
The Curcumin Analogue 1,5-Bis(2-hydroxyphenyl)-1,4-pentadiene-3-one Induces Apoptosis and Downregulates E6 and E7 Oncogene Expression in HPV16 and HPV18-Infected Cervical Cancer Cells

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
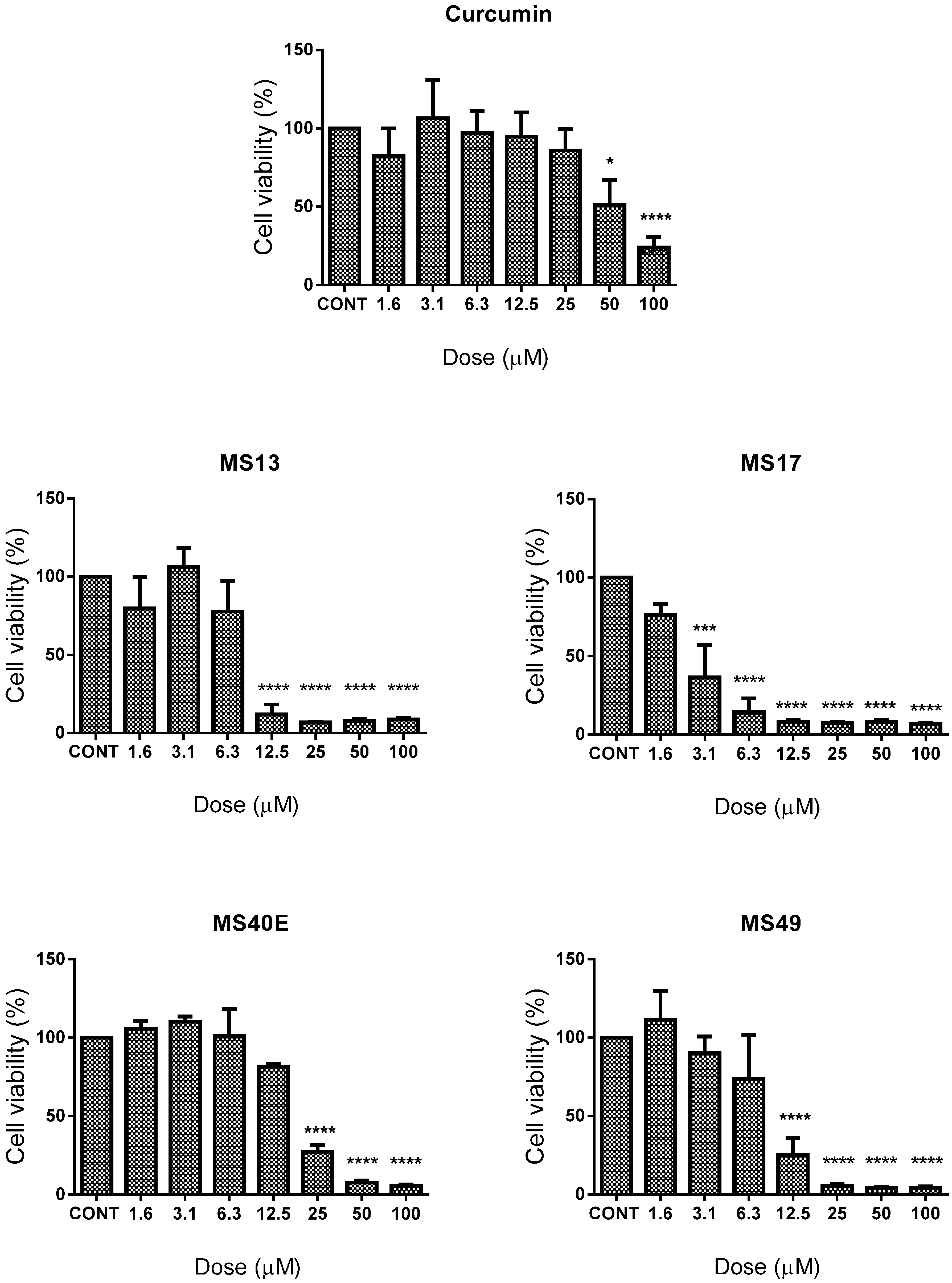
2.1. Screening and Cytotoxicity of Diarylpentanoids
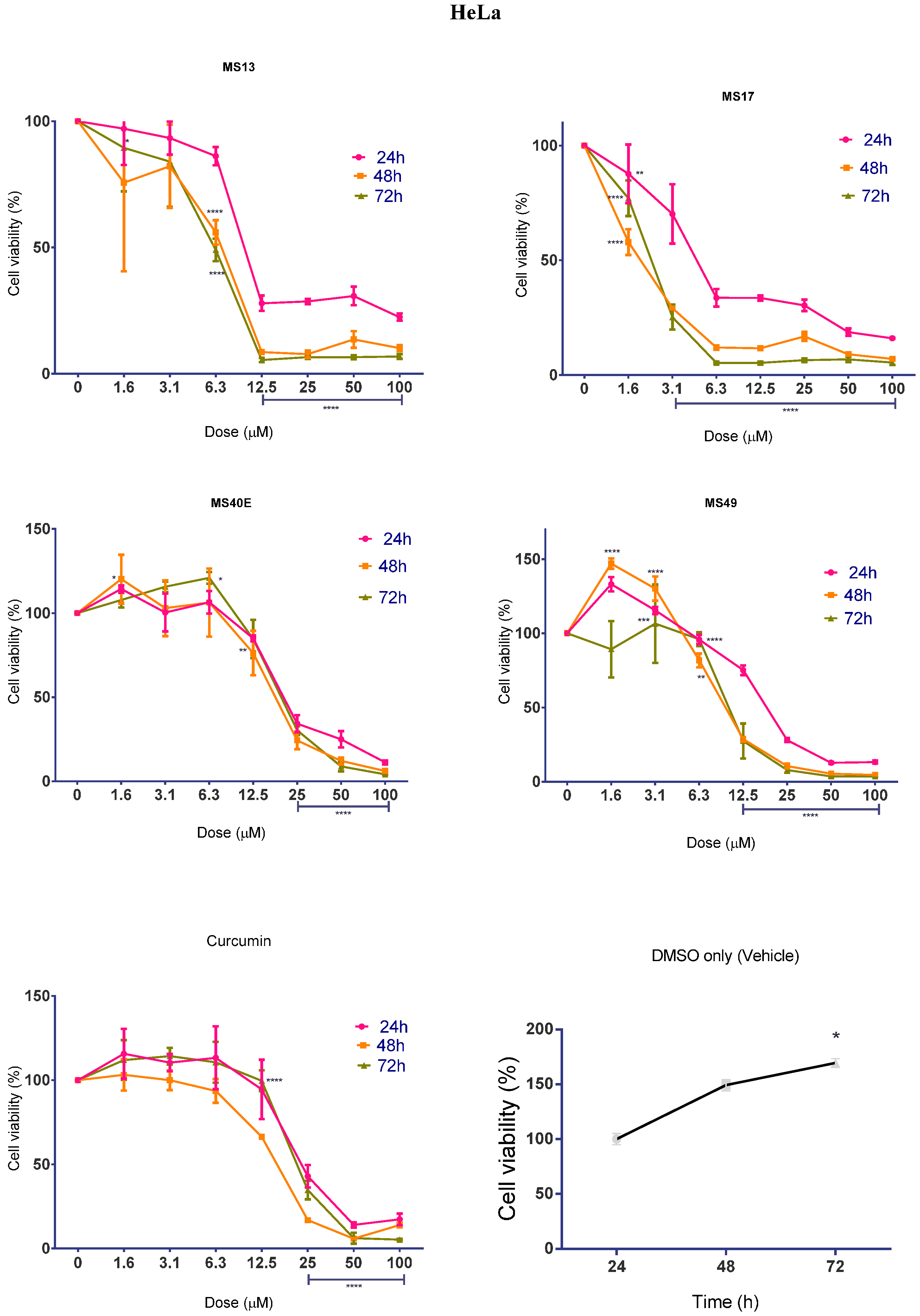
