
Synthesis of Peptide Nucleic Acids Containing a Crosslinking Agent and Evaluation of Their Reactivities

Abstract
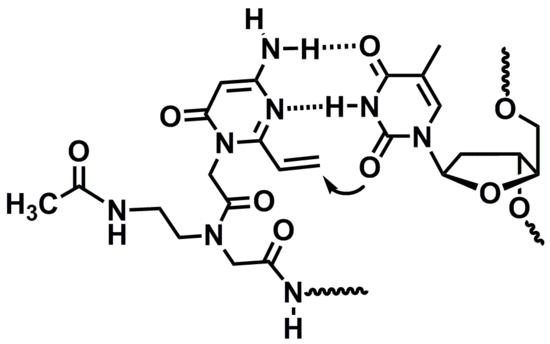
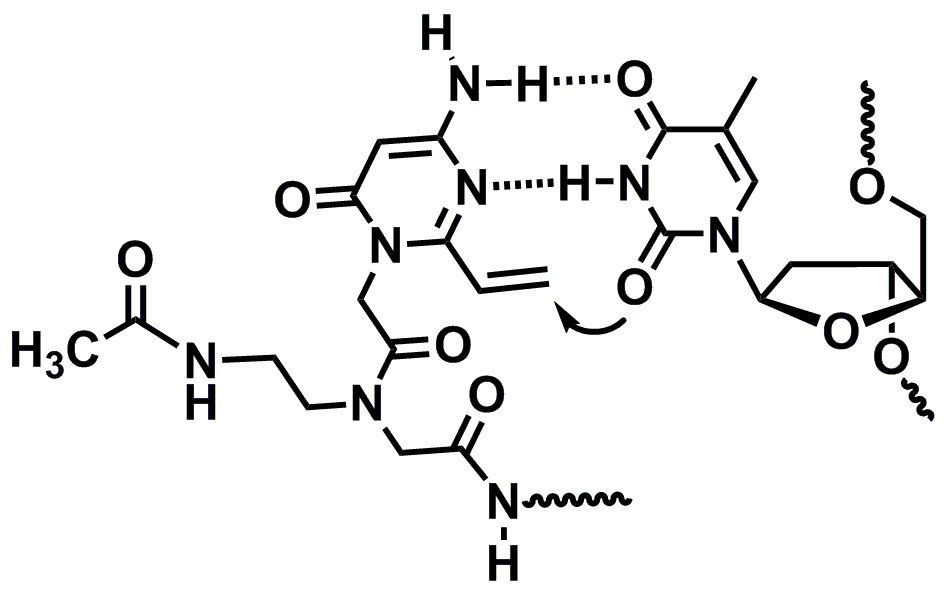
:
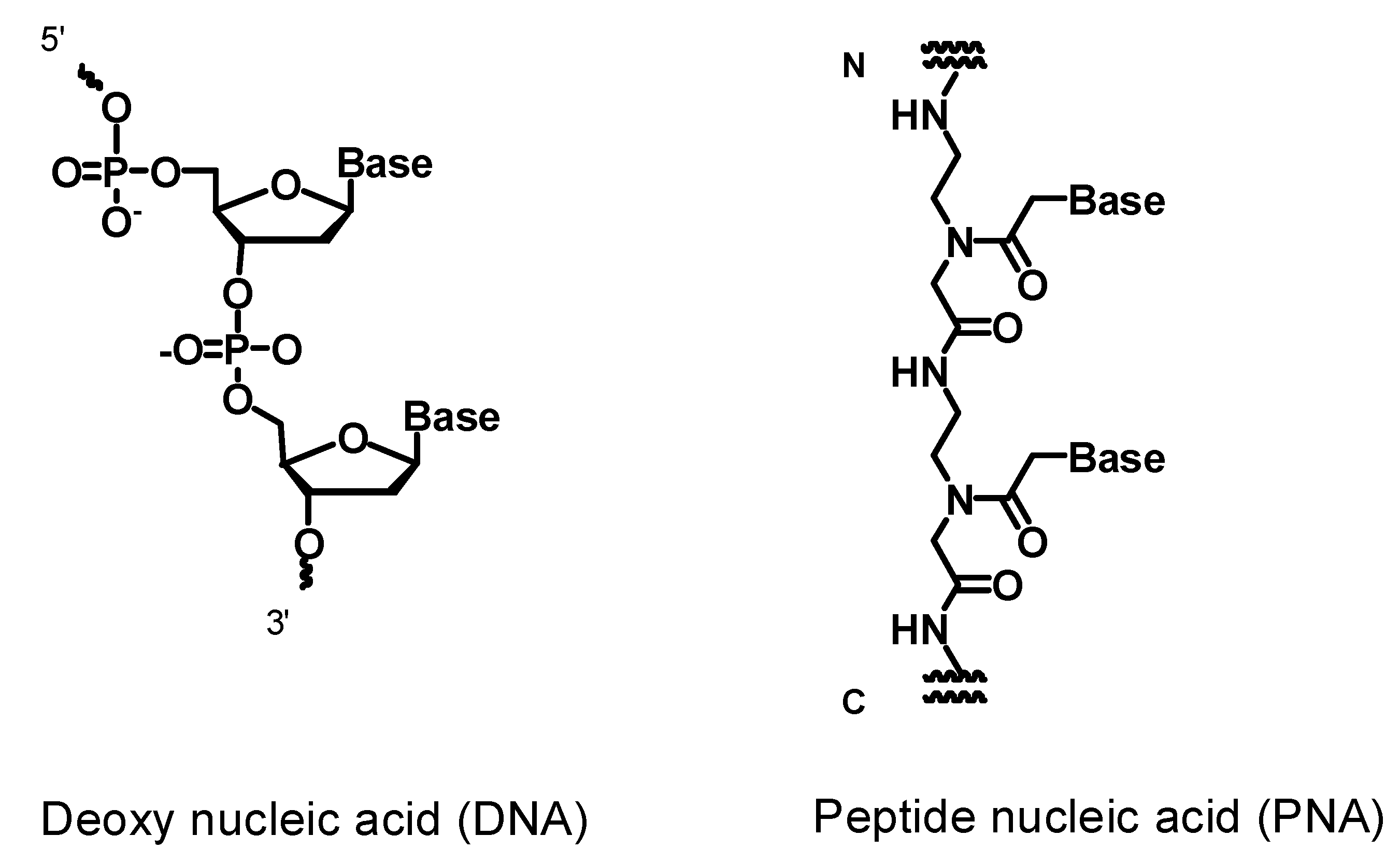
1. Introduction


2. Results and Discussion
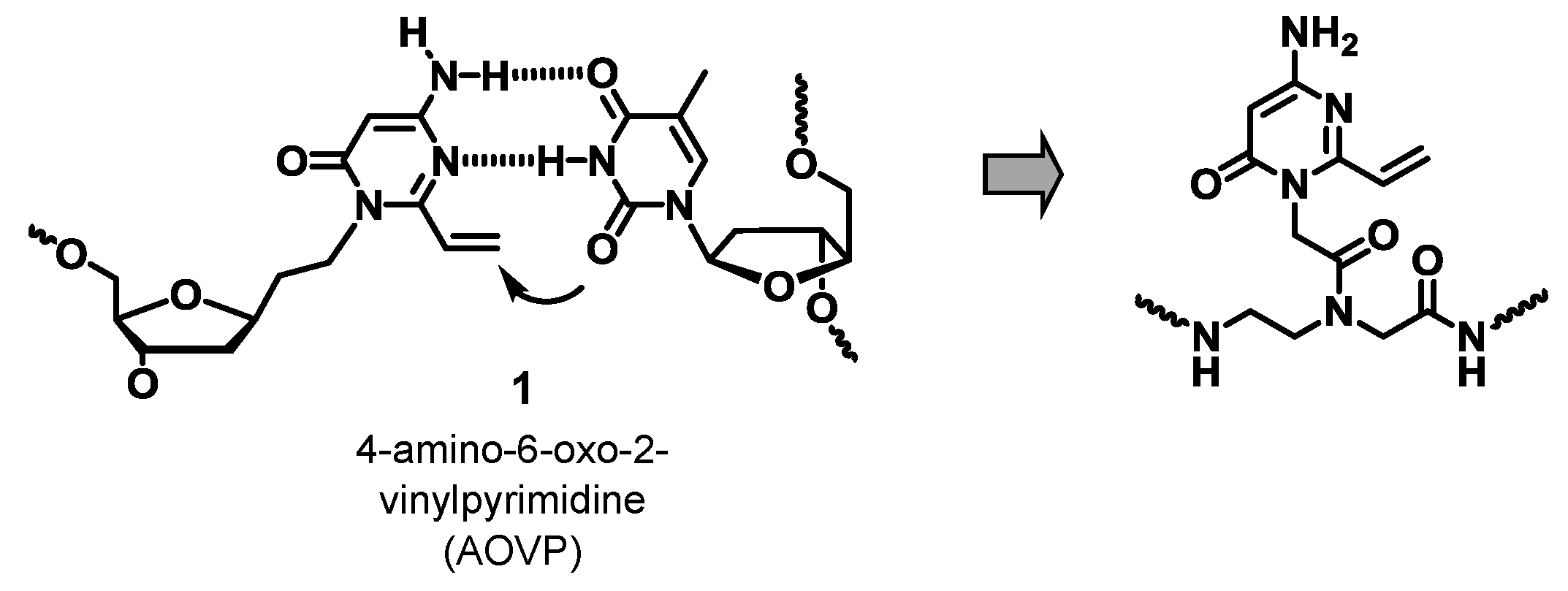
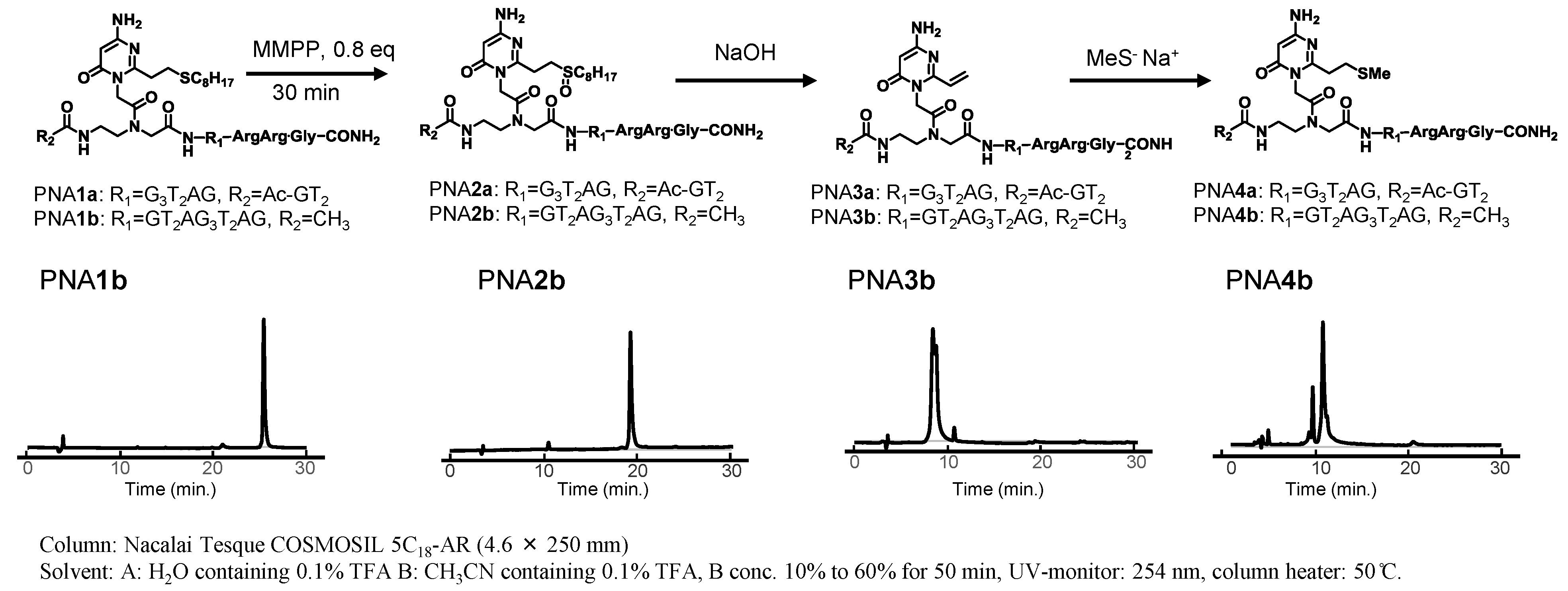
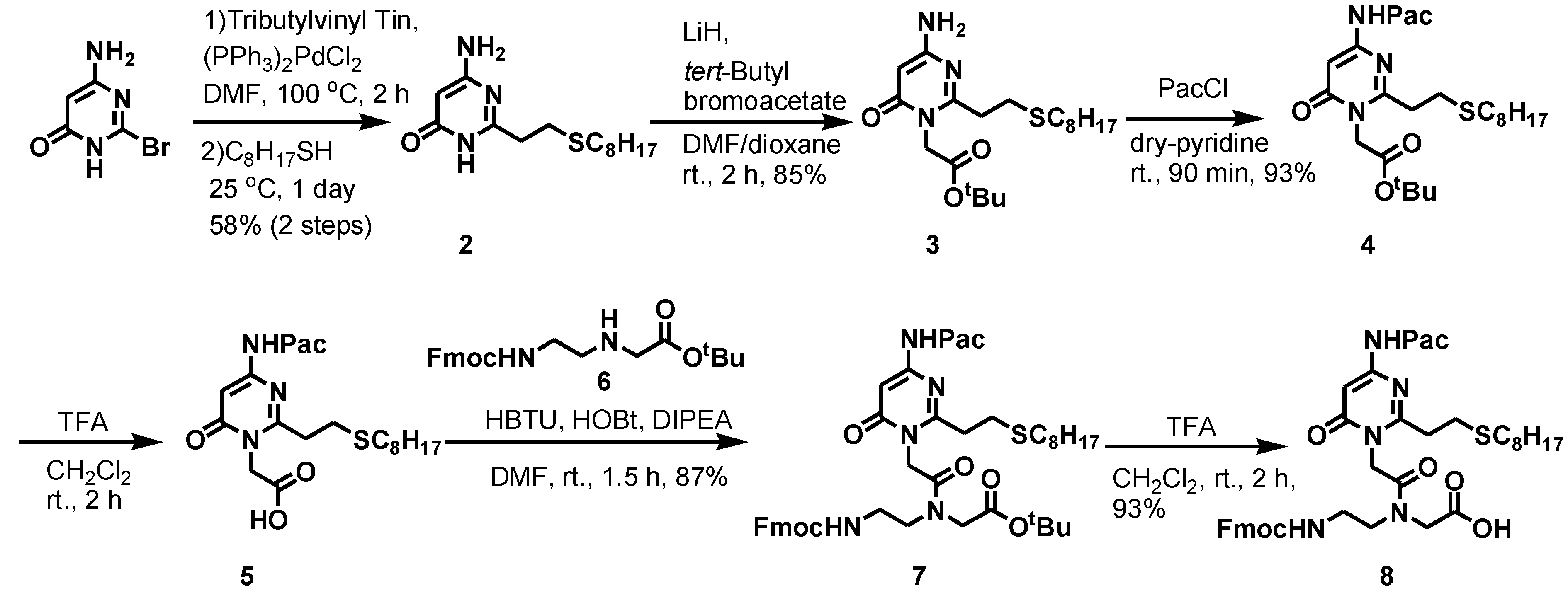
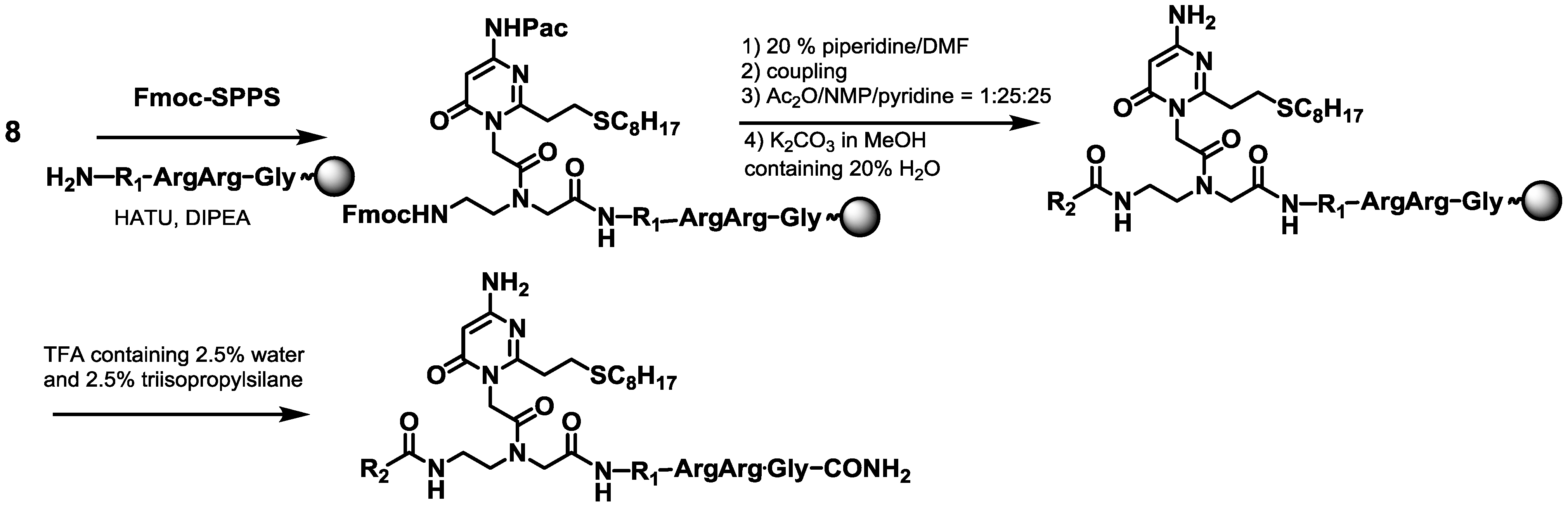
2.1. Synthesis of PNAs Containing 4-Amino-6-oxo-2-vinyl Pyrimidine (AOVP)
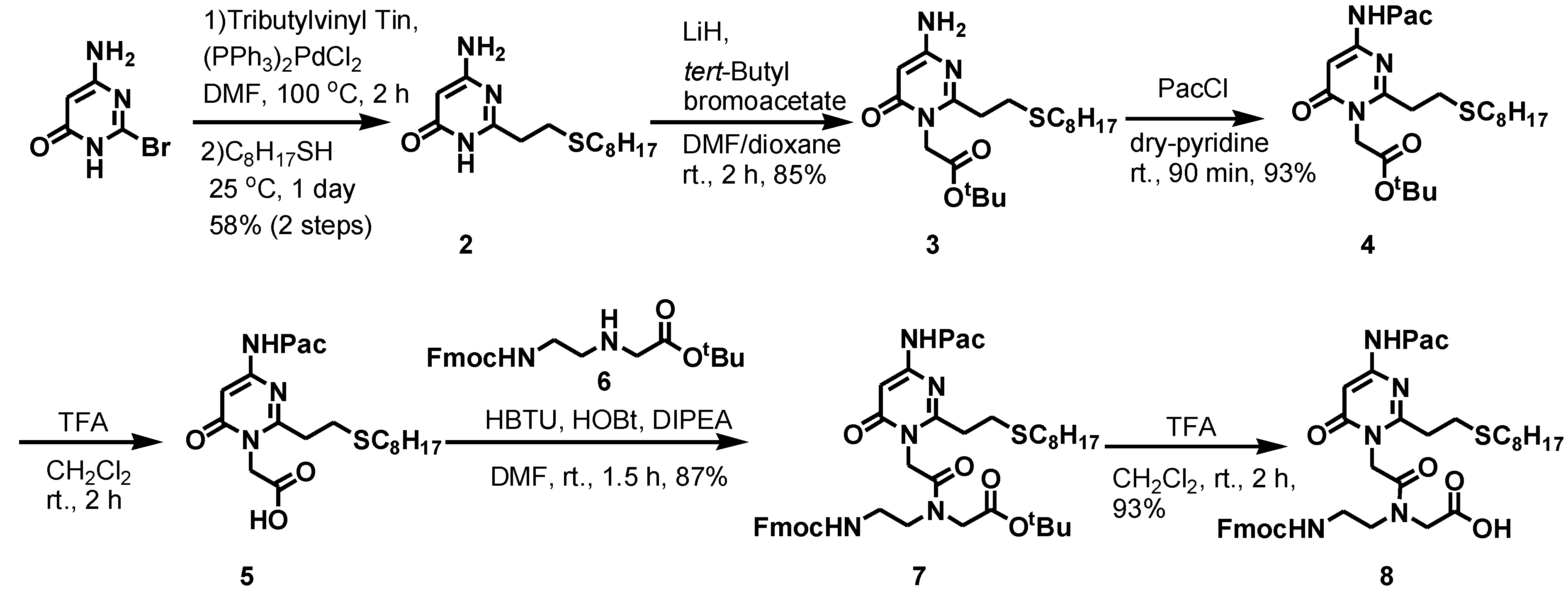


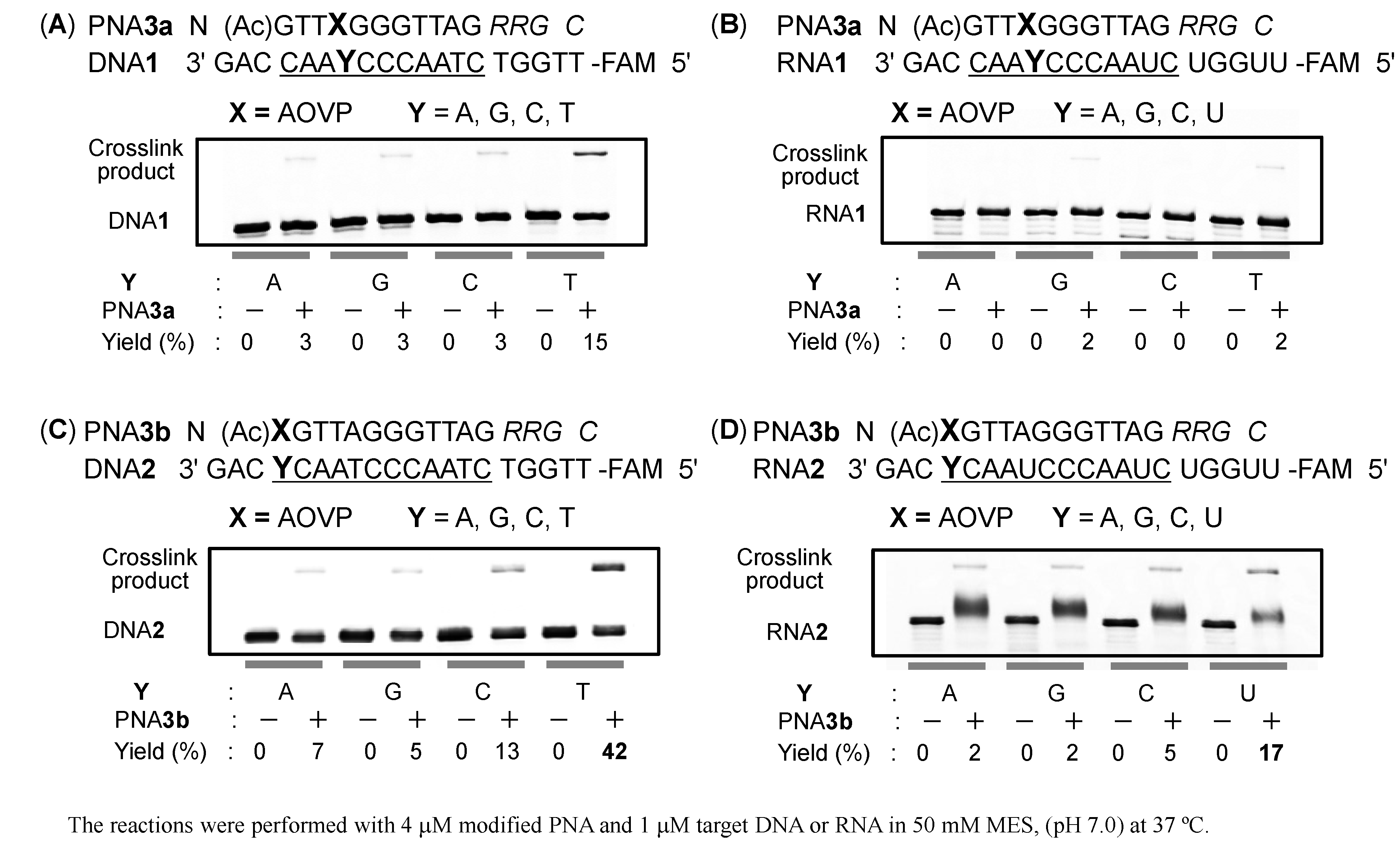
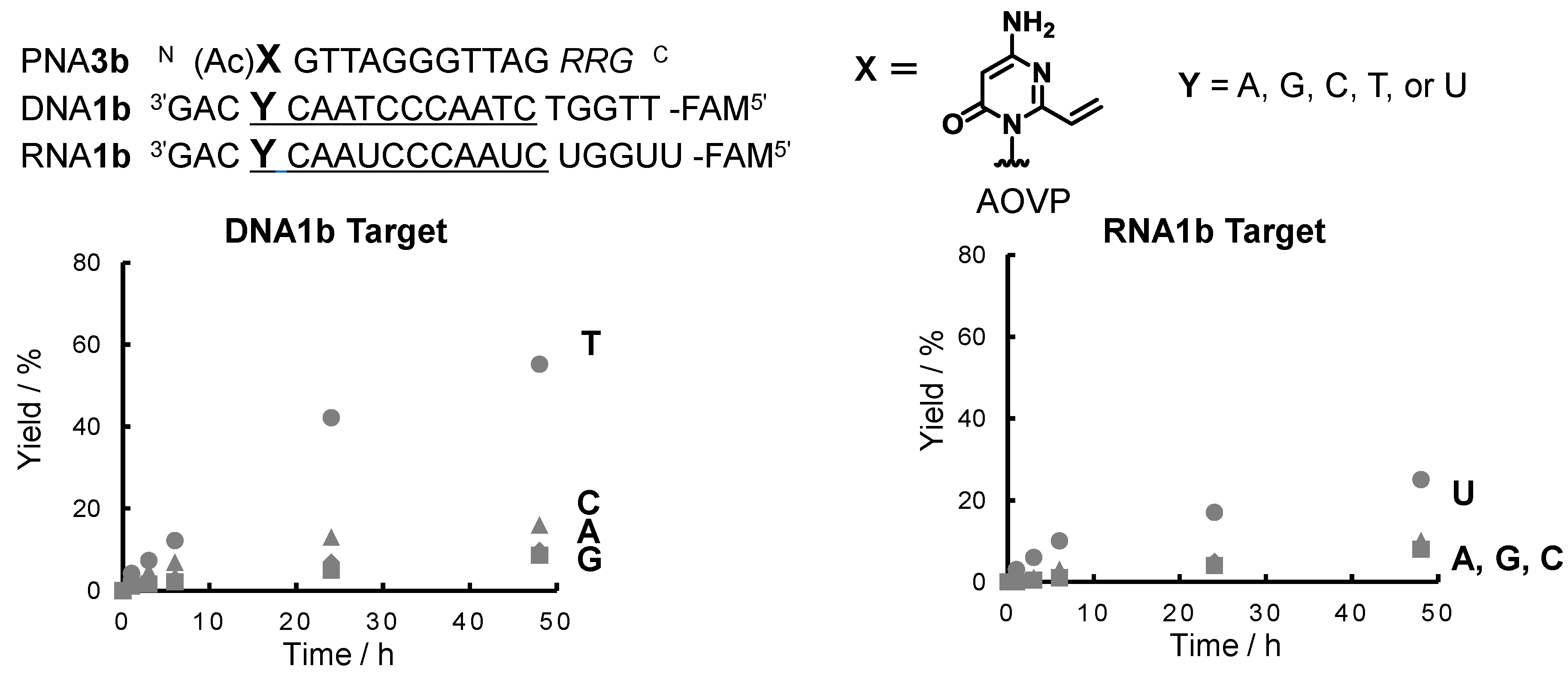
2.2. Evaluation of the Crosslinking Reactivity of the PNA Containing AOVP



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PNA4a | PNA4b | ||||
|---|---|---|---|---|---|
| Target | Tm (°C) | ∆Tm/°C | Target | Tm (°C) | ∆Tm/°C |
| DNA1 (Y=G) | 69.7 | −7.6 | DNA2 (Y=G) | 79.2 | −1.1 |
| DNA1 (Y=A) | 68.0 | −9.3 | DNA2 (Y=A) | 79.2 | −1.1 |
