
Synthesis, Characterization and Biological Evaluation of Some Quinoxaline Derivatives: A Promising and Potent New Class of Antitumor and Antimicrobial Agents

Abstract
:1. Introduction
2. Results and Discussion
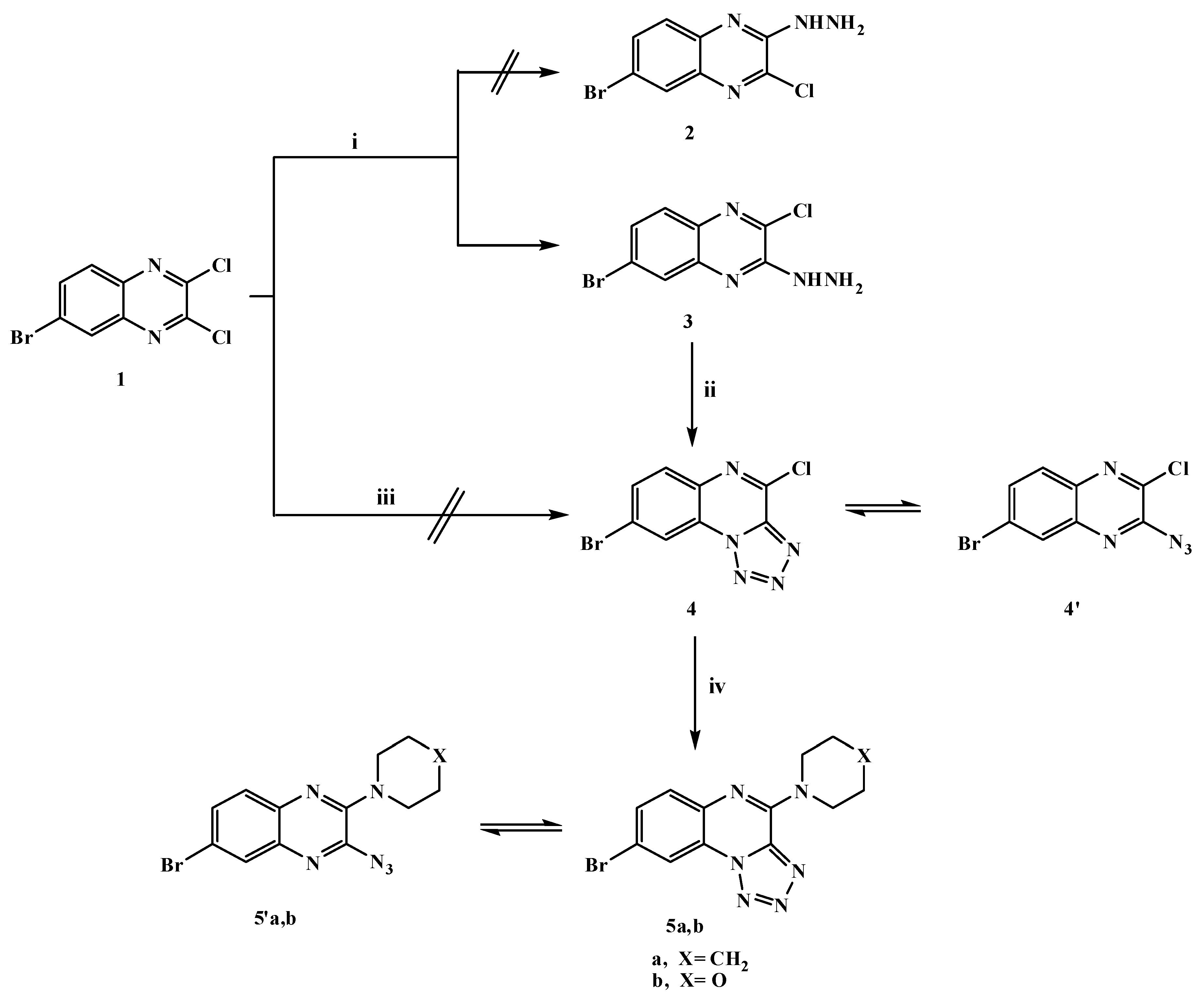
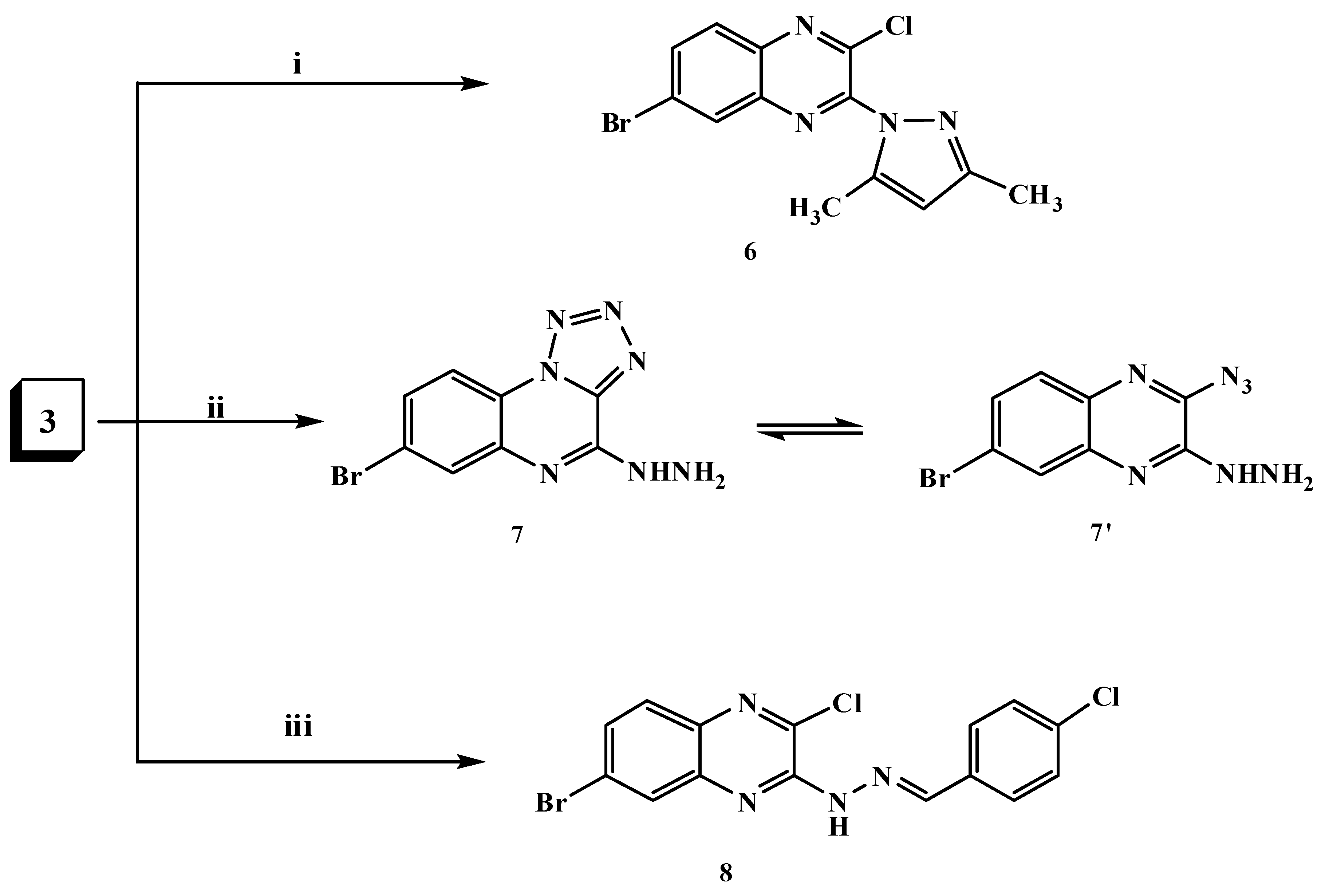
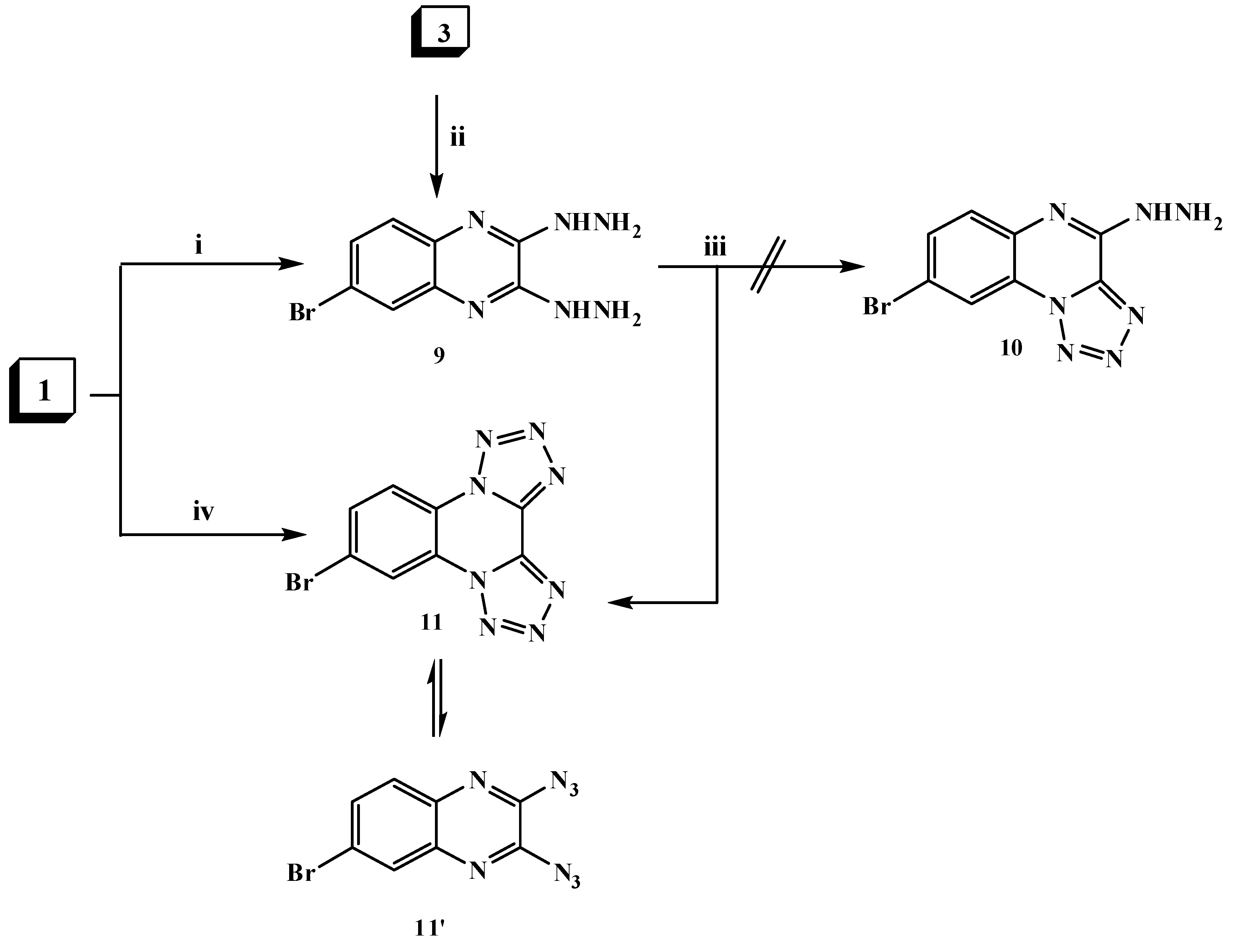
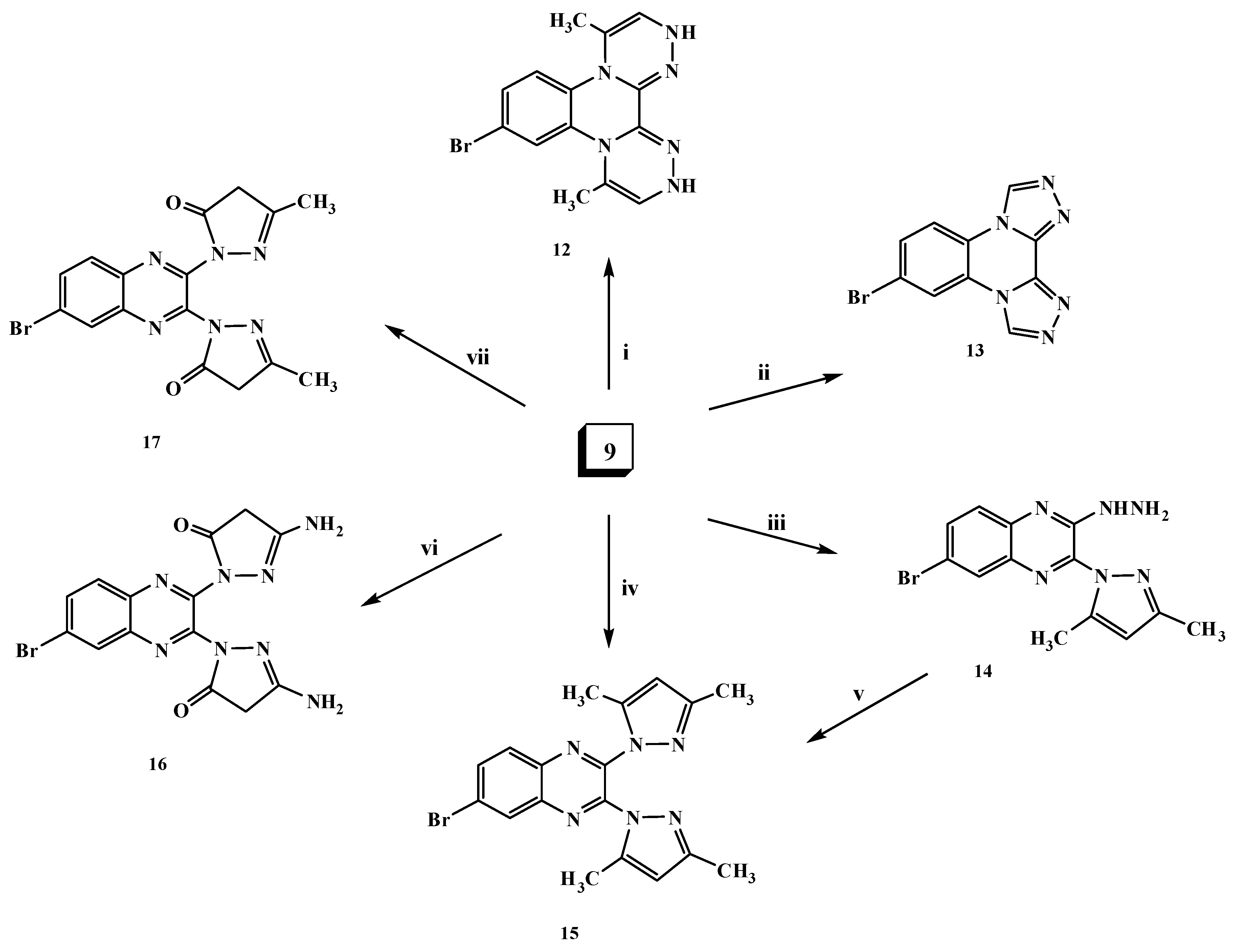
2.1. Chemistry




2.2. Pharmacological Screening
2.2.1. In Vitro Anticancer Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 (μg/mL) | |||
|---|---|---|---|---|
| MCF-7 | NCI-H460 | SF-268 | WI 38 | |
| 3 | 36.02 ± 7.33 c | 26.74 ± 2.18 b | 30.64 ± 2.39 b | 32.16 ± 6.54 |
| 4 | 0.02 ± 0.006 a | 0.01 ± 0.004 a | 0.06 ± 0.003 a | non-cytotoxic |
| 5a | 0.01 ± 0.001 a | 0.02 ± 0.006 a | 0.02 ± 0.008 a | non-cytotoxic |
| 5b | 0.02 ± 0.002 a | 0.01 ± 0.002 a | 0.06 ± 0.008 a | non-cytotoxic |
| 6 | 22.41 ± 10.4 b | 30.48 ± 10.8 b | 26.51 ± 2.87 b | 28.25 ± 0.87 |
| 7 | 26.4 ± 2.10 b | 12.42 ± 3.01 b | 10.63 ± 2.83 b | non-cytotoxic |
| 8 | 42.16 ± 2.46 c | 26.60 ± 2.63 b | 35.32 ± 12.81 c | 10.59 ± 5.51 |
| 9 | 37.07 ± 7.34 c | 16.37 ± 2.32 b | 38.94 ± 2.63 c | 30.62 ± 6.21 |
| 11 | 1.18 ± 1.03 a | 2.83 ± 0.53 a | 2.86 ± 4.92 a | 56.85 ± 4.05 |
| 12 | 35.22 ± 4.18 c | 34.03 ± 6.05 c | 22.10 ± 2.81 b | 22.97 ± 8.2 |
| 13 | 37.64 ± 6.72 c | 36.05 ± 5.23 c | 29.35 ± 7.01 c | 18.62 ± 1.21 |
| 14 | 30.32 ± 3.86 b | 38.32 ± 2.35 c | 42.06 ± 5.58 c | 58.70 ± 8.65 |
| 15 | 28.42 ± 5.80 b | 22.73 ± 8.12 b | 30.24 ± 2.04 b | 18.16 ± 4.03 |
| 16 | 12.82 ± 1.46 b | 22.95 ± 0.46 b | 49.85 ± 8.64 c | 30.03 ± 2.36 |
| 17 | 21.23 ± 0.14 b | 15.81 ± 0.10 b | 21.33 ± 2.12 b | 21.40 ± 2.02 |
| DMSO | 0 | 0 | 0 | 0 |
| Doxorubicin | 0.04 ± 0.008 | 0.09 ± 0.008 | 0.09 ± 0.007 | non-cytotoxic |
2.2.2. Antimicrobial Activity
Antibacterial Activity
Antifungal Activity
| Compound No. | Inhibition Zone Diameter (IZ) (mm) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gram Positive Bacteria | Gram Negative Bacteria | Fungi | ||||||||
