
Towards the Synthesis of Graphene Azide from Graphene Oxide

Abstract
:1. Introduction

2. Results and Discussion

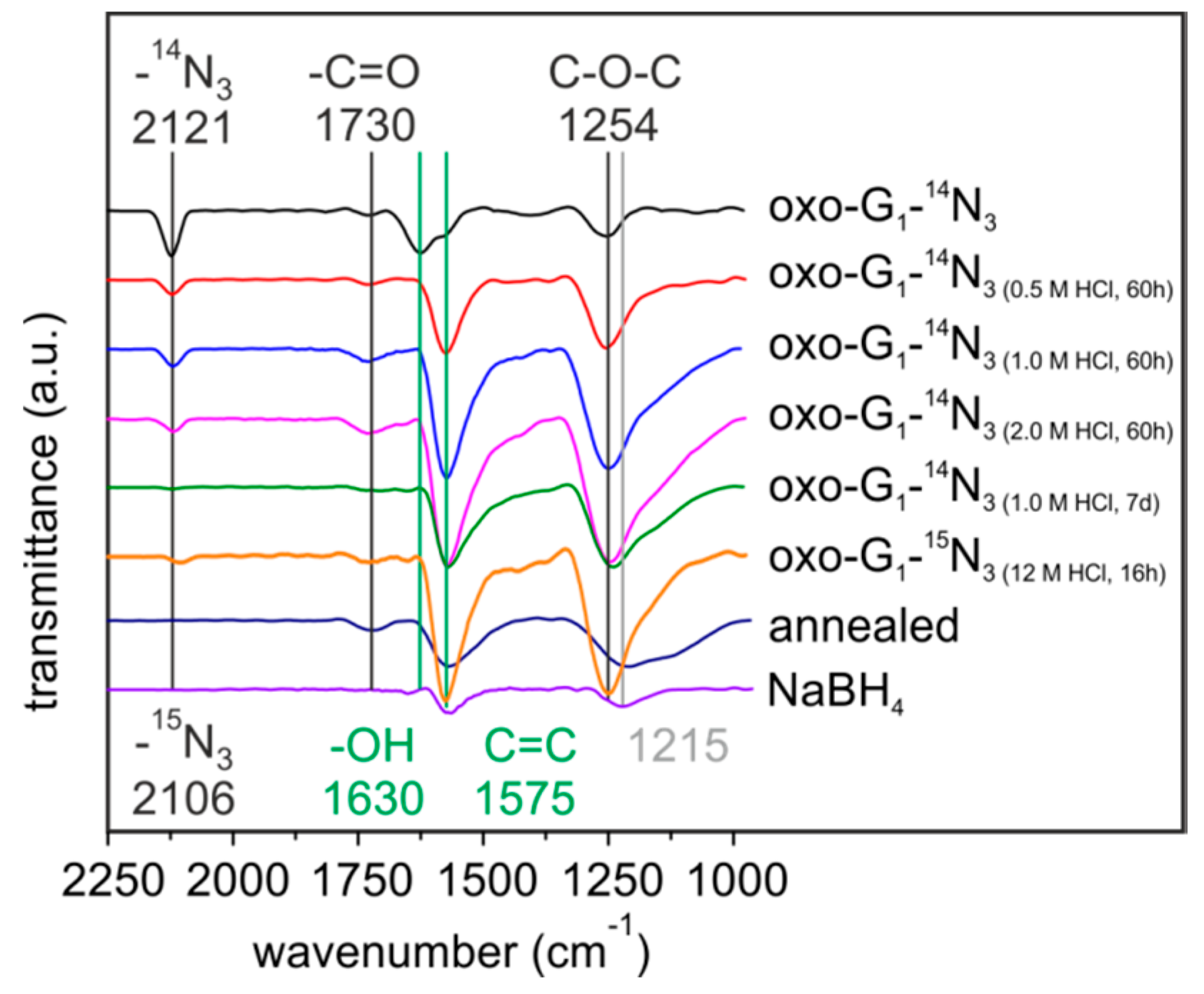{kind=link}
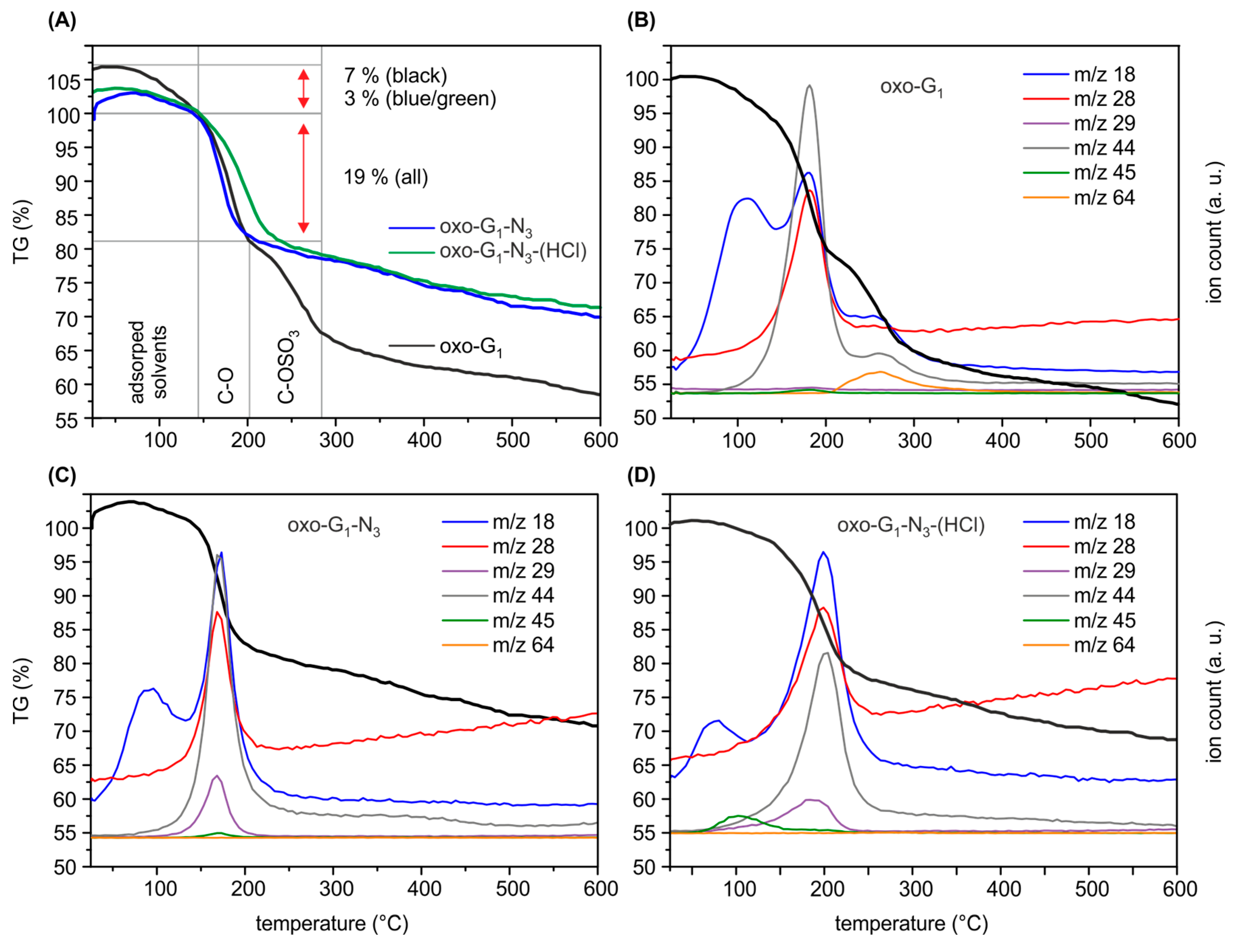{kind=link}
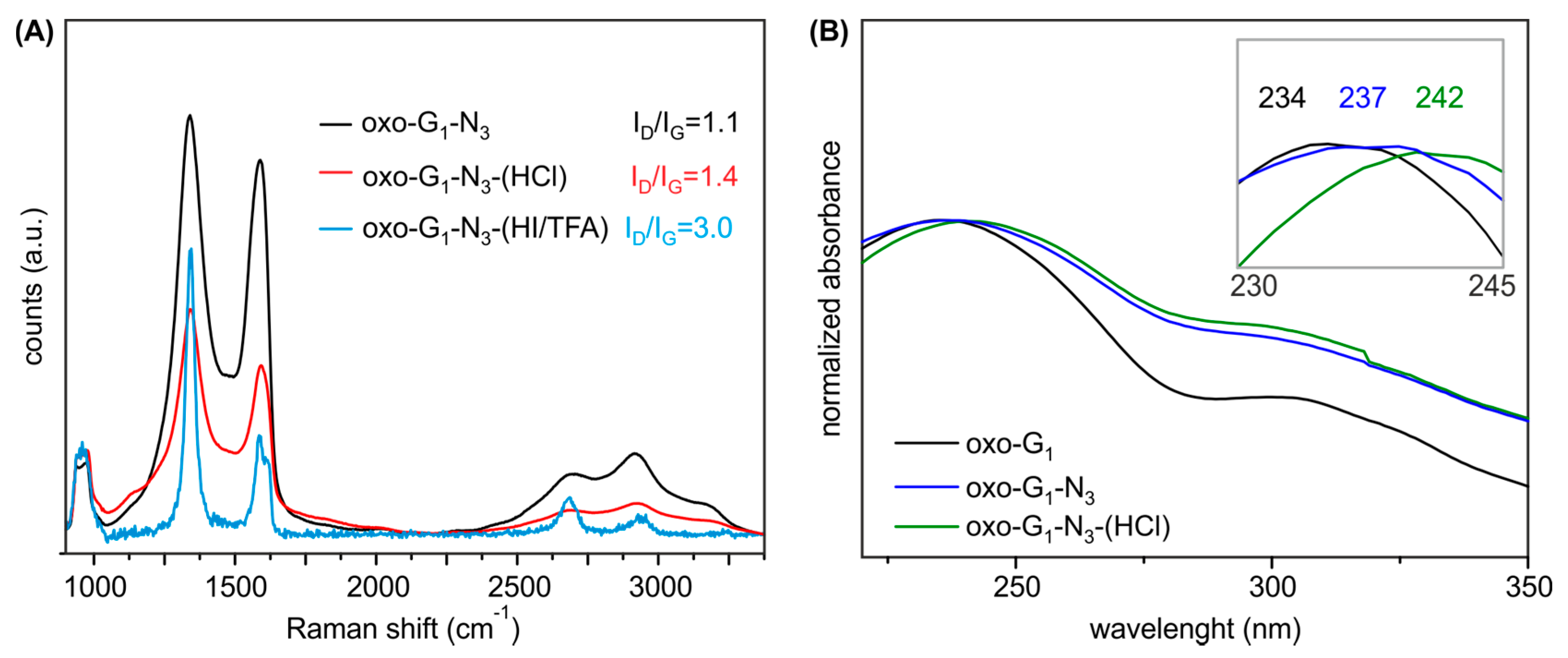{kind=link}
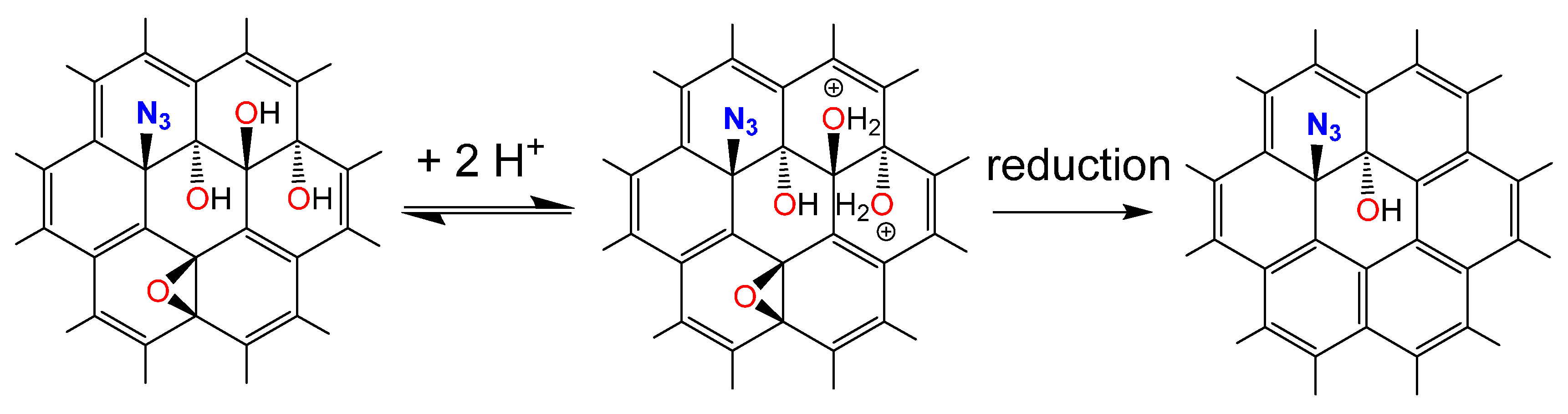{kind=link}
| Sample Name | C (%) | N (%) | H (%) | S (%) | O * (%) |
|---|---|---|---|---|---|
| oxo-G1 | 45.9 | 0.0 | 2.4 | 4.3 | 47.4 |
| oxo-G1-N3 | 44.8 | 3.3 | 2.6 | 1.1 | 48.2 |
| oxo-G1-N3-(2 M HCl) | 54.8 | 4.5 | 2.6 | 0.8 | 37.3 |
| oxo-G1-N3-(12 M HCl) | 57.7 | 4.1 | 2.4 | 0.4 | 35.4 |



3. Materials and Methods
3.1. Chemicals and Instrumentation
3.2. Synthesis of Oxo-G1 and Oxo-G1-N3
3.3. Preparation of Oxo-G1-N3-(HCl)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Bubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.; Geim, A.K. Fine structure constant defines visual transparency of graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [PubMed]
- Morozov, S.V.; Novoselov, K.S.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant intrinsic carrier mobilities in graphene and its bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.V.; Novoselov, K.S.; Morozov, S.V.; Peres, N.M.; dos Santos, J.M.; Nilsson, J.; Guinea, F.; Geim, A.K.; Neto, A.H. Biased bilayer graphene: Semiconductor with a gap tunable by the electric field effect. Phys. Rev. Lett. 2007, 99, 216802. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nature Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Murali, S.; Zhu, Y.; Ruoff, R.S.; Bielawski, C.W. Reduction of graphite oxide using alcohols. J. Mater. Chem. 2011, 21, 3443–3447. [Google Scholar] [CrossRef]
