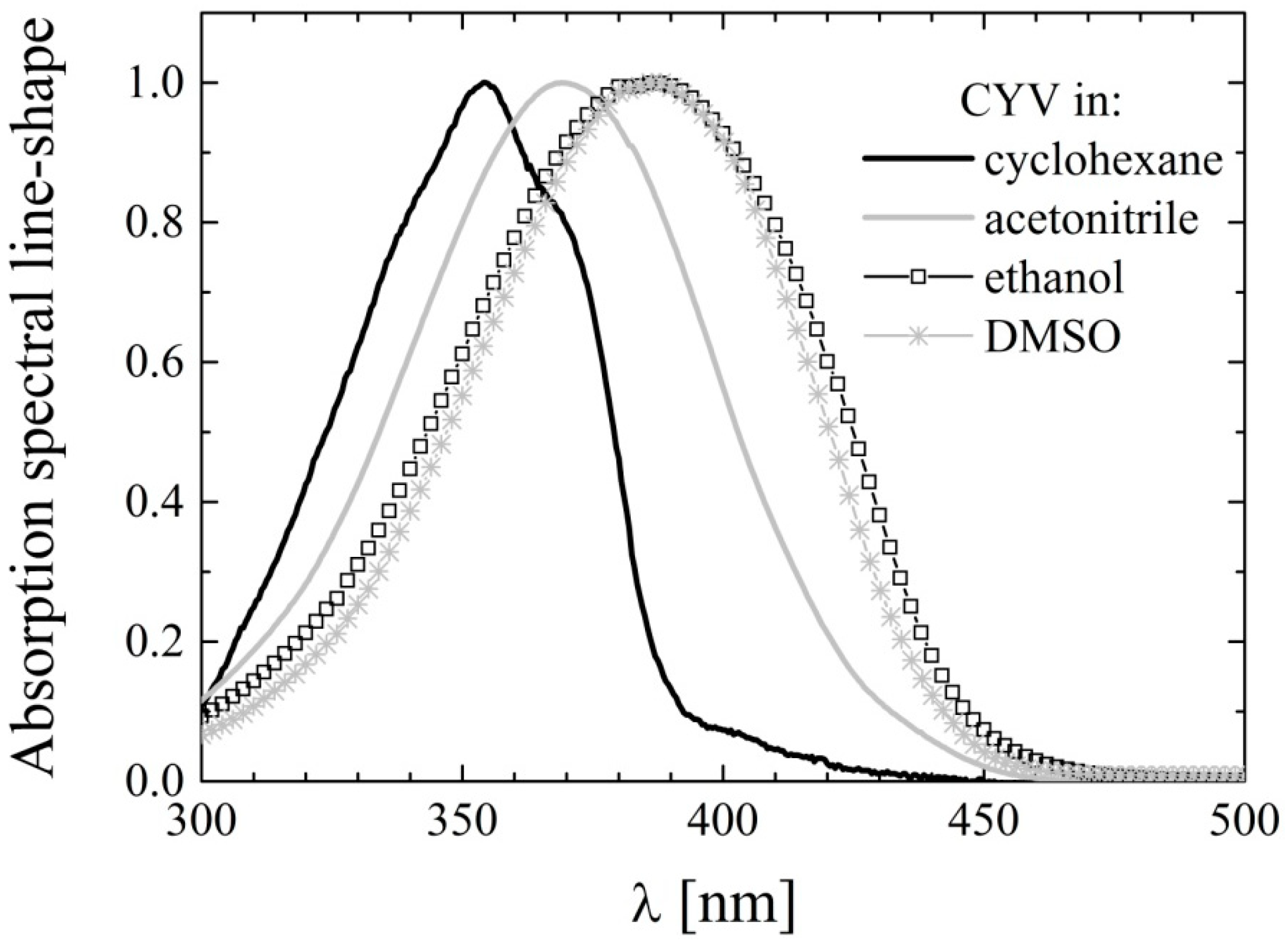
2.2. UV-Vis Absorption Spectroscopy
| Solvent | λAbs (nm) | λCURC (nm) | |
|---|---|---|---|
| Non Polar | Cyclohexane | 355 | 408,429 |
| Polar weakly H-bonding | Chloroform | 372 | 419 |
| Ethyl acetate | 367 | 419 | |
| Acetone | 370 | 420 | |
| Acetonitrile | 370 | 419 | |
| Strong H-bond acceptors | DMFA | 383 | 431 |
| DMSO | 386 | 434 | |
| Alcohols | Ethanol | 387 | 430 |
| Methanol | 385 | 423 | |

2.3. Steady-State Fluorescence
| Solvent | λAbs (nm) (ΦFluor) | λCURC (nm) (ΦCURC) | |
|---|---|---|---|
| Non polar | Cyclohexane | 481 ([4 ± 1] × 10−5) | 502, 471, 443 (0.006 ± 0.003) |
| Polar weakly H-bonding | Chloroform | 478 | 503 |
| Ethyl acetate | 487 | 494 | |
| Acetone | 502 | 510 | |
| Acetonitrile | 496 ([4 ± 1] × 10−5) | 521 (0.156 ± 0.003) | |
| Strong H-bond acceptors | DMFA | 514 | 536 |
| DMSO | 511 ([4.4 ± 0.5] × 10−4) | 550 (0.026 ± 0.002) | |
| Alcohols | Ethanol | 509 ([1.2 ± 0.1] × 10−3) | 553 (0.033 ± 0.003) |
| Methanol | 518 ([1.4 ± 0.2] × 10−3) | 566 (0.028 ± 0.002) | |

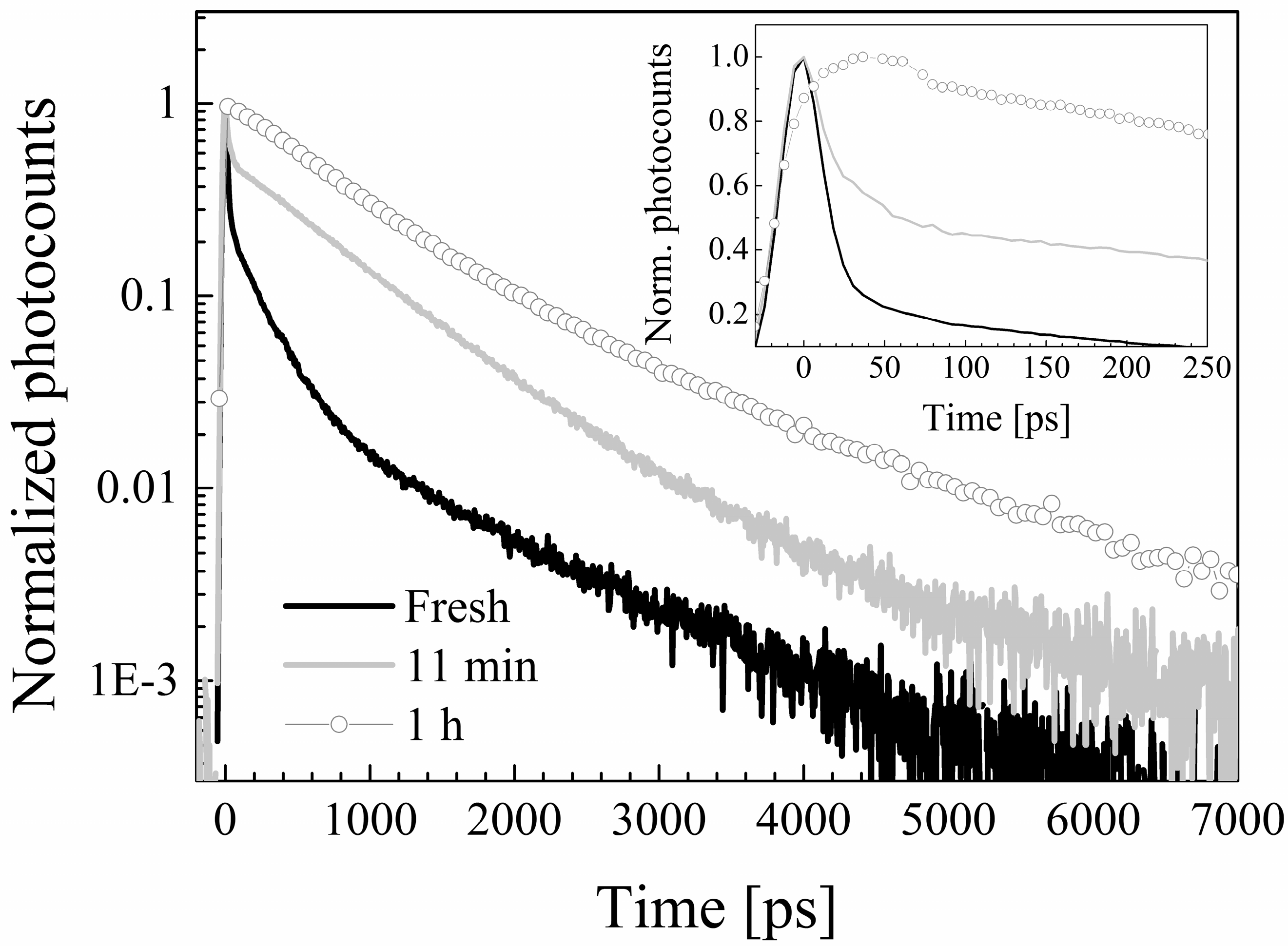
2.4. Photodegradation Studies


2.5. Time-Resolved Fluorescence
| Solvent | τ1 [ps] (A1) | τ2 [ps] (A2) | τ3 [ps] (A3) | τ4 [ps] (A4) | τ5 [ps] (A5) | τAv [ps] |
|---|---|---|---|---|---|---|
| Cyclohexane (exc. 355 nm) | - | 188 ± 5 (0.89) | - | - | 2416 ± 68 (0.11) | 433 |
| Chloroform | 11 ± 2 (0.84) | 203 ± 4 (0.14) | 807 ± 42 (0.02) | 2194 ± 212 (<0.01) | 391 (75) | |
| Ethyl acetate | - | - | - | 1074 ± 9 (0.57) | 4088 ± 38 (0.43) | 2370 |
| Ethyl acetate (exc. 355 nm) | - | - | - | 1004 ± 40 (0.56) | 3804 ± 370 (0.44) | 2236 |
| Acetone | 34 ± 2 (0.80) | - | - | 965 ± 6 (0.09) | 3852 ± 23 (0.11) | 2553 (538) |
| Acetonitrile | 10 ± 1 (0.86) | 222 ± 0 (0.14) | - | 2795 ± 160 (<0.01) | 394 (67) | |
| DMFA | 24 ± 1 (0.58) | - | 190 ± 12 (0.08) | 765 ± 8 (0.19) | 4500 ± 12 (0.15) | 1989 (849) |
| DMSO | 38 ± 1 (0.71) | - | 208 ± 2 (0.26) | 1021 ± 57 (0.02) | 5032 ± 91 (0.01) | 430 (152) |
| Ethanol | 35 ± 2 (0.63) | - | 232 ± 3 (0.14) | 1340 ± 15 (0.03) | 5419 ± 19 (0.20) | 3126 (1179) |
| Ethanol (exc. 355 nm) | 36 ± 1 (0.78) | - | 248 ± 6 (0.12) | 1394 ± 30 (0.06) | 5500 ± 100 (0.04) | 361 (1515) |