| Bacillus subtilis | Staphylococcus aureus | Streptococcus faecalis | Escherichia coli | Neisseria gonorrhoeae | Pseudomonas aeruginosa | Salmonella typhimurium | Aspergillus flavus | Aspergillus fumigatus | Candida albicans | |
| 3 | 15 | 14 | 14 | 13 | 14 | 14 | 13 | R | R | R |
| 4 | 20 | 26 | 18 | 17 | 21 | 21 | 17 | 13 | 10 | 20 |
| 5a | 13 | 11 | 9 | 13 | R | 11 | 14 | R | 12 | 12 |
| 5b | 17 | 17 | 11 | 11 | R | 10 | 13 | R | 12 | 9 |
| 6 | 11 | 9 | 11 | 11 | 11 | 9 | 8 | R | R | R |
| 7 | 11 | 10 | 11 | 10 | 10 | 10 | 9 | R | R | R |
| 8 | 9 | 9 | 10 | 11 | 9 | 9 | 7 | R | R | R |
| 9 | 28 | 27 | 27 | 29 | 28 | 31 | 17 | R | 5 | 11 |
| 11 | 19 | 17 | 15 | 15 | R | 14 | 18 | R | R | R |
| 12 | 18 | 19 | 18 | 17 | R | 20 | 18 | R | R | R |
| 13 | 17 | 16 | 20 | 17 | R | 19 | 17 | R | R | R |
| 14 | 12 | 12 | 12 | 12 | 12 | 12 | 10 | R | R | R |
| 15 | 10 | 9 | 9 | 10 | 9 | 9 | 10 | R | R | R |
| 16 | 10 | 12 | 11 | 10 | 11 | 11 | 10 | R | R | R |
| 17 | 7 | 8 | 10 | 11 | 6 | 8 | 5 | R | R | R |
| DMSO | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ampicillin | 20 | 22 | 19 | - | - | - | - | - | - | - |
| Gentamicin | - | - | - | 20 | 18 | 17 | 23 | - | - | - |
| Amphotericin B | - | - | - | - | - | - | - | 17 | 23 | 19 |
| Compound No. | Minimum Inhibitory Concentration (MIC) (µg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| Gram positive Bacteria | Gram Negative Bacteria | Fungi | ||||||
| Bacillus subtilis | Staphylococcus aureus | Streptococcus faecalis | Escherichia coli | Pseudomonas aeruginosa | Salmonella typhimurium | Aspergillus fumigatus | Candida albicans | |
| 3 | 1.95 | 3.9 | 15.63 | 3.9 | 125 | 1.95 | - | - |
| 4 | 0.98 | 3.9 | 0.98 | 3.9 | >125 | 0.98 | - | 3.9 |
| 5a | 31.25 | 62.5 | 62.5 | 125 | 125 | 31.25 | 31.25 | 62.5 |
| 5b | 1.95 | 1.95 | 3.9 | 1.95 | >125 | 1.95 | 1.95 | 3.9 |
| 9 | 0.98 | 1.95 | 0.98 | 1.95 | 125 | 0.98 | 0.98 | 0.98 |
