
Anti-Inflammatory Effects of Vinpocetine in Atherosclerosis and Ischemic Stroke: A Review of the Literature

{kind=link}
{kind=link}
{kind=link}
Abstract
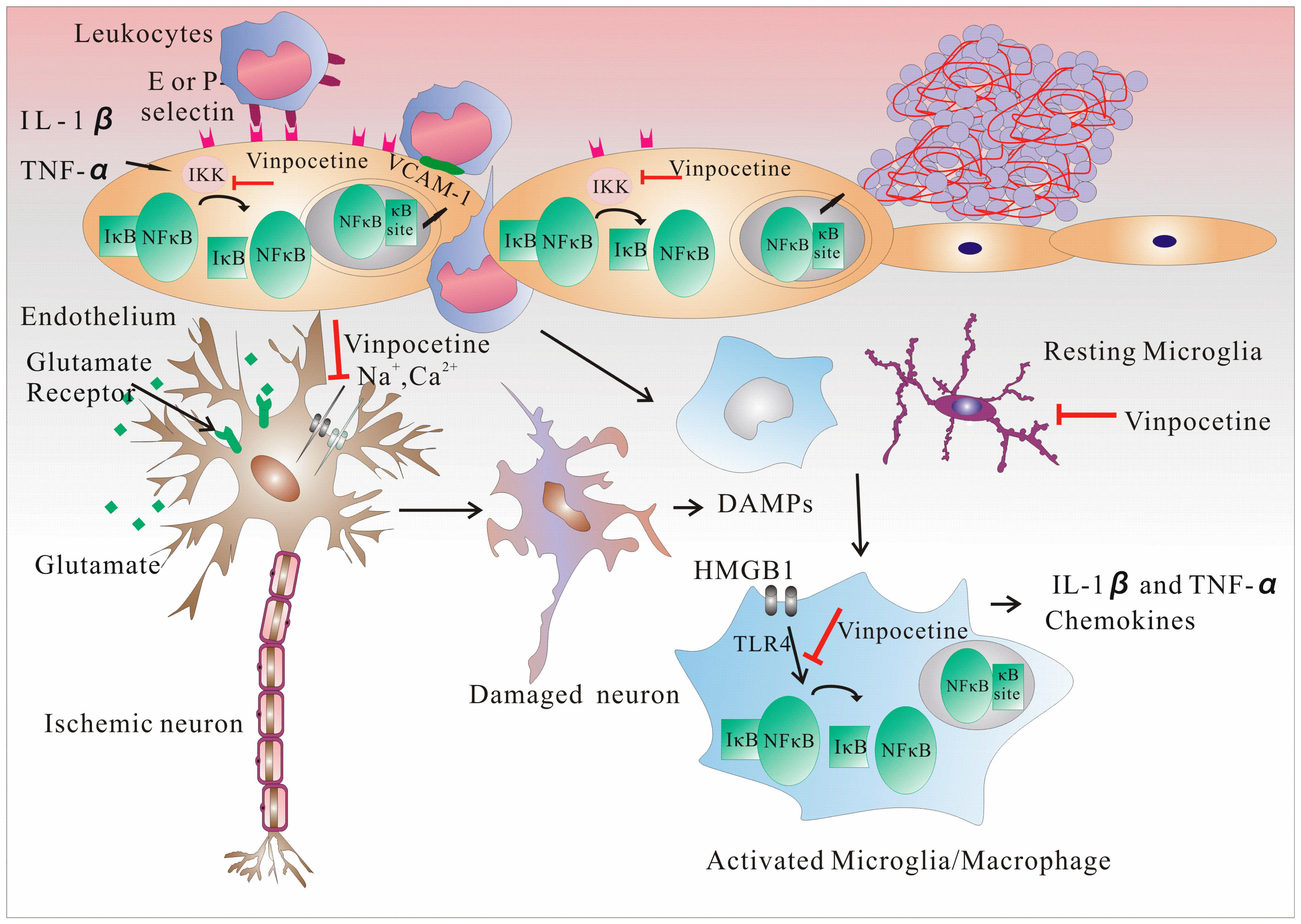
:1. Introduction

2. Anti-Inflammatory Effects of Vinpocetine in Atherosclerosis and Ischemic Stroke
2.1. Anti-Inflammatory Effects of Vinpocetine in Atherosclerosis
2.1.1. Vinpocetine Inhibits the Progression of Atherosclerosis
2.1.2. Vinpocetine and the Adaptive Immune Response in Atherosclerosis

2.2. Anti-Inflammatory Effects of Vinpocetine in Ischemic Stroke
2.2.1. Vinpocetine Inhibits Early Inflammation in Ischemic Stroke by Inhibiting IKK/NF-κB

2.2.2. Vinpocetine Inhibits the Proliferation of Microglia by Inhibiting IKK/NF-κB
2.2.3. Vinpocetine and the Adaptive Immune Response after Ischemic Stroke
3. Conclusions
Acknowledgments
Author Contributions
Abbreviation
| AP-1 | activator protein-1 |
| CNS | central nervous system |
| DAMPs | damaged neurons release danger-associated molecular patterns |
| ERK1/2 | extracellular signal-regulated protein kinases 1 and 2 |
| HUVECs | human umbilical vein endothelial cells |
| HSP | heat shock protein |
| HMGB1 | high-mobility group box 1 protein |
| IL | interleukin |
| I/R | ischemia and reperfusion |
| TNF | tumor necrosis factor |
| IκB | inhibitor κB |
| IKK | IκB kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| NF-κB | nuclear factor κ-light-chain-enhancer of activated B cells κB |
| NAD | nicotinamide adenine dinucleotide |
| ox-LDL | oxidized low-density lipoprotein |
| PDE1 | phosphodiesterase type 1 |
| PAMPs | pathogen-associated molecular patterns |
| PDGF | platelet-derived growth factor |
| TLRs | Toll-like receptors |
| ROS | reactive oxygen species |
| TSPO | translocator protein |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VSMCs | vascular smooth muscle cells |
Conflicts of Interest
References
- WHO. The Top 10 Causes of Death. Available online: http://www.who.int/mediacentre/factsheets/ (accessed on 8 June 2014).