| Methanol | 23 ± 1 (0.82) | - | 223 ± 2 (0.16) | 1374 ± 134 (0.01) | 5470 ± 173 (0.01) | 578 (123) |
| Methanol (exc. 355 nm) | 18 ± 2 (0.74) | - | 227 ± 18 (0.10) | 1350 ± 46 (0.08) | 5500 ± 150 (0.08) | 2195 (584) |

2.6. Spectrofluorimetric Detection of Photosensitized Reactive Oxygen Species Generation

3. Experimental Section
3.1. Chemicals and Samples Preparation
3.2. IR Absorption Spectra
3.3. UV-Vis Absorption and Fluorescence Spectra, Fluorescence Quantum Yields
3.4. Fluorescence-Decay Measurements
3.5. Photodegradation Quantum Yields and Half-Lives
3.6. Isolation of Photodecomposition Products
3.7. Spectrofluorimetric Detection of Photosensitized Reactive Oxygen Species Generation
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tønnesen, H.H.; Màsson, M.; Loftsson, T. Studies of curcumin and curcuminoids. XXVII. Cyclodextrin complexation: Solubility, chemical and photochemical stability. Int. J. Pharm. 2002, 244, 127–135. [Google Scholar] [CrossRef]
- Sagi, S.S.K.; Mathew, T.; Patir, H. Prophylactic administration of curcumin abates the incidence of hypobaric hypoxia induced pulmonary edema in rats: A molecular approach. J. Pulm. Respir. Med. 2014, 4, 1000164. [Google Scholar]
- Marchiani, A.; Rozzo, C.; Fadda, A.; Delogu, G.; Ruzza, P. Curcumin and curcumin-like molecules: From spice to drugs. Curr. Med. Chem. 2014, 21, 204–222. [Google Scholar]
- Chen, X.; Chenna, V.; Maitra, A.; Devaraj, S. Nanocurcumin attenuates inflammation by decreasing Toll-like receptor 2 and 4 expression and activity and promoting an anti-inflammatory macrophage phenotype. FASEB J. 28 (Suppl.), 830–822.
- Priyadarsini, K.I.; Maity, D.K.; Naik, G.H.; Kumar, M.S.; Unnikrishnan, M.K.; Satav, J.G.; Mohan, H. Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 2003, 35, 475–484. [Google Scholar]
- Ak, T.; Gulcin, I. Antioxidant and radical scavenging properties of curcumin. Chem.-Biol. Interact. 2008, 174, 27–37. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Diduk, R.; Ramirez-Silva, M.T.; Alarcòn-Angeles, G.; Rojas Hernandez, A. Role of the reacting free radicals on the antioxidant mechanism of curcumin. Chem. Phys. 2009, 363, 13–23. [Google Scholar]