| 10 | 0.98 | 0.98 | 0.98 | 3.9 | 125 | 1.95 | - | - |
| 11 | 0.98 | 7.81 | 62.5 | 62.5 | 125 | 7.81 | - | - |
| 13 | 1.95 | 7.81 | 62.5 | 7.81 | 125 | 3.9 | - | - |
| 14 | 1.95 | 1.95 | 1.95 | 3.9 | 125 | 1.95 | - | - |
| 16 | 1.95 | 1.95 | 31.25 | 3.9 | 125 | 1.95 | - | - |
3. Experimental Section
3.1. General Information
3.1.1. 6-Bromo-2-chloro-3-hydrazinylquinoxaline (3)
3.1.2. 8-Bromo-4-chlorotetrazolo[1,5-a]quinoxaline (4)
3.1.3. General Procedure for Synthesis of 8-Bromo-4-(substituted amino)tetrazolo[1,5-a]quinoxaline (5a,b)
8-Bromo-4-(piperidin-1-yl)tetrazolo[1,5-a]quinoxaline (5a)
8-Bromo-4-(morpholin-4-yl)tetrazolo[1,5-a]quinoxaline (5b)
3.1.4. 6-Bromo-2-chloro-3-(3,5-dimethyl-1H-pyrazol-1-yl)quinoxaline (6)
3.1.5. 7-Bromo-4-hydrazinyltetrazolo[1,5-a]quinoxaline (7)
3.1.6. 6-Bromo-2-chloro-3-[2-(4-chlorobenzylidene)hydrazinyl]quinoxaline (8)
3.1.7. 6-Bromo-2,3-dihydrazinylquinoxaline (9)
3.1.8. 9-Bromoditetrazolo[1,5-a:5′,1′-c]quinoxaline (11)
3.1.9. 7-Bromo-4,11-dimethyl-2,13-dihydrobis[1,2,4]triazino[4,3-a:3',4'-c]quinoxaline (12)
3.1.10. 9-Bromo-di-1,2,4-triazolo[1,5-a:5′,1′-c]quinoxaline (13)
3.1.11. 6-Bromo-3-(3,5-dimethyl-1H-pyrazol-1-yl)-2-hydrazinyl quinoxaline (14)
3.1.12. 6-Bromo-2,3-bis(3,5-dimethyl-1H-pyrazol-1-yl)quinoxaline (15)
3.1.13. 6-Bromo-2,3-bis(3-amino-1H-pyrazol-5(4H)-one)quinoxaline (16)
3.1.14. 6-Bromo-2,3-bis(3-methyl-1H-pyrazol-5(4H)-one)quinoxaline (17)
3.2. Pharmacological Evaluation
3.2.1. Anticancer Activity
Cell Cultures
Cancer Cell Growth Assay
3.2.2. Antimicrobial Activity
Preparation of Microbial Suspensions
Determination of Antimicrobial Activity by Disk Diffusion Method
Determination of Minimum Inhibitory Concentration (MIC)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sawadogo, W.R.; Boly, R.; Cerella, C.; Teiten, M.H.; Dicato, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2012. Molecules 2015, 20, 7097–7142. [Google Scholar] [CrossRef] [PubMed]