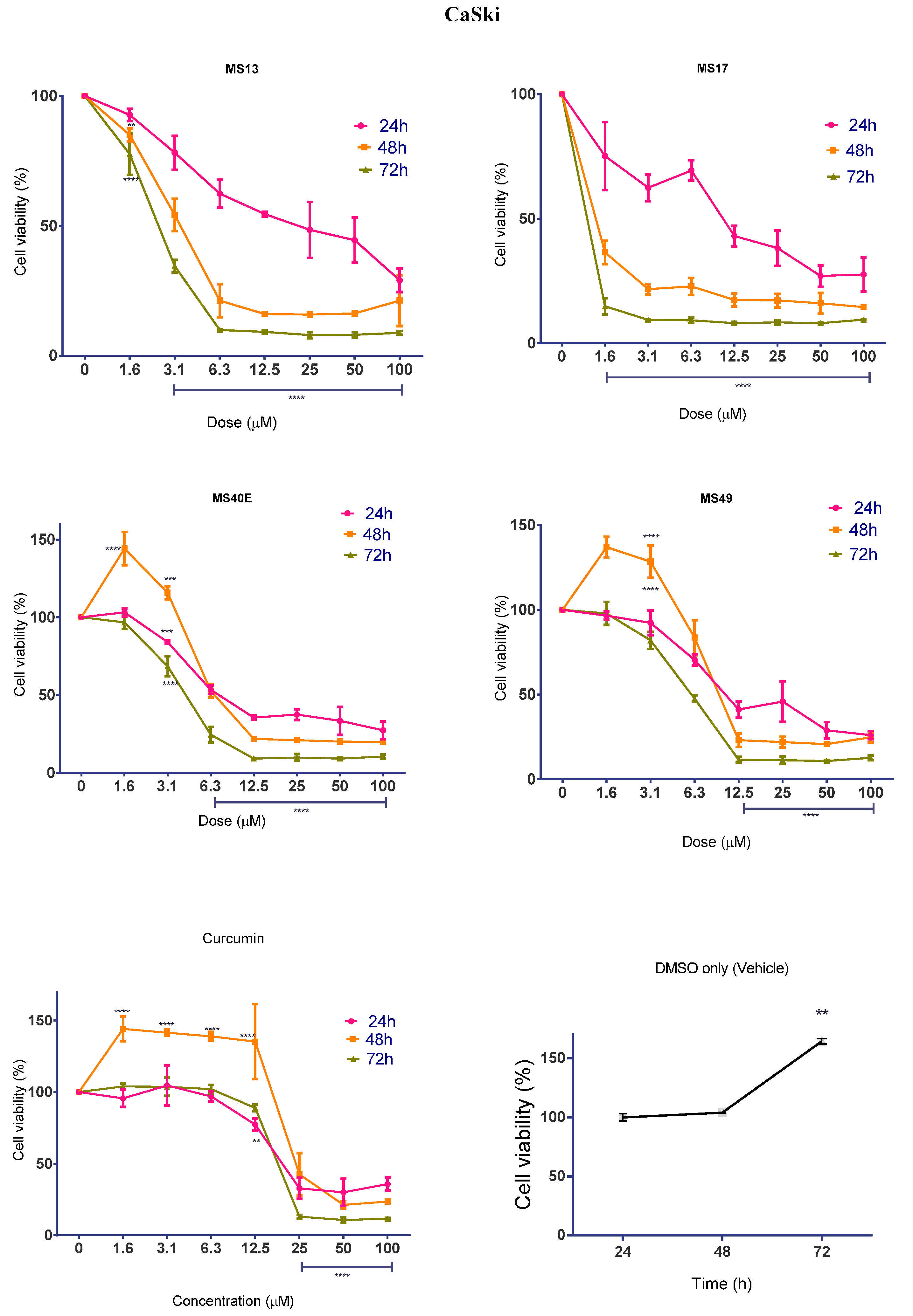
2.1.1. Diarylpentanoids Induce Cytotoxic Effects on HeLa and CaSki Cell Growth

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 (Mean ± S.E.M.) | Selective Index | |||
|---|---|---|---|---|---|
| HeLa | CaSki | MRC9 | HeLa | CaSki | |
| MS13 | 6.7 ± 2.4 | 2.8 ± 0.4 | 9.7 ± 2.4 | 145 | 346 |
| MS17 | 2.6 ± 0.9 | 1.03 ± 0.5 | 4.6 ± 1.2 | 177 | 447 |
| MS40E | 15.5 ± 1.9 | 3.5 ± 0.4 | 31.7 ± 3.2 | 204 | 906 |
| MS49 | 8.3 ± 4.6 | 6.0 ± 2.9 | 16.4 ± 4.4 | 199 | 273 |
| Curcumin | 26.7 ± 14.3 | 15.8 ± 3.1 | 26.6 ± 3.2 | 100 | 168 |


2.1.2. Diarylpentanoids Inhibit HeLa and CaSki Cell Proliferation


2.2. Apoptotic Activity Induced by MS17
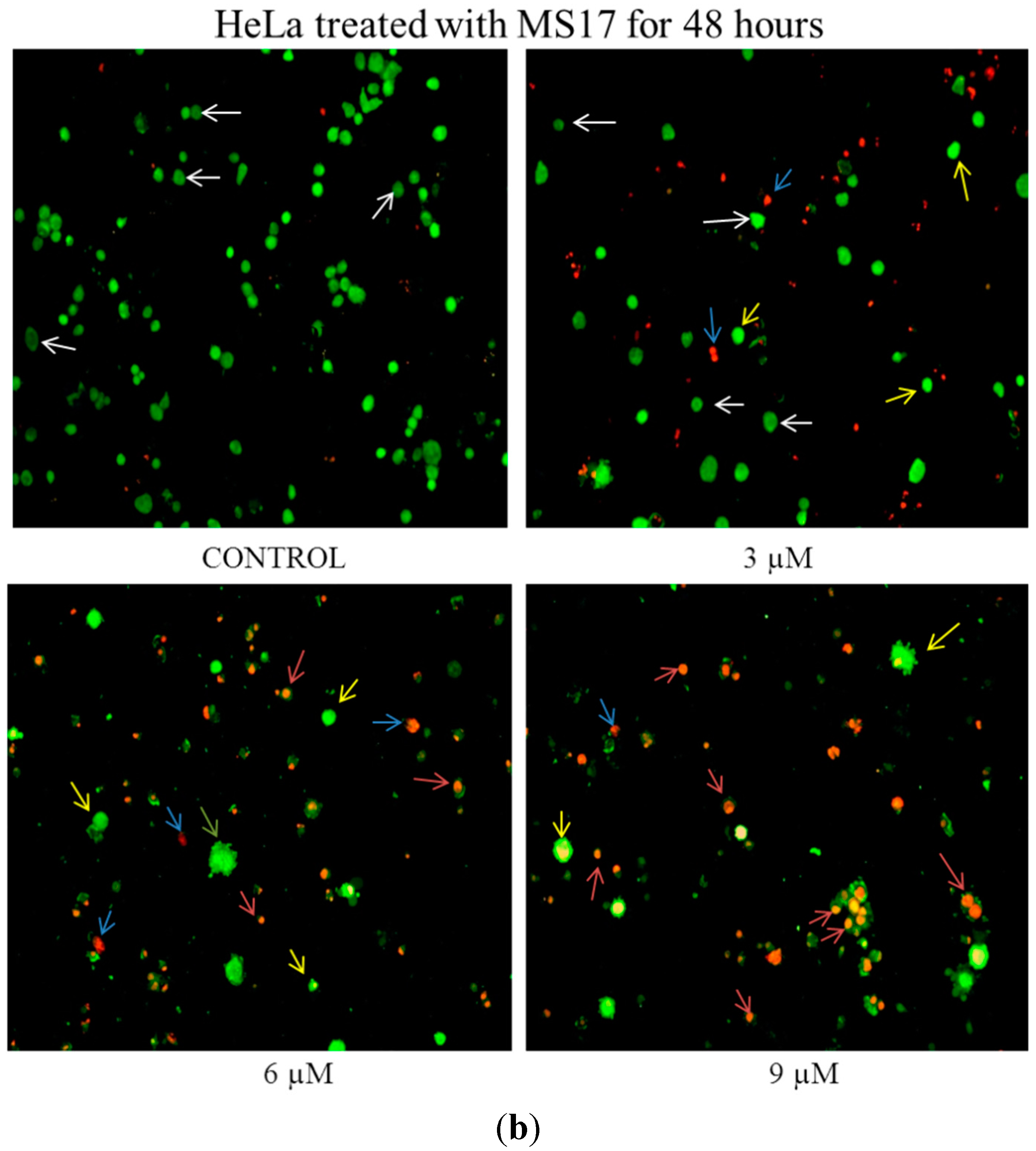