- Jayaprakasha, G.K.; Rao, L.J.; Sakarian, K.K. Antioxidant activities of curcumin, demethoxycurcumin and bisdemethoxycurcumin. Food Chem. 2006, 98, 720–724. [Google Scholar]
- Heger, M.; van Golen, R.F.; Michel, M.C. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer. Pharmacol. Rev. 2014, 66, 222–307. [Google Scholar]
- Leu, T.H.; Maa, M.C. The molecular mechanisms for the antitumorigenic effect of curcumin. Curr. Med. Chem. 2002, 2, 357–370. [Google Scholar]
- Woo, J.; Kim, Y.; Choi, Y.; Kim, D.; Lee, K.; Bae, J.H.; Chang, D.S.; Jeong, Y.J.; Lee, Y.H.; Park, J.; et al. Molecular mechanisms of curcumin-induced cyclotoxicity: Induction of apoptosis through generation of reactive oxygen species, down-regulation of Bcl-XL and IAP, the release of cytochrome c and inhibition of Akt. Carcinogenesis 2003, 24, 1199–1208. [Google Scholar] [CrossRef]
- Moos, P.J.; Edes, K.; Mullally, J.; Fitzpatrick, J. Curcumin impairs tumor suppressor p53 function in colon cancer cells. Carcinogenesis 2004, 25, 1611–1617. [Google Scholar]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef]
- Egan, M.E.; Pearson, M.; Weiner, S.A.; Rajendarn, V.; Rubin, D.; Glochner-Pagel, J.; Canney, S.; Du, K.; Lukacs, G.L.; Caplan, M.F. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 2004, 304, 600–602. [Google Scholar]
- Aggrawal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar]
- Kong, L.; Priyadarsini, K.I.; Zhang, H.Y. A theoretical investigation on intramolecular hydrogen-atom transfer in curcumin. J. Mol. Struct. Theochem 2004, 685, 111–116. [Google Scholar]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef]
- Mague, J.T.; Alworth, W.L.; Payton, F.L. Curcumin and derivatives. Acta Cryst. C 2004, 60, 608–610. [Google Scholar]
- Tønnesen, H.H.; Karlsen, J.; Mostad, A. Structural studies of curcuminoids. I. The crystal structure of curcumin. Acta Chem. Scand. B 1982, 36, 475–479. [Google Scholar]
- Cai, Y.Z.; Sun, M.; Xing, J.; Luo, Q.; Corke, H. Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medical plants. Life Sci. 2006, 78, 2872–2888. [Google Scholar]