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [PubMed]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics–2013 update: A report from the american heart association. Circulation 2013, 127, e6–e245. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. Toast. Trial of org 10172 in acute stroke treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis–An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation and immune response in atherosclerosis. Curr. Atheroscler. Rep. 1999, 1, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E. Injury and repair mechanisms in ischemic stroke: Considerations for the development of novel neurotherapeutics. Curr. Opin. Investig. Drugs 2009, 10, 644–654. [Google Scholar] [PubMed]
- Macrez, R.; Ali, C.; Toutirais, O.; le Mauff, B.; Defer, G.; Dirnagl, U.; Vivien, D. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol. 2011, 10, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Engelhardt, B.; Koistinaho, J.; Lindvall, O.; Meairs, S.; Mohr, J.P.; Planas, A.; Rothwell, N.; Schwaninger, M.; Schwab, M.E.; et al. Improving outcome after stroke: Overcoming the translational roadblock. Cerebrovasc. Dis. 2008, 25, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.N.; Buggey, H.F.; Denes, A.; Allan, S.M. Systemic immune activation shapes stroke outcome. Mol. Cell. Neurosci. 2013, 53, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Dabek, J.; Kulach, A.; Gasior, Z. Nuclear factor κ-light-chain-enhancer of activated b cells (NF-κB): A new potential therapeutic target in atherosclerosis? Pharmacol. Rep. 2010, 62, 778–783. [Google Scholar] [CrossRef]
- Xu, L.; Zhan, Y.; Wang, Y.; Feuerstein, G.Z.; Wang, X. Recombinant adenoviral expression of dominant negative IκBα protects brain from cerebral ischemic injury. Biochem. Biophys. Res. Commun. 2002, 299, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Potrovita, I.; Tarabin, V.; Herrmann, O.; Beer, V.; Weih, F.; Schneider, A.; Schwaninger, M. Neuronal activation of NF-κB contributes to cell death in cerebral ischemia. J. Cereb. Blood Flow Metab. 2005, 25, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Ning, M.; Zhou, Y.; Chen, G.; Mei, X. Preparation and in vitro/in vivo evaluation of vinpocetine elementary osmotic pump system. Adv. Pharmacol. Sci. 2011, 2011. [Google Scholar] [CrossRef]
- Vinpocetine. Monograph. Altern. Med. Rev. 2002, 7, 240–243.
