
Extremely Randomized Machine Learning Methods for Compound Activity Prediction


Abstract
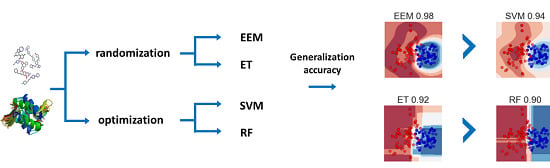
:
1. Introduction
2. Experimental Section
2.1. Datasets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target/Dataset | True Actives | True Inactives | DUD 1 | DUD 2 |
|---|---|---|---|---|
| 5-HT | 1835 | 851 | 1697 | 3388 |
| 5-HT | 1210 | 926 | 1072 | 2136 |
| 5-HT | 1490 | 341 | 1443 | 2883 |
| 5-HT | 704 | 339 | 633 | 1264 |
| M | 759 | 938 | 317 | 631 |
| H | 635 | 545 | 556 | 1107 |
| HIV | 101 | 914 | 83 | 163 |
| Method | Optimized Parameters With Range | |
|---|---|---|
| EEM | ||
| SVM | ||
| ET | no of trees | |
| RF | no of trees | |
2.2. Methods
| Method | Training Complexity | Classifying Complexity |
|---|---|---|
| EEM | ||
| SVM | ||
| ET [2] | ||
| RF [2] |

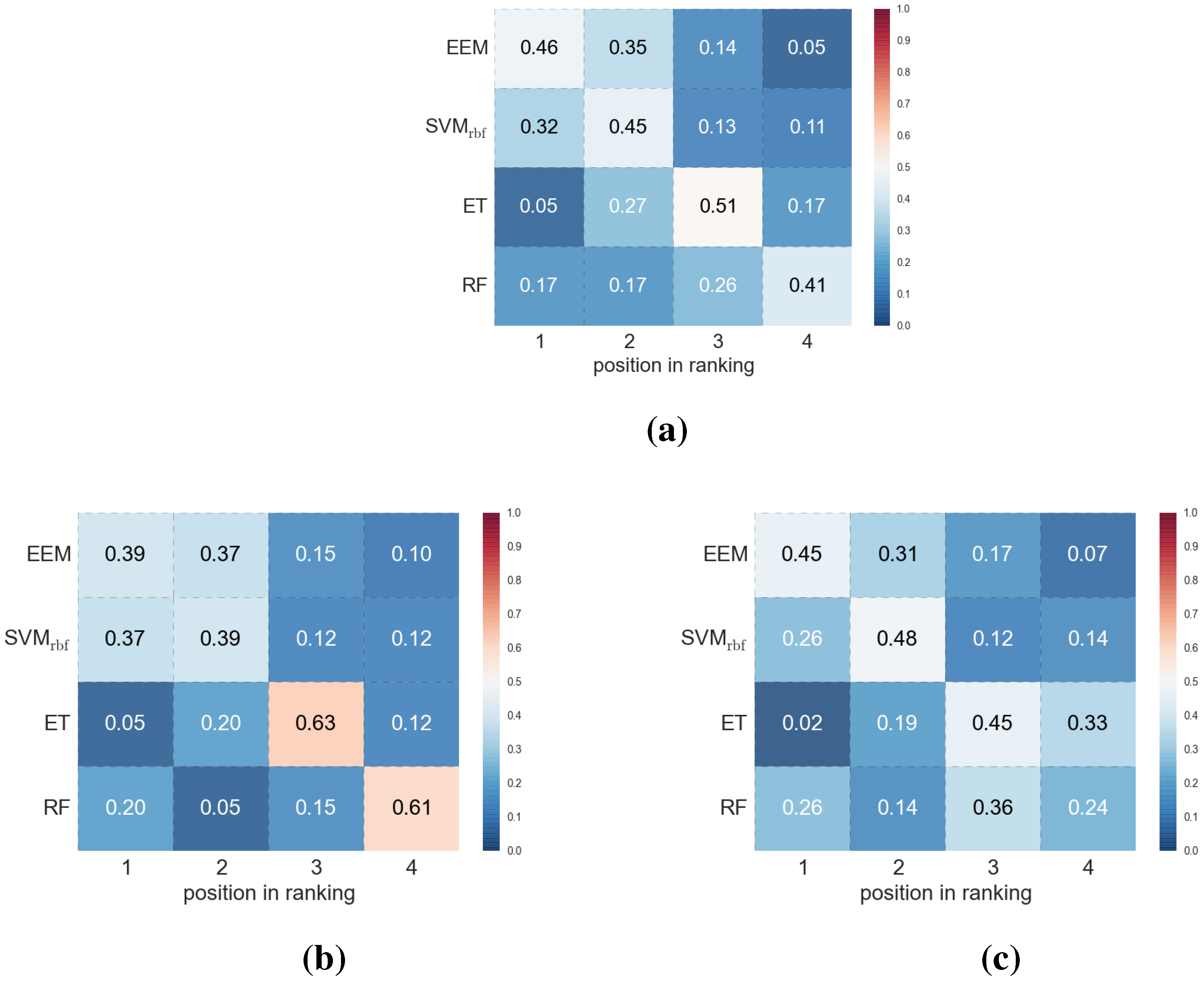
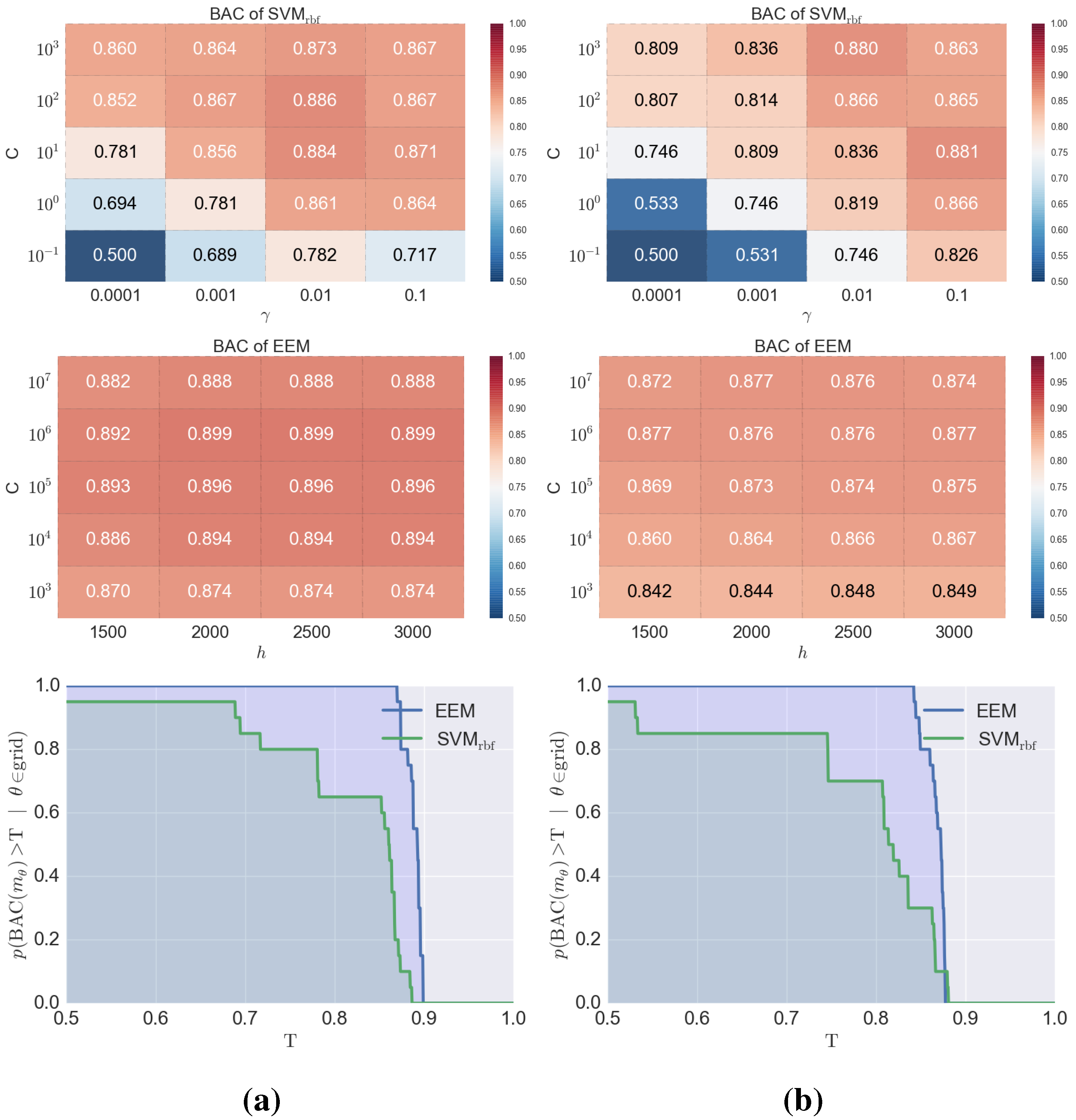
3. Results and Discussion
| EstateFP | EEM | SVM | ET | RF | SubFP | EEM | SVM | ET | RF |
|---|---|---|---|---|---|---|---|---|---|
| 5-HT | 0.936 | 0.928 | 0.932 | 0.935 | 5-HT | 0.968 | 0.964 | 0.967 | 0.964 |
| 5-HT | 0.913 | 0.920 | 0.923 | 0.927 | 5-HT | 0.951 | 0.945 | 0.935 | 0.940 |
| 5-HT | 0.964 | 0.961 | 0.967 | 0.965 | 5-HT | 0.984 | 0.980 | 0.982 | 0.981 |
| 5-HT | 0.925 | 0.920 | 0.925 | 0.922 | 5-HT | 0.976 | 0.974 | 0.976 | 0.976 |
| M | 0.925 | 0.917 | 0.916 | 0.912 | M | 0.967 | 0.965 | 0.965 | 0.960 |
| H | 0.922 | 0.918 | 0.927 | 0.925 | H | 0.970 | 0.968 | 0.967 | 0.965 |
| HIV | 0.968 | 0.983 | 0.971 | 0.971 | HIV | 0.980 | 0.980 | 0.985 | 0.985 |
| PubchemFP | EEM | SVM | ET | RF | ExtFP | EEM | SVM | ET | RF |
| 5-HT | 0.999 | 0.999 | 0.999 | 0.999 | 5-HT | 0.986 | 0.982 | 0.979 | 0.975 |
| 5-HT | 0.999 | 0.999 | 1.000 | 1.000 | 5-HT | 0.982 | 0.980 | 0.981 | 0.979 |
| 5-HT | 0.999 | 0.999 | 0.999 | 0.999 | 5-HT | 0.991 | 0.992 | 0.989 | 0.988 |
| 5-HT | 0.999 | 0.999 | 0.999 | 0.999 | 5-HT | 0.981 | 0.982 | 0.978 | 0.977 |