- Chen, W.F.; Deng, S.L.; Zhou, B.; Yang, L.; Liu, Z.L. Curcumin and its analogues as potent inhibitors of low density lipoprotein oxidation: H-atom abstraction from the phenolic groups and possible involvement of the 4-hydroxy-3-methoxyphenil groups. Free Radic. Biol. Med. 2006, 40, 526–535. [Google Scholar]
- Somparn, P.; Phisalaphong, C.; Nakornchai, S.; Unchern, S.; Morales, N.P. Comparative antioxidant activities of curcumin and its demethoxy and hydrogenated derivatives. Biol. Pharm. Bull. 2007, 30, 74–78. [Google Scholar]
- Dairam, A.; Limson, J.L.; Watkins, G.M.; Antunes, E.; Daya, S. Curcuminoids, curcumin, and demethoxycurcumin reduce lead-induced memory deficits in male wistar rats. J. Agric. Food Chem. 2007, 55, 1039–1044. [Google Scholar] [CrossRef]
- Dahl, T.A.; Mcgowan, W.M.; Shand, M.A.; Srinivasan, V.S. Photokilling of bacteria by the natural dye curcumin. Arch. Microbiol. 1989, 151, 183–185. [Google Scholar]
- Tønnesen, H.H.; de Vries, H.; Karlsen, J.; van Henegouwen, G.B. Studies on curcumin and curcuminoids IX: Investigation of the photobiological activity of curcumin using bacterial indicator systems. J. Pharm. Sci. 1987, 76, 371–373. [Google Scholar]
- Haukvik, T.; Bruzell, E.; Kristensen, S.; Tønnesen, H.H. Photokilling of bacteria by curcumin in different aqueous preparations. Studies on curcumin and curcuminoids. XXXVII. Pharmazie 2009, 64, 666–673. [Google Scholar]
- Haukvik, T.; Bruzell, E.; Kristensen, S.; Tønnesen, H.H. Photokilling of bacteria by curcumin in selected polyethylene glycol 400 (PEG 400) preparations studies on curcumin and curcuminoids XLI. Pharmazie 2010, 65, 600–606. [Google Scholar]
- Hegge, A.B.; Andersen, T.; Melvik, J.E.; Bruzell, E.; Kristensen, S.; Tønnesen, H.H. Formulation and bacterial phototoxicity of curcumin loaded alginate foams for wound applications studies on curcumin and curcumioids. XLII. J. Pharm. Sci. 2011, 100, 174–185. [Google Scholar] [CrossRef]
- Haukvik, T.; Bruzell, E.; Kristensen, S.; Tønnesen, H.H. A Screening for antibacterial phototoxic effects of curcumin derivatives studies on curcumin and curcuminoids. XLIII. Pharmazie 2011, 66, 69–77. [Google Scholar]
- Hegge, A.B.; Nielsen, T.T.; Larsen, K.L.; Bruzell, E.; Tønnesen, H.H. Impact of curcumin supersaturation in antibacterial photodynamic therapy (aPDT)-Effect of cyclodextrin type and amount, studies on curcumin and curcuminoids XLV. J. Pharm. Sci. 2012, 101, 1524–1537. [Google Scholar]