- Jeon, K.I.; Xu, X.; Aizawa, T.; Lim, J.H.; Jono, H.; Kwon, D.S.; Abe, J.; Berk, B.C.; Li, J.D.; Yan, C. Vinpocetine inhibits NF-κB-dependent inflammation via an ikk-dependent but pde-independent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 9795–9800. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995, 75, 725–748. [Google Scholar] [PubMed]
- Zhao, Y.Y.; Yu, J.Z.; Li, Q.Y.; Ma, C.G.; Lu, C.Z.; Xiao, B.G. Tspo-specific ligand vinpocetine exerts a neuroprotective effect by suppressing microglial inflammation. Neuron Glia Biol. 2011, 7, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Torres, K.J.; Gottle, P.; Kremer, D.; Rivera, J.F.; Aguirre-Cruz, L.; Corona, T.; Hartung, H.P.; Kury, P. Vinpocetine inhibits oligodendroglial precursor cell differentiation. Cell. Physiol. Biochem. 2012, 30, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Moroni, F.; Magnoni, M.; Camici, P.G. The role of T and B cells in human atherosclerosis and atherothrombosis. Clin. Exp. Immunol. 2014. [Google Scholar] [CrossRef]
- Dong, Z.M.; Chapman, S.M.; Brown, A.A.; Frenette, P.S.; Hynes, R.O.; Wagner, D.D. The combined role of p- and e-selectins in atherosclerosis. J. Clin. Investig. 1998, 102, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenberg, J.F.; Smeets, E.F.; Neefjes, J.J.; Shaffer, M.A.; Cinek, T.; Jeunhomme, T.M.; Ahern, T.J.; Buurman, W.A. E-selectin and intercellular adhesion molecule-1 are released by activated human endothelial cells in vitro. Immunology 1992, 77, 543–549. [Google Scholar] [PubMed]
- Poole, J.C.; Florey, H.W. Changes in the endothelium of the aorta and the behaviour of macrophages in experimental atheroma of rabbits. J. Pathol. Bacteriol. 1958, 75, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Cybulsky, M.I. NF-κB: Pivotal mediator or innocent bystander in atherogenesis? J. Clin. Investig. 2001, 107, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Berdeaux, A.; Loueslati, E.; Gerard, J.L.; Pussard, E.; Giudicelli, J.F. Evaluation of the natriuretic and beta-adrenoceptor-blocking effects of tienoxolol in normal volunteers. Fundam. Clin. Pharmacol. 1988, 2, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Zhang, Y.; Georgescu, S.P.; Johnson, K.L.; Kong, D.; Galper, J.B. Human umbilical vein endothelial cells and human dermal microvascular endothelial cells offer new insights into the relationship between lipid metabolism and angiogenesis. Stem Cell Rev. 2006, 2, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Peng, W.; Li, H.; Lu, Y.; Wang, K.; Fan, F.; Li, S.; Xu, Y. Inhibitory effects of vinpocetine on the progression of atherosclerosis are mediated by AKT/NF-κB dependent mechanisms in apoe-/-mice. PLoS One 2013, 8, e82509. [Google Scholar] [CrossRef]
- Cai, Y.; Knight, W.E.; Guo, S.; Li, J.D.; Knight, P.A.; Yan, C. Vinpocetine suppresses pathological vascular remodeling by inhibiting vascular smooth muscle cell proliferation and migration. J. Pharmacol. Exp. Ther. 2012, 343, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Gerthoffer, W.T. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Bonoczk, P.; Gulyas, B.; Adam-Vizi, V.; Nemes, A.; Karpati, E.; Kiss, B.; Kapas, M.; Szantay, C.; Koncz, I.; Zelles, T.; et al. Role of sodium channel inhibition in neuroprotection: Effect of vinpocetine. Brain Res. Bull. 2000, 53, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, G.; Nagy, Z.; Balkay, L.; Boros, I.; Emri, M.; Lehel, S.; Marian, T.; Molnar, T.; Szakall, S.; Tron, L.; et al. Effects of vinpocetine on the redistribution of cerebral blood flow and glucose metabolism in chronic ischemic stroke patients: A pet study. J. Neurol. Sci. 2005, 229–230, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, I.; Gronholdt, M.L.; Soderberg, I.; Ares, M.P.; Nordestgaard, B.G.; Bentzon, J.F.; Fredrikson, G.N.; Nilsson, J. Humoral immune response against defined oxidized low-density lipoprotein antigens reflects structure and disease activity of carotid plaques. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, A.H.; Binder, C.J.; Tsimikas, S.; Witztum, J.L. Adaptive immunity in atherogenesis: New insights and therapeutic approaches. J. Clin. Investig. 2013, 123, 27–36. [Google Scholar] [CrossRef] [PubMed]
- De Boer, O.J.; van der Wal, A.C. FOXP3+ regulatory T cells in vulnerable atherosclerotic plaques. Int. J. Cardiol. 2010, 145, 161. [Google Scholar]