| M | 0.996 | 0.994 | 0.998 | 0.995 | M | 0.976 | 0.973 | 0.971 | 0.965 |
| H | 1.000 | 1.000 | 0.999 | 0.999 | H | 0.972 | 0.977 | 0.967 | 0.962 |
| HIV | 1.000 | 0.994 | 0.995 | 0.990 | HIV | 0.984 | 0.990 | 0.980 | 0.980 |
| KlekFP | EEM | SVM | ET | RF | MACCSFP | EEM | SVM | ET | RF |
| 5-HT | 0.992 | 0.988 | 0.986 | 0.983 | 5-HT | 0.984 | 0.981 | 0.978 | 0.976 |
| 5-HT | 0.991 | 0.986 | 0.980 | 0.975 | 5-HT | 0.982 | 0.977 | 0.975 | 0.972 |
| 5-HT | 0.999 | 0.999 | 1.000 | 1.000 | 5-HT | 0.988 | 0.988 | 0.981 | 0.979 |
| 5-HT | 0.987 | 0.989 | 0.981 | 0.980 | 5-HT | 0.982 | 0.975 | 0.979 | 0.975 |
| M | 0.977 | 0.974 | 0.964 | 0.956 | M | 0.975 | 0.975 | 0.965 | 0.962 |
| H | 0.987 | 0.981 | 0.986 | 0.984 | H | 0.975 | 0.975 | 0.974 | 0.974 |
| HIV | 0.984 | 0.980 | 0.984 | 0.989 | HIV | 0.989 | 0.984 | 0.984 | 0.978 |
| True Inactives | EEM | SVM | ET | RF | trueInact/DUDs | EEM | SVM | ET | RF |
|---|---|---|---|---|---|---|---|---|---|
| 5-HT | 0.882 | 0.875 | 0.862 | 0.852 | 5-HT | 0.918 | 0.919 | 0.917 | 0.918 |
| 5-HT | 0.875 | 0.885 | 0.883 | 0.881 | 5-HT | 0.904 | 0.899 | 0.901 | 0.895 |
| 5-HT | 0.901 | 0.895 | 0.888 | 0.885 | 5-HT | 0.965 | 0.967 | 0.965 | 0.962 |
| 5-HT | 0.876 | 0.868 | 0.847 | 0.825 | 5-HT | 0.924 | 0.921 | 0.907 | 0.907 |
| M | 0.888 | 0.882 | 0.885 | 0.887 | M | 0.899 | 0.890 | 0.890 | 0.893 |
| H | 0.919 | 0.913 | 0.908 | 0.911 | H | 0.928 | 0.923 | 0.926 | 0.927 |
| HIV | 0.911 | 0.920 | 0.867 | 0.859 | HIV | 0.899 | 0.919 | 0.867 | 0.858 |
| DUD 1 | EEM | SVM | ET | RF | DUD 2 | EEM | SVM | ET | RF |
| 5-HT | 0.999 | 0.999 | 0.999 | 0.999 | 5-HT | 0.999 | 0.999 | 0.999 | 0.999 |
| 5-HT | 0.999 | 0.999 | 1.000 | 1.000 | 5-HT | 1.000 | 0.999 | 1.000 | 1.000 |
| 5-HT | 0.999 | 0.999 | 1.000 | 1.000 | 5-HT | 0.999 | 0.999 | 1.000 | 1.000 |
| 5-HT | 0.999 | 0.999 | 0.999 | 0.999 | 5-HT | 0.999 | 0.999 | 0.999 | 0.999 |
| M | 0.996 | 0.994 | 0.998 | 0.995 | M | 0.996 | 0.994 | 0.996 | 0.996 |