- Hegge, A.B.; Bruzell, E.; Kristensen, S.; Tønnesen, H.H. Photoinactivation of Staphylococcus epidermidis biofilms and suspensions by the hydrophobic photosensitizer curcumin-Effect of selected nanocarrier. Studies on curcumin and curcuminoides XLVII. Eur. J. Pharm. Sci. 2012, 47, 65–74. [Google Scholar] [CrossRef]
- Singh, R.; Kristensen, S.; Tønnesen, H.H.; Berg, K. The influence of Pluronics on dark cytotoxicity, photocytotoxicity, localization and uptake of curcumin in cancer cells. Studies on curcumin and curcuminoids XLIX. Photochem. Photobiol. Sci. 2013, 12, 559–575. [Google Scholar]
- Hegge, A.B.; Vukicevic, M.; Bruzell, E.; Kristensen, S.; Tønnesen, H.H. Solid dispersions for preparation of phototoxic supersaturated solutions for antimicrobial photodynamic therapy (aPDT). Studies on curcumin and curcuminoids L. Eur. J. Pharm. Biopharm. 2013, 83, 95–105. [Google Scholar] [CrossRef]
- Dahl, T.A.; Bilski, P.; Reszka, K.J.; Chignell, C.F. Photocytotoxicity of curcumin. Photochem. Photobiol. 1994, 59, 290–294. [Google Scholar]
- Bruzell, E.; Morisbak, E.; Tønnesen, H.H. Studies on curcumin and curcuminoids. XXIX. Photoinduced cytotoxicity of curcumin in selected aqueous preparations. Photochem. Photobiol. Sci. 2005, 4, 523–530. [Google Scholar] [CrossRef]
- Nardo, L.; Paderno, R.; Andreoni, A.; Màsson, M.; Haukvik, T.; Tønnesen, H.H. Role of H-bond formation in the photoreactivity of curcumin. Spectroscopy 2008, 22, 187–198. [Google Scholar]
- Nardo, L.; Andreoni, A.; Bondani, M.; Màsson, M.; Tønnesen, H.H. Photophysical properties of a symmetrical, non-substituted curcumin analogue. J. Photochem. Photobiol. Sci. B Biol. 2009, 97, 77–86. [Google Scholar]
- Nardo, L.; Andreoni, A.; Haukvik, T.; Màsson, M.; Tønnesen, H.H. Photophysical properties of bis-demethoxy-curcumin. J. Fluorescence 2011, 21, 627–635. [Google Scholar]
- Nardo, L.; Andreoni, A.; Bondani, M.; Màsson, M.; Tønnesen, H.H. Photophysical properties of dimethoxycurcumin and bis-dehydroxycurcumin. J. Fluorescence 2012, 22, 597–608. [Google Scholar]