- Yi, M.; Shen, Z. A review on mechanical exfoliation for the scalable production of graphene. J. Mater. Chem. A 2015, 3, 11700–11715. [Google Scholar] [CrossRef]
- Mei, K.C.; Rubio, N.; Costa, P.M.; Kafa, H.; Abbate, V.; Festy, F.; Bansal, S.S.; Hider, R.C.; Al-Jamal, K.T. Synthesis of double-clickable functionalised graphene oxide for biological applications. Chem. Commun. 2015, 51, 14981–14984. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Samulski, E.T. Synthesis of water soluble graphene. Nano Lett. 2008, 8, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Eigler, S.; Hirsch, A. Chemistry with graphene and graphene oxide-challenges for synthetic chemists. Angew. Chem. Int. Ed. 2014, 53, 7720–7738. [Google Scholar] [CrossRef] [PubMed]
- Eigler, S.; Hu, Y.; Ishii, Y.; Hirsch, A. Controlled functionalization of graphene oxide with sodium azide. Nanoscale 2013, 5, 12136–12139. [Google Scholar] [CrossRef] [PubMed]
- Eigler, S.; Grimm, S.; Hof, F.; Hirsch, A. Graphene oxide: A stable carbon framework for functionalization. J. Mater. Chem. A 2013, 1, 11559–11562. [Google Scholar] [CrossRef]