| DNA1 (Y=C) | 66.1 | −11.2 | DNA2 (Y=C) | 78.6 | −1.7 |
| DNA1 (Y=T) | 68.1 | −9.2 | DNA2 (Y=T) | 77.9 | −2.4 |
| RNA1 (Y=G) | 71.3 | −11.7 | RNA2 (Y=G) | 83.2 | −1.3 |
| RNA1 (Y=A) | 72.9 | −10.1 | RNA2 (Y=A) | 84.1 | −0.4 |
| RNA1 (Y=C) | 69.1 | −13.9 | RNA2 (Y=C) | 83.2 | −1.3 |
| RNA1 (Y=U) | 69.1 | −13.9 | RNA2 (Y=U) | 82.9 | −1.6 |
3. Experimental Section
3.1. General
3.2. Chemistry
3.2.1. Synthesis of 1-(tert-Butoxycarbonylmethyl)-4-amino-6-oxo-2-(2-octylthioethyl)pyrimidine (3)
3.2.2. Synthesis of 1-(tert-Butoxycarbonylmethyl)-4-phenoxyacetylamino-6-oxo-2-(2-octylthioethyl)pyrimidine (4)
3.2.3. Synthesis of 1-(Carbonylmethyl)-4-phenoxyacetylamino-6-oxo-2-(2-octylthioethyl)pyrimidine (5)
3.2.4. Synthesis of tert-Butyl-N-[2-(N-9-fluorenylmethoxycarbonyl)aminoethyl]-N-[[4-phenoxyacetyl-amino-6-oxo-2-(2-octylthioethyl)pyrimidin-1-yl]acetyl]glycinate (7)
3.2.5. Synthesis of N-[2-(N-9-Fluorenylmethoxycarbonyl)aminoethyl]-N-[[4-phenoxyacetyl-amino-6-oxo-2-(2-octylthioethyl)pyrimidin-1-yl]acetyl]glycine (8)