- Nardo, L.; Maspero, A.; Selva, M.; Bondani, M.; Palmisano, G.; Ferrari, E.; Saladini, M. Excited-state dynamics of bis-dehydroxycurcumin carboxylic acid, a water-soluble derivative of the photosensitizer curcumin. J. Phys. Chem. A 2012, 116, 9321–9330. [Google Scholar]
- French, H.S.; Holden, M.G.T. Absorption Spectra of Certain α,β-Unsaturated Ketones, including Benzal Compounds. J. Am. Chem. Soc. 1945, 67, 1239–1242. [Google Scholar]
- Huitric, A.C.; Kumler, W.D. The dipole moments, spectra and structure of some new 2-phenyl-, 2-benzyl-, 2-(p-halobenzy1idene)- and 2,6-Bis-(p-halobenzy1idene)-cyclohexanones. J. Am. Chem. Soc. 1956, 78, 614–622. [Google Scholar] [CrossRef]
- Issa, R.M.; Etaiw, S.H.; Issa, I.M.; El-Shafie, A.K. Electronic absorption spectra of some diarylidene-cyclopentanones and-cyclohexanones. Acta Chim. (Budapest) 1976, 89, 381–391. [Google Scholar]
- Connors, R.E.; Ucak-Astarlioglu, M.G. Electronic absorption and fluorescence properties of 2,5-diarylidene-cyclopentanones. J. Phys. Chem. A 2003, 107, 7684–7691. [Google Scholar]
- Ucak-Astarlioglu, M.G.; Connors, R.E. Absorption and fluorescence of 2,5-diarylidenecyclopentanones in acidic media: Evidence for excited-state proton transfer. J. Phys. Chem. A 2005, 109, 8275–8279. [Google Scholar]
- Itokawa, H.; Shi, Q.; Akiyama, T.; Morris-Natschke, S.L.; Lee, K.-H. Recent advantages in the investigation of curcuminoids. Chin. Med. 2008, 3, 11. [Google Scholar]
- Artico, M.; di Santo, R.; Costi, R.; Novellino, E.; Greco, G.; Massa, S.; Tramontano, E.; Marongiu, M.E.; de Montis, A.; la Colla, P. Geometrically and conformationally restrained cinnamoyl compounds as inhibitors of HIV-1 integrase: Synthesis, biological evaluation, and molecular modeling. J. Med. Chem. 1998, 41, 3948–3960. [Google Scholar]
- Kamlet, M.J.; Abboud, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, π*, α, and, β, and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar]
- Chignell, C.F.; Bilski, P.; Reszka, K.J.; Motton, A.G.; Sik, R.H.; Dahl, T.A. Spectral and photochemical properties of curcumin. Photochem. Photobiol. 1994, 59, 295–302. [Google Scholar]
- Shen, L.; Ji, H.F. Theoretical study on physicochemical properties of curcumin. Spectrochim. Acta A 2007, 67, 619–623. [Google Scholar]