- Compton, O.C.; Dikin, D.A.; Putz, K.W.; Brinson, L.C.; Nguyen, S.T. Electrically conductive “alkylated” graphene paper via chemical reduction of amine-functionalized graphene oxide paper. Adv. Mater. 2009, 22, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Schafer, R.A.; Englert, J.M.; Wehrfritz, P.; Bauer, W.; Hauke, F.; Seyller, T.; Hirsch, A. On the way to graphane-pronounced fluorescence of polyhydrogenated graphene. Angew. Chem. Int. Ed. 2013, 52, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Pumera, M.; Wong, C.H. Graphane and hydrogenated graphene. Chem. Soc. Rev. 2013, 42, 5987–5995. [Google Scholar] [CrossRef] [PubMed]
- Jankovsky, O.; Simek, P.; Klimova, K.; Sedmidubsky, D.; Matejkova, S.; Pumera, M.; Sofer, Z. Towards graphene bromide: Bromination of graphite oxide. Nanoscale 2014, 6, 6065–6074. [Google Scholar] [CrossRef] [PubMed]
- Poh, H.L.; Simek, P.; Sofer, Z.; Pumera, M. Halogenation of graphene with chlorine, bromine, or iodine by exfoliation in a halogen atmosphere. Chemistry 2013, 19, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Eigler, S.; Enzelberger-Heim, M.; Grimm, S.; Hofmann, P.; Kroener, W.; Geworski, A.; Dotzer, C.; Rockert, M.; Xiao, J.; Papp, C.; et al. Wet chemical synthesis of graphene. Adv. Mater. 2013, 25, 3583–3587. [Google Scholar] [CrossRef] [PubMed]
- Haubner, K.; Murawski, J.; Olk, P.; Eng, L.M.; Ziegler, C.; Adolphi, B.; Jaehne, E. The route to functional graphene oxide. Chem. Phys. Chem. 2010, 11, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bai, H.; Lu, G.; Li, C.; Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 2008, 130, 5856–5857. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.I.; Villar-Rodil, S.; Martinez-Alonso, A.; Tascon, J.M. Graphene oxide dispersions in organic solvents. Langmuir 2008, 24, 10560–10564. [Google Scholar] [CrossRef] [PubMed]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Synthesis and exfoliation of isocyanate-treated graphene oxide nanoplatelets. Carbon 2006, 44, 3342–3347. [Google Scholar] [CrossRef]
- Bourlinos, A.B.; Gournis, D.; Petridis, D.; Szabo, T.; Szeri, A.; Dekany, I. Graphite oxide: Chemical reduction to graphite and surface modification with primary aliphatic amines and amino acids. Langmuir 2003, 19, 6050–6055. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Alemany, L.B.; Tour, J.M. Graphene oxide. Origin of acidity, its instability in water, and a new dynamic structural model. ACS Nano 2013, 7, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Eigler, S.; Dotzer, C.; Hof, F.; Bauer, W.; Hirsch, A. Sulfur species in graphene oxide. Chem. Eur. J. 2013, 19, 9490–9496. [Google Scholar] [CrossRef] [PubMed]
- Cançado, L.G.; Jorio, A.; Ferreira, E.H.M.; Stavale, F.; Achete, C.A.; Capaz, R.B.; Moutinho, M.V.O.; Lombardo, A.; Kulmala, T.S.; Ferrari, A.C. Quantifying defects in graphene via Raman spectroscopy at different excitation energies. Nano Lett. 2011, 11, 3190–3196. [Google Scholar] [CrossRef] [PubMed]
- Eigler, S.; Hof, F.; Enzelberger-Heim, M.; Grimm, S.; Müller, P.; Hirsch, A. Statistical-Raman-microscopy and atomic-force-microscopy on heterogeneous graphene obtained after reduction of graphene oxide. J. Phys. Chem. C 2014, 118, 7698–7704. [Google Scholar] [CrossRef]