- IARC. Cancer Incidence and Mortality Worlwide; International Agency for Research on Cancer: Lyon, France, 2011. [Google Scholar]
- WHO. Global Status Report on Noncommunicable Diseases 2010; WHO: Geneva, Switzerland, 2011; pp. 11–15. [Google Scholar]
- Işikdağ, I.; Özkay, Y.; Incesu, Z. Synthesis and anticancer activity of some bisquinoxaline derivatives. Turk J. Pharm. Sci. 2011, 8, 179–188. [Google Scholar]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-Carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Ismail, M.M.F.; El-Gaby, M.S.; Ammar, Y.A. Synthesis and antimicrobial activities of some novel quinoxalinone derivatives. Molecules 2000, 5, 864–873. [Google Scholar] [CrossRef]
- Wadavrao, S.B.; Ghogare, R.S.; Narsaiah, A.V. A simple and efficient protocol for the synthesis of quinoxalines catalyzed by pyridine. Org. Commun. 2013, 6, 23–30. [Google Scholar]
- Ismail, M.M.F.; Nofal, S.M.; Ibrahim, M.K.; El-Zahaby, H.S.A.; Ammar, Y.A. 3-Ethoxy-carbonylmethylenequinoxaline-2-one in heterocyclic synthesis (part 2): Synthesis and pharmacological evaluation of new 6,7-dimethyl quinoxalines as potential nonulcerogenic, anti-inflamatory and analgesic agents. Afinidad 2006, 63, 689–696. [Google Scholar]
- Ismail, M.M.F.; Ammar, Y.A; Ibrahim, M.K.; El-Zahaby, H.S.A. Synthesis and pharmacological evaluation of novel quinoxalines as potential nonulcerogenic, anti-inflammatory and analgesic agents. Arzneimittelforschung 2004, 55, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.A.R.; Bomfima, I.D.S.; Cavalcanti, B.C.; Pessoa, C.D.Ó.; Wardell, J.L.; Wardell, S.M.; Pinheiro, A.C.; Kaiser, C.R.; Nogueira, T.C.M.; Low, J.N.; et al. Design, synthesis and biological evaluation of (E)-2-(2-arylhydrazinyl)quinoxalines, a promising and potent new class of anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.F.; Amin, K.M.; Noaman, E.; Soliman, D.H.; Ammar, Y.A. New quinoxaline-1,4-di-N-oxides: Anticancer and hypoxia-selective therapeutic agents. Eur. J. Med. Chem. 2010, 45, 2733–2738. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; Ismail, M.M.F.; Noaman, E.; Soliman, D.H.; Ammar, Y.A. New quinoxaline-1,4-di-N-oxides (part 1): Hypoxia-selective cytotoxine and anticancer agents derived from quinoxaline-1,4-di-N-oxides. Bioorg. Med. Chem. 2006, 14, 6917–6923. [Google Scholar] [CrossRef] [PubMed]
- Farrag, A.A.; Ammar, Y.A.; El-Sehemi, A.G.; Thabet, H.Kh.; Hassan, N.A.; Samy, A.Kh. Synthesis and pharmacological screening of novel sulfamoylphenyl carbamoylquinoxaline derivatives as anti-inflammatory, analgesic and antitumour agets. J. Chem. Res. 2011, 163, 163–166. [Google Scholar] [CrossRef]
- Suresh, M.; Lavanya, P.; Sudhakar, D.; Vasu, K.; Rao, C.V. Synthesis and biological activity of 8-chloro-[1,2,4]triazolo[4,3-a]quinoxalines. J. Chem. Pharm. Res. 2010, 2, 497–504. [Google Scholar]
- Noolvi, M.N.; Patel, H.M.; Bhardwaj, V.; Chauhan, A. Synthesis and in vitro antitumor activity of substituted quinazoline and quinoxaline derivatives: Search for anticancer agent. Eur. J. Med. Chem. 2011, 46, 2327–2346. [Google Scholar] [CrossRef] [PubMed]
- Hassam, S.Y. Synthesis, antibacterial and antifungal activity of some new pyrazoline and pyrazole derivatives. Molecules 2013, 18, 2683–2711. [Google Scholar] [CrossRef] [PubMed]
- Mielcke, T.R.; Mascarello, A.; Fillipi-Chiela, E.; Zanin, R.F.; Lenz, G.; Leal, P.C.; Chiaradia, L.D.; Yunes, R.A.; Nunes, R.J.; Battastinie, A.M.O.; et al. Activity of novel quinoxaline-derived chalcones on in vitro glioma cell proliferation. Eur. J. Med. Chem. 2012, 48, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Su, Y.; Liu, X.; Yan, J.; Ye, Y.; Zhang, L.; Xu, J.; Weng, S.; Li, Y.; Liu, T.; et al. Discovery of novel morpholino–quinoxalines as PI3Kα inhibitors by pharmacophore-based screening. Med. Chem. Comm. 2012, 3, 659–662. [Google Scholar] [CrossRef]