3.2.6. Synthesis of PNA1
3.2.7. Synthesis of PNA3
3.2.8. Synthesis of PNA4
3.3. General Procedure for the Crosslinking Reaction
3.4. Melting Temperature (Tm) Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Dispacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Demidov, V.V.; Potaman, V.N.; Frankkamenetskii, M.D.; Egholm, M.; Buchard, O.; Sonnichsen, S.H.; Nielsen, P.E. Stability of Peptide Nucleic-Acids in Human Serum and Cellular-Extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Peptide Nucleic Acids (PNA) in Chemical Biology and Drug Discovery. Chem. Biodivers. 2010, 7, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Targeted Gene Repair Facilitated by Peptide Nucleic Acids (PNA). Chembiochem 2010, 11, 2073–2076. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Gene Targeting and Expression Modulation by Peptide Nucleic Acids (PNA). Curr. Pharm. Des. 2010, 16, 3118–3123. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, S.; Karim, S.; Ali, A. Peptide nucleic acid (PNA)—A review. J. Chem. Technol. Biotechnol. 2006, 81, 892–899. [Google Scholar] [CrossRef]
- Parkash, B.; Ranjan, A.; Tiwari, V.; Gupta, S.K.; Kaur, N.; Tandon, V. Inhibition of 5'-UTR RNA Conformational Switching in HIV-1 Using Antisense PNAs. PLoS One 2012, 7, e49310. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kittaka, A. Chiral Peptide Nucleic Acids with a Substituent in the N-(2-Aminoethy)glycine Backbone. Molecules 2013, 18, 287–310. [Google Scholar] [CrossRef]
- Xia, X.; Piao, X.; Bong, D. Bifacial Peptide Nucleic Acid as an Allosteric Switch for Aptamer and Ribozyme Function. J. Am. Chem. Soc. 2014, 136, 7265–7268. [Google Scholar] [CrossRef] [PubMed]
- Devi, G.; Yuan, Z.; Lu, Y.; Zhao, Y.; Chen, G. Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes. Nucl. Acids Res. 2014, 42, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A Simple Cytosine to G-Clamp Nucleobase Substitution Enables Chiral gamma-PNAs to Invade Mixed-Sequence Double-Helical B-form DNA. Chembiochem 2008, 9, 2388–2391. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Sahu, B.; Rapireddy, S.; Bahal, R.; Wheeler, S.E.; Procopio, E.M.; Kim, J.; Joyce, S.C.; Contrucci, S.; Wang, Y.; et al. Antitumor Effects of EGFR Antisense Guanidine-Based Peptide Nucleic Acids in Cancer Models. ACS Chem. Biol. 2013, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.G.; Fabani, M.M.; Vigorito, E.; Williams, D.; Al-Obaidi, N.; Wojciechowski, F.; Hudson, R.H.E.; Seitz, O.; Gait, M.J. Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs. Nucleic Acids Res. 2012, 40, 2152–2167. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Peptide nucleic acid. A molecule with two identities. Acc. Chem. Res. 1999, 32, 624–630. [Google Scholar] [CrossRef]
- Gupta, A.; Lee, L.-L.; Roy, S.; Tanious, F.A.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. Strand Invasion of DNA Quadruplexes by PNA: Comparison of Homologous and Complementary Hybridization. Chembiochem 2013, 14, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Nagatsugi, F.; Sasaki, S. Synthesis of Reactive Oligonucleotides for Gene Targeting and Their Application to Gene Expression Regulation. Bull.Chem. Soc. Jpn. 2010, 83, 744–755. [Google Scholar] [CrossRef]
- Fujimoto, K.; Yamada, A.; Yoshimura, Y.; Tsukaguch, T.; Sakamoto, T. Details of the Ultrafast DNA Photo-Cross-Linking Reaction of 3-Cyanovinylcarbazole Nucleoside: Cis-Trans Isomeric Effect and the Application for SNP-Based Genotyping. J. Am. Chem. Soc. 2013, 135, 16161–16167. [Google Scholar] [CrossRef] [PubMed]
- Op de Beeck, Madder, A. Unprecedented C-Selective Interstrand Cross-Linking through in Situ Oxidation of Furan-Modified Oligodeoxynucleotides. J. Am. Chem. Soc. 2011, 133, 796–807. [Google Scholar] [Green Version]