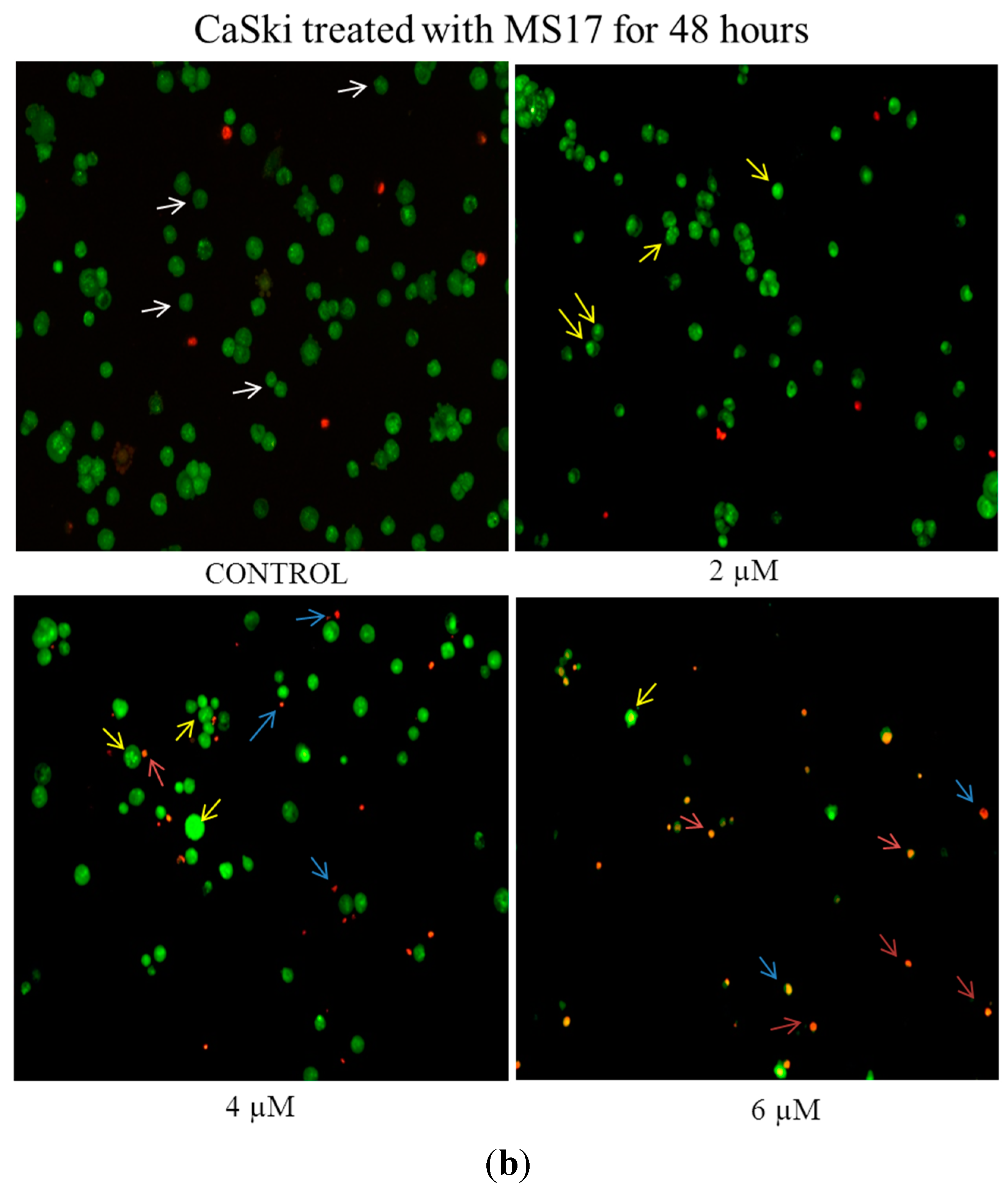
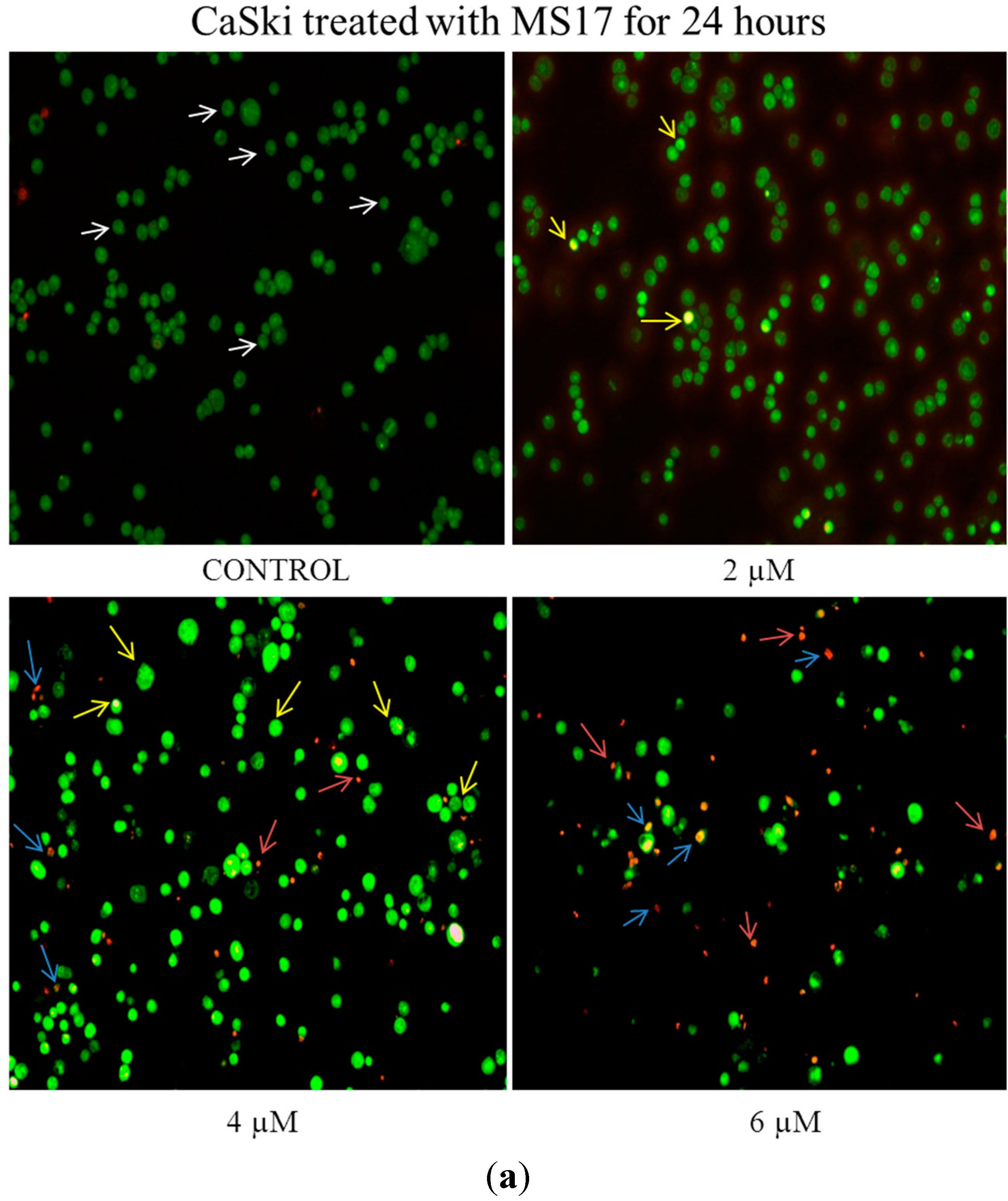
2.2.1. Morphological Observation of Treated Cancer Cells Using Fluorescence Microscopy