- Seitz, L.E.; Suling, W.J.; Reynold, R.C. Synthesis and antimycobacterial activity of pyrazine and quinoxaline derivatives. J. Med. Chem. 2002, 45, 5604–5606. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Kim, Y.H.; Park, J.Y.; Kim, S.K. Synthesis and biological activity of new quinoxaline antibiotics of echinomycin analogues. Bioorg. Med. Chem. Lett. 2004, 14, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Aravind, K.; Ganesh, A.; Ashok, D. Microwave assisted synthesis, characterization and antibacterial activity of quinoxaline derivatives. J. Chem. Pharm. Res. 2013, 5, 48–52. [Google Scholar]
- Sajjadifar, S.; Nezhad, E.R. Qinoxaline III. synthesis of quinoxaline derivatives over highly efficient and reusable bronsted acidic ionic liquids. Int. J. ChemTech Res. 2013, 5, 2041–2050. [Google Scholar]
- Thomas, K.R.J.; Velusamy, M.; Lin, J.T.; Chuen, C.H.; Tao, Y.T. Chromophore-labeled qquinoxaline derivatives as efficient electroluminescent materials. Chem. Mater. 2005, 17, 1860–1866. [Google Scholar] [CrossRef]
- Toshima, K.; Takano, R.; Ozawa, T.; Matsumara, S. Molecular design and evaluation of quinoxaline-carbohydrate hybrids as novel and efficient photo-induced GG-selective DNA cleaving agents. Chem. Commun. 2002, 3, 212–213. [Google Scholar] [CrossRef]
- Dailey, S.; Feast, W.J.; Peace, R.J.; Sage, I.C.; Till, S.; Wood, E.L. Synthesis and device characterization of side-chain polymer electron transport materials for organic semiconducting applications. J. Mater. Chem. 2001, 11, 2238–2243. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Saxena, A.; De, A.; Mozumdar, S. Ni-nanoparticles: An efficient catalyst for the synthesis of quinoxalines. Catal. Commun. 2008, 9, 778–784. [Google Scholar] [CrossRef]
- Moustafa, O.S.; Badr, M.Z.A.; El-Emary, T.I. New fused quinoxalines: Synthesis and reactions of pyrimidothienoquinoxaline and oxadizolylthieno-quinoxalines. Bull. Korean Chem. Soc. 2002, 23, 567–570. [Google Scholar] [CrossRef]