- Kim, Y.; Hong, I.S. PNA/DNA interstrand cross-links from a modified PNA base upon photolysis or oxidative conditions. Bioorg.Med. Chem. Lett. 2008, 18, 5054–5057. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rokita, S.E. Inducible Alkylation of DNA by a Quinone Methide-Peptide Nucleic Acid Conjugate. Biochemistry 2012, 51, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Zhilina, Z.V.; Ziemba, A.J.; Nielsen, P.E.; Ebbinghaus, S.W. PNA-nitrogen mustard conjugates are effective suppressors of HER-2/neu and biological tools for recognition of PNA/DNA interactions. Bioconjugate Chem. 2006, 17, 214–222. [Google Scholar] [CrossRef]
- Hattori, K.; Hirohama, T.; Imoto, S.; Kusano, S.; Nagatsugi, F. Formation of highly selective and efficient interstrand cross-linking to thymine without photo-irradiation. Chem. Commun. 2009, 45, 6463–6465. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Pitts, A.E.; Katipally, R.R.; Jia, X.Y.; Rutter, J.P.; Davies, B.A.; Shay, J.W.; Wright, W.E.; Corey, D.R. Identification of determinants for inhibitor binding within the RNA active site of human telomerase using PNA scanning. Biochemistry 1997, 36, 11873–11880. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds (2–4) are available from the authors. On the other compounds, please contact with the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akisawa, T.; Ishizawa, Y.; Nagatsugi, F. Synthesis of Peptide Nucleic Acids Containing a Crosslinking Agent and Evaluation of Their Reactivities. Molecules 2015, 20, 4708-4719. https://doi.org/10.3390/molecules20034708
Akisawa T, Ishizawa Y, Nagatsugi F. Synthesis of Peptide Nucleic Acids Containing a Crosslinking Agent and Evaluation of Their Reactivities. Molecules. 2015; 20(3):4708-4719. https://doi.org/10.3390/molecules20034708
Chicago/Turabian StyleAkisawa, Takuya, Yuki Ishizawa, and Fumi Nagatsugi. 2015. "Synthesis of Peptide Nucleic Acids Containing a Crosslinking Agent and Evaluation of Their Reactivities" Molecules 20, no. 3: 4708-4719. https://doi.org/10.3390/molecules20034708