- El Sayed, M.A. Spin orbit coupling and the radiationless processes in nitrogen heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar]
- Lilletvedt, M.; Tønnesen, H.H.; Høgset, A.; Kristensen, S.; Nardo, L. Time-domain evaluation of drug-solvent interactions of the photosensitizers TPCS2a and TPPS2a as part of physicochemical characterization. J. Photochem. Photobiol. A Chem. 2010, 214, 40–47. [Google Scholar]
- Maspero, A.; Giovenzana, G.B.; Masciocchi, N.; Palmisano, G.; Comotti, A.; Sozzani, P.; Bassanetti, I.; Nardo, L. Mohlau’s anthradipyrazole revisited: A new look at an old molecular system. Cryst. Growth Des. 2013, 13, 4948–4956. [Google Scholar]
- Zhang, X.-F.; Li, X. The photostability and fluorescence properties of diphenylisobenzofuran. J. Lumin. 2011, 131, 2263–2266. [Google Scholar]
- Gomes, A.; Fernandes, E.; Lima, J.L.F.C. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar]
- Berlman, I.B. Handbook of Fluorescence Spectra of Aromatic Molecules; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Moore, D.E. Standardization of kinetic studies of photodegradation reactions. In Photostability of Drugs and Drug Formulations; Tønnesen, H.H., Ed.; CRC Press: Boca Raton, FL, USA, 2004; pp. 49–53. [Google Scholar]
- Sample Availability: Samples of CURC are available from the authors. The CYV specimens used in this work are commercially available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lamperti, M.; Maspero, A.; Tønnesen, H.H.; Bondani, M.; Nardo, L. Elucidation of the Relationships between H-Bonding Patterns and Excited State Dynamics in Cyclovalone. Molecules 2014, 19, 13282-13304. https://doi.org/10.3390/molecules190913282
Lamperti M, Maspero A, Tønnesen HH, Bondani M, Nardo L. Elucidation of the Relationships between H-Bonding Patterns and Excited State Dynamics in Cyclovalone. Molecules. 2014; 19(9):13282-13304. https://doi.org/10.3390/molecules190913282
Chicago/Turabian StyleLamperti, Marco, Angelo Maspero, Hanne H. Tønnesen, Maria Bondani, and Luca Nardo. 2014. "Elucidation of the Relationships between H-Bonding Patterns and Excited State Dynamics in Cyclovalone" Molecules 19, no. 9: 13282-13304. https://doi.org/10.3390/molecules190913282