- Crossley, M.J.; Johnston, L.A. Laterally-extended porphyrin systems incorporating a switchable unit. Chem. Commun. 2002, 21, 1122–1123. [Google Scholar] [CrossRef]
- Sessler, J.L.; Maeda, H.; Mizuno, T.; Lynch, V.M.; Furuta, H. Quinoxaline-oligopyrroles: improved pyrrole-based anion receptors. Chem. Commun. 2002, 21, 862–863. [Google Scholar] [CrossRef]
- Sascha, O.; Rudiger, F. Quinoxalinodehydroannulenes: A novel class of carbon-rich materials. Synlett 2004, 9, 1509–1513. [Google Scholar]
- Islami, M.R.; Hassani, Z. One-pot and efficient protocol for synthesis of quinoxaline derivatives. Arkivoc 2008, 15, 280–287. [Google Scholar]
- Wagle, S.; Adhikari, A.V.; Kumari, N.S. Synthesis of some new 4-styryltetrazolo[1,5-a]quinoxaline and 1-substituted-4-styryl[1,2,4]triazolo[4,3-a]quinoxaline derivatives as potent anticonvulsants. Eur. J. Med. Chem. 2009, 44, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, T.E.; Kurasawa, Y. Synthesis of 1,3,4-Thiadiazino[5,6-b] quinoxalines with Antimicrobial Activity. J. Korean Chem. Soc. 2001, 45, 325–333. [Google Scholar]
- Natarajan, U.; Kaliappan, I.; Singh, N.K. A facile design and efficient synthesis of schiff’s bases of tetrazolo[1,5-a]quinoxalines as potential anti-inflammatory and anti-microbial agents. Pharm. Chem. 2010, 2, 159–167. [Google Scholar]
- Suryawanshi, M.R.; Kulkarni, V.M.; Mahadik, K.R.; Bhosale, Sh.H. Synthesis, pharmacological evaluation and QSAR study of 2-aryl-1H-[1,3,4]oxadiazino[5,6-b] quinoxalines as antidepressants. Arch. Appl. Sci. Res. 2011, 3, 380–391. [Google Scholar]
- Podsiad1y, R.; Szymczak, A.M.; Podemska, K. The synthesis of novel, visible-wavelength, oxidizable polymerization sensitizers based on the 8-halogeno-5,12-dihydroquinoxalino[2,3-b]quinoxaline skeleton. Dyes Pigm. 2009, 82, 365–371. [Google Scholar] [CrossRef]