| H | 1.000 | 1.000 | 0.999 | 0.999 | H | 1.000 | 1.000 | 1.000 | 0.999 |
| HIV | 1.000 | 0.994 | 0.995 | 0.990 | HIV | 0.995 | 0.995 | 0.990 | 0.990 |




4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pao, Y.H.; Park, G.H.; Sobajic, D.J. Learning and generalization characteristics of the random vector functional-link net. Neurocomputing 1994, 6, 163–180. [Google Scholar] [CrossRef]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Czarnecki, W.M.; Tabor, J. Extreme Entropy Machines: Robust information theoretic classification. Pattern Anal. Appl. 2015, 1–18. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Sencanski, M.; Sukalovic, V.; Shakib, K.; Soskic, V.; Dosen-Micovic, L.; Kostic-Rajacic, S. Molecular Modelling of 5HT2A Receptor—Arylpiperazine Ligands Interactions. Chem. Biol. Drug Des. 2014, 83, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Serotonin 5-HT2C receptors as a target for the treatment of depressive and anxious states: Focus on novel therapeutic strategies. Therapie 2005, 60, 441–460. [Google Scholar] [CrossRef] [PubMed]
- Upton, N.; Chuang, T.T.; Hunter, A.J.; Virley, D.J. 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.J.; Hedlund, P.B. The 5-HT7 Receptor in Learning and Memory. Hippocampus 2012, 22, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, R.L.; Gelfand, E.W.; Dunford, P.J. The role of histamine H1 and H4 receptors in allergic inflammation: The search for new antihistamines. Nat. Rev. Drug Discov. 2008, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.; Simms, J.; Sexton, P.M.; Christopoulos, A. Structure-Function Studies of Muscarinic Acetylcholine Receptors. Handb. Exp. Pharmacol. 2012, 208, 29–48. [Google Scholar] [PubMed]
- Craigie, R. HIV Integrase, a Brief Overview from Chemistry to Therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Instant JChem 15.3.30.0, ChemAxon. Available online: http://www.chemaxon.com (accessed on 10 August 2015).