- Englert, J.M.; Vecera, P.; Knirsch, K.C.; Schafer, R.A.; Hauke, F.; Hirsch, A. Scanning-Raman-microscopy for the statistical analysis of covalently functionalized graphene. ACS Nano 2013, 7, 5472–5482. [Google Scholar] [CrossRef] [PubMed]
- Halbig, C.E.; Nacken, T.J.; Walter, J.; Damm, C.; Eigler, S.; Peukert, W. Quantitative investigation of the fragmentation process and defect density evolution of oxo-functionalized graphene due to ultrasonication and milling. Carbon 2016, 96, 897–903. [Google Scholar] [CrossRef]
- Eigler, S.; Grimm, S.; Enzelberger-Heim, M.; Müller, P.; Hirsch, A. Graphene oxide: Efficiency of reducing agents. Chem. Commun. 2013, 49, 7391–7393. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available, but can easily prepared as described in literature above. We can’t guarantee long time stability due composition.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halbig, C.E.; Rietsch, P.; Eigler, S. Towards the Synthesis of Graphene Azide from Graphene Oxide. Molecules 2015, 20, 21050-21057. https://doi.org/10.3390/molecules201219747
Halbig CE, Rietsch P, Eigler S. Towards the Synthesis of Graphene Azide from Graphene Oxide. Molecules. 2015; 20(12):21050-21057. https://doi.org/10.3390/molecules201219747
Chicago/Turabian StyleHalbig, Christian E., Philipp Rietsch, and Siegfried Eigler. 2015. "Towards the Synthesis of Graphene Azide from Graphene Oxide" Molecules 20, no. 12: 21050-21057. https://doi.org/10.3390/molecules201219747