- Khalil, Z.H.; Geies, A.A. Synthesis and reactions of some thieno[2,3-d]pyrimidine derivatives. Phosphorus Sulfur Silicon 1991, 60, 223–231. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenne, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paul, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [PubMed]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fourth Informational Supplement. CLSI Document M100-S24; CLSI: Wayne, PA, USA, 2014. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Disk Susceptibility Testing, Approved 11th ed.; CLSI Document M02-A11; CLSI: Wayne, PA, USA, 2012. [Google Scholar]
- Clinical and Laboratory Standards Institute. Method for Antifungal Disk Diffusion Susceptibility Testing of Yeasts, Approved Guideline-2nd ed.; CLSI Document M44-A2; CLSI: Wayne, PA, USA, 2009. [Google Scholar]
- Liebowitz, L.D.; Ashbee, H.R.; Evans, E.G.; Chong, Y.; Mallatova, N.; Zaidi, M.; Gibbs, D.; Global Antifungal surveillance group. A two year global evaluation of the susceptibility of Candida species to fluconazole by disk diffusion. Diagn. Microbiol. Infect. Dis. 2001, 40, 27–33. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved-Standard 9th ed.; CLSI Document M07-A9; CLSI: Wayne, PA, USA, 2012. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Marhabi, A.R.; Abbas, H.-A.S.; Ammar, Y.A. Synthesis, Characterization and Biological Evaluation of Some Quinoxaline Derivatives: A Promising and Potent New Class of Antitumor and Antimicrobial Agents. Molecules 2015, 20, 19805-19822. https://doi.org/10.3390/molecules201119655
Al-Marhabi AR, Abbas H-AS, Ammar YA. Synthesis, Characterization and Biological Evaluation of Some Quinoxaline Derivatives: A Promising and Potent New Class of Antitumor and Antimicrobial Agents. Molecules. 2015; 20(11):19805-19822. https://doi.org/10.3390/molecules201119655
Chicago/Turabian StyleAl-Marhabi, Aisha R., Hebat-Allah S. Abbas, and Yousry A. Ammar. 2015. "Synthesis, Characterization and Biological Evaluation of Some Quinoxaline Derivatives: A Promising and Potent New Class of Antitumor and Antimicrobial Agents" Molecules 20, no. 11: 19805-19822. https://doi.org/10.3390/molecules201119655