- Burioni, R.; Canducci, F.; Saita, D.; Perotti, M.; Mancini, N.; de Marco, D.; Clementi, N.; Chieffo, A.; Denaro, M.; Cianflone, D.; et al. Antigen-driven evolution of b lymphocytes in coronary atherosclerotic plaques. J. Immunol. 2009, 183, 2537–2544. [Google Scholar] [CrossRef]
- Gerondakis, S.; Fulford, T.S.; Messina, N.L.; Grumont, R.J. NF-κB control of T cell development. Nat. Immunol. 2014, 15, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Magnus, T.; Wiendl, H.; Kleinschnitz, C. Immune mechanisms of stroke. Curr. Opin. Neurol. 2012, 25, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Bereczki, D.; Fekete, I. Vinpocetine for acute ischaemic stroke. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Liu, N.; Wei, W.Z.W.; Wei, Y.; Yin, W. Efficacy and safety of vinpocetine in treatment of patients with acute cerebral infarction. Chin. J. Neuromed. 2014, 13, 4. [Google Scholar]
- Patyar, S.; Prakash, A.; Modi, M.; Medhi, B. Role of vinpocetine in cerebrovascular diseases. Pharmacol. Rep. 2011, 63, 618–628. [Google Scholar] [CrossRef]
- Karpati, E.; Szporny, L. General and cerebral haemodynamic activity of ethyl apovincaminate. Arzneimittel-Forschung 1976, 26, 1908–1912. [Google Scholar] [PubMed]
- Santos, M.S.; Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Synaptosomal response to oxidative stress: Effect of vinpocetine. Free Radic. Res. 2000, 32, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Gavish, M. The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol. Ther. 2006, 110, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Tarnok, K.; Kiss, E.; Luiten, P.G.; Nyakas, C.; Tihanyi, K.; Schlett, K.; Eisel, U.L. Effects of vinpocetine on mitochondrial function and neuroprotection in primary cortical neurons. Neurochem. Int. 2008, 53, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Marsh, B.J.; Williams-Karnesky, R.L.; Stenzel-Poore, M.P. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 2009, 158, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [PubMed]
- Zotti, T.; Uva, A.; Ferravante, A.; Vessichelli, M.; Scudiero, I.; Ceccarelli, M.; Vito, P.; Stilo, R. TRAF7 protein promotes Lys-29-linked polyubiquitination of IκB kinase (IKKγ)/NF-κB essential modulator (NEMO) and p65/Rela protein and represses NF-κB activation. J. Biol. Chem. 2011, 286, 22924–22933. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J.P.; Drukarch, B.; van Dam, A.M. (Peri)vascular production and action of pro-inflammatory cytokines in brain pathology. Clin. Sci. 2007, 112, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, H.; Tryc, A.B.; Goldbecker, A.; Ma, Y.T.; Tountopoulou, A.; Hahn, A.; Dengler, R.; Lichtinghagen, R.; Weissenborn, K. The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cerebrovasc. Dis. 2010, 30, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Yenari, M.A. Post-ischemic inflammation: Molecular mechanisms and therapeutic implications. Neurol. Res. 2004, 26, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early detrimental t-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.C.; Candelario-Jalil, E.; Langbein, J.; Wendeburg, L.; Bhatia, H.S.; Schlachetzki, J.C.; Biber, K.; Fiebich, B.L. Pharmacological inhibition of akt and downstream pathways modulates the expression of COX-2 and mPGES-1 in activated microglia. J. Neuroinflamm. 2012, 9. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Kraft, P.; Dreykluft, A.; Hagedorn, I.; Gobel, K.; Schuhmann, M.K.; Langhauser, F.; Helluy, X.; Schwarz, T.; Bittner, S.; et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood 2013, 121, 679–691. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Yang, L. Anti-Inflammatory Effects of Vinpocetine in Atherosclerosis and Ischemic Stroke: A Review of the Literature. Molecules 2015, 20, 335-347. https://doi.org/10.3390/molecules20010335
Zhang L, Yang L. Anti-Inflammatory Effects of Vinpocetine in Atherosclerosis and Ischemic Stroke: A Review of the Literature. Molecules. 2015; 20(1):335-347. https://doi.org/10.3390/molecules20010335
Chicago/Turabian StyleZhang, Linjie, and Li Yang. 2015. "Anti-Inflammatory Effects of Vinpocetine in Atherosclerosis and Ischemic Stroke: A Review of the Literature" Molecules 20, no. 1: 335-347. https://doi.org/10.3390/molecules20010335