- Yap, C.W. PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.H.; Kier, L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Model. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Klekota, J.; Roth, F.P. Chemical substructures that enrich for biological activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.; Baber, J.C.; Feher, M. Novel 2D fingerprints for ligand-based virtual screening. J. Chem. Inf. Model. 2006, 46, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Czarnecki, W.M. Weighted Tanimoto Extreme Learning Machine with Case Study in Drug Discovery. IEEE Comput. Intell. Mag. 2015, 10, 19–29. [Google Scholar] [CrossRef]
- Fernández-Delgado, M.; Cernadas, E.; Barro, S.; Amorim, D. Do we need hundreds of classifiers to solve real world classification problems? J. Mach. Learn. Res. 2014, 15, 3133–3181. [Google Scholar]
- Fern, X.Z.; Brodley, C.E. Random projection for high dimensional data clustering: A cluster ensemble approach. In Proceedings of the ICML-2003, Washington, DC, USA, 21 August 2003; Volume 3, pp. 186–193.
- Arriaga, R.I.; Vempala, S. An algorithmic theory of learning: Robust concepts and random projection. Mach. Learn. 2006, 63, 161–182. [Google Scholar] [CrossRef]
- Sample Availability: not apply.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecki, W.M.; Podlewska, S.; Bojarski, A.J. Extremely Randomized Machine Learning Methods for Compound Activity Prediction. Molecules 2015, 20, 20107-20117. https://doi.org/10.3390/molecules201119679
Czarnecki WM, Podlewska S, Bojarski AJ. Extremely Randomized Machine Learning Methods for Compound Activity Prediction. Molecules. 2015; 20(11):20107-20117. https://doi.org/10.3390/molecules201119679
Chicago/Turabian StyleCzarnecki, Wojciech M., Sabina Podlewska, and Andrzej J. Bojarski. 2015. "Extremely Randomized Machine Learning Methods for Compound Activity Prediction" Molecules 20, no. 11: 20107-20117. https://doi.org/10.3390/molecules201119679