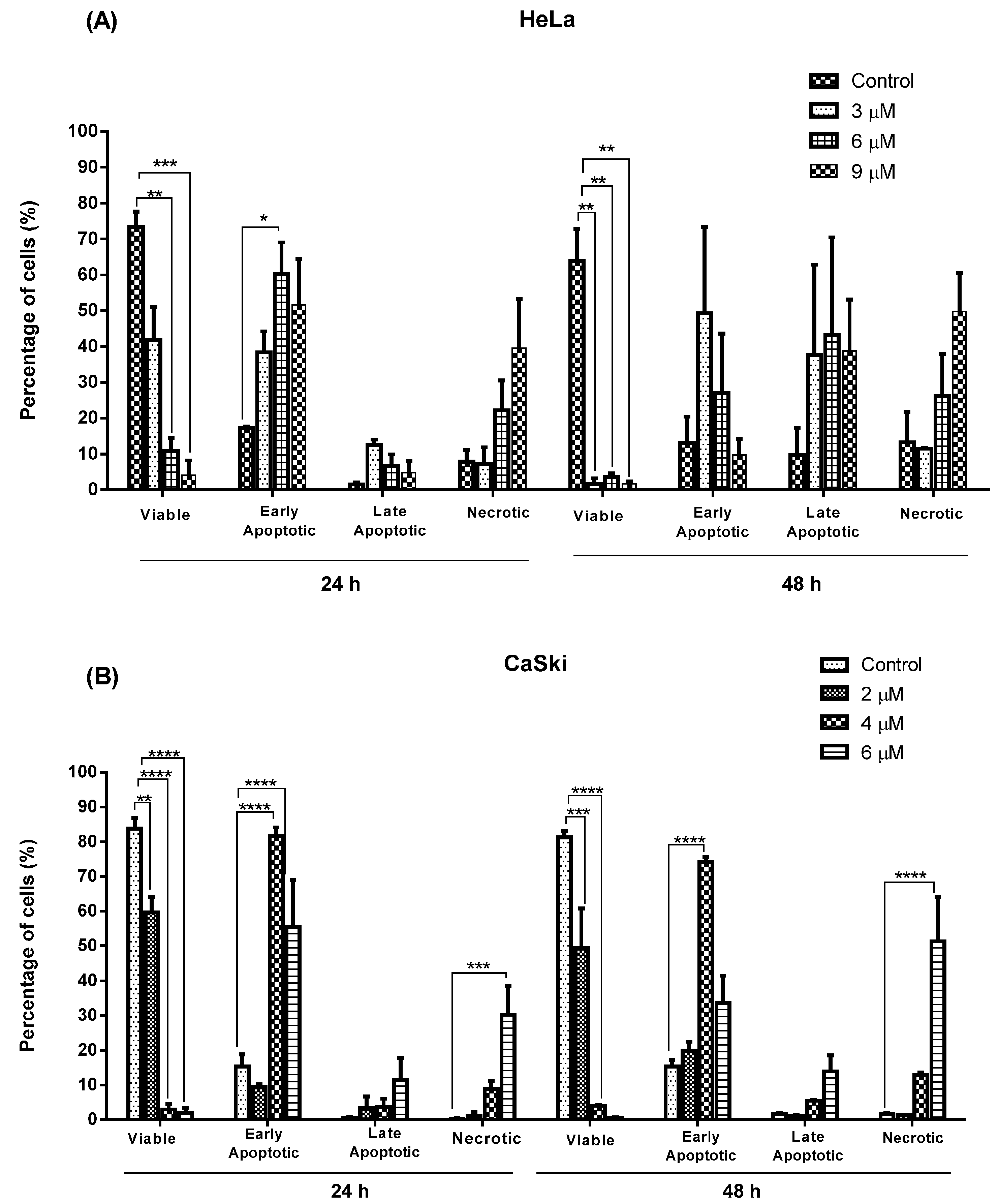
2.2.2. Quantification of Apoptotic and Necrotic Cell Percentage


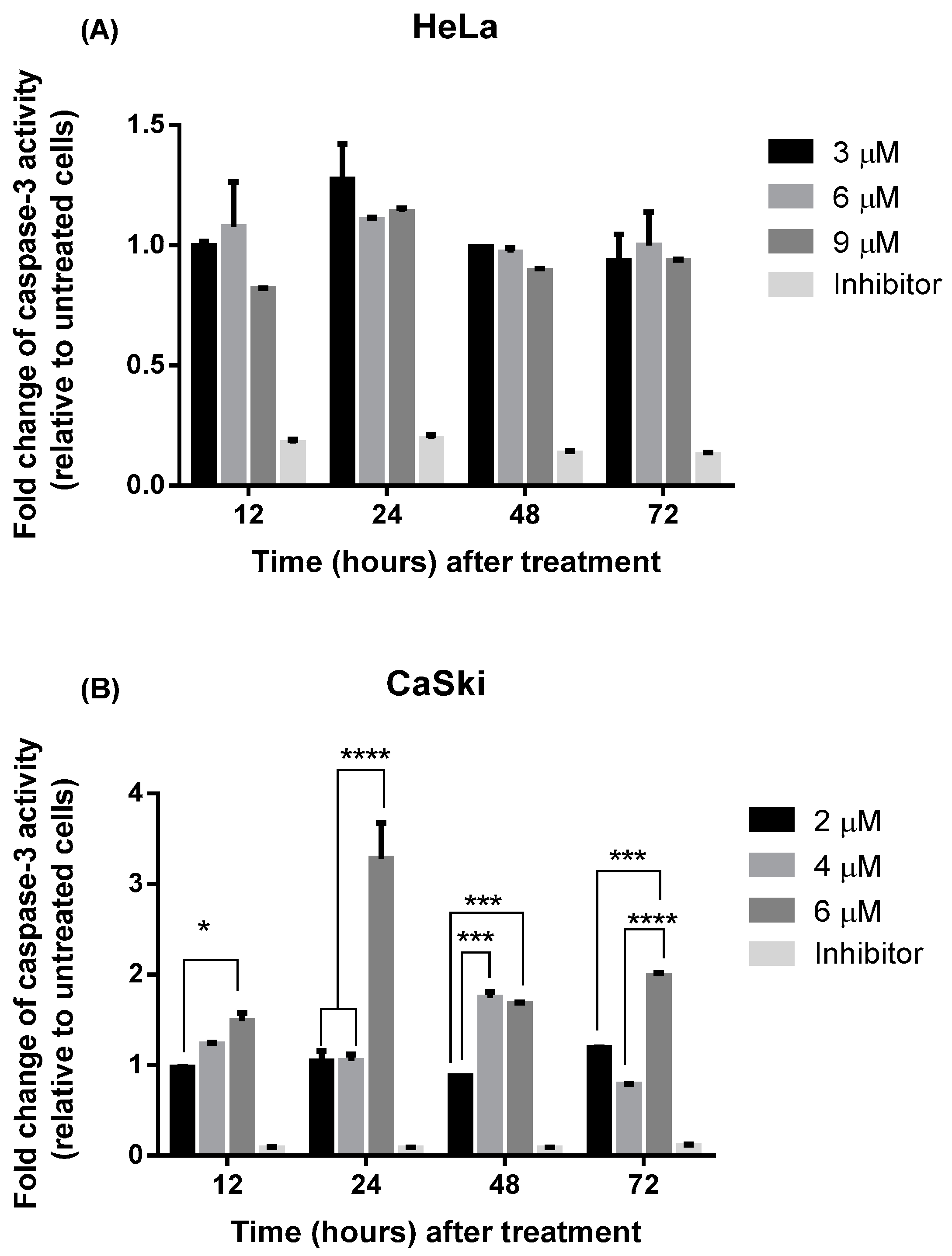
2.2.3. MS17 Increases Caspase-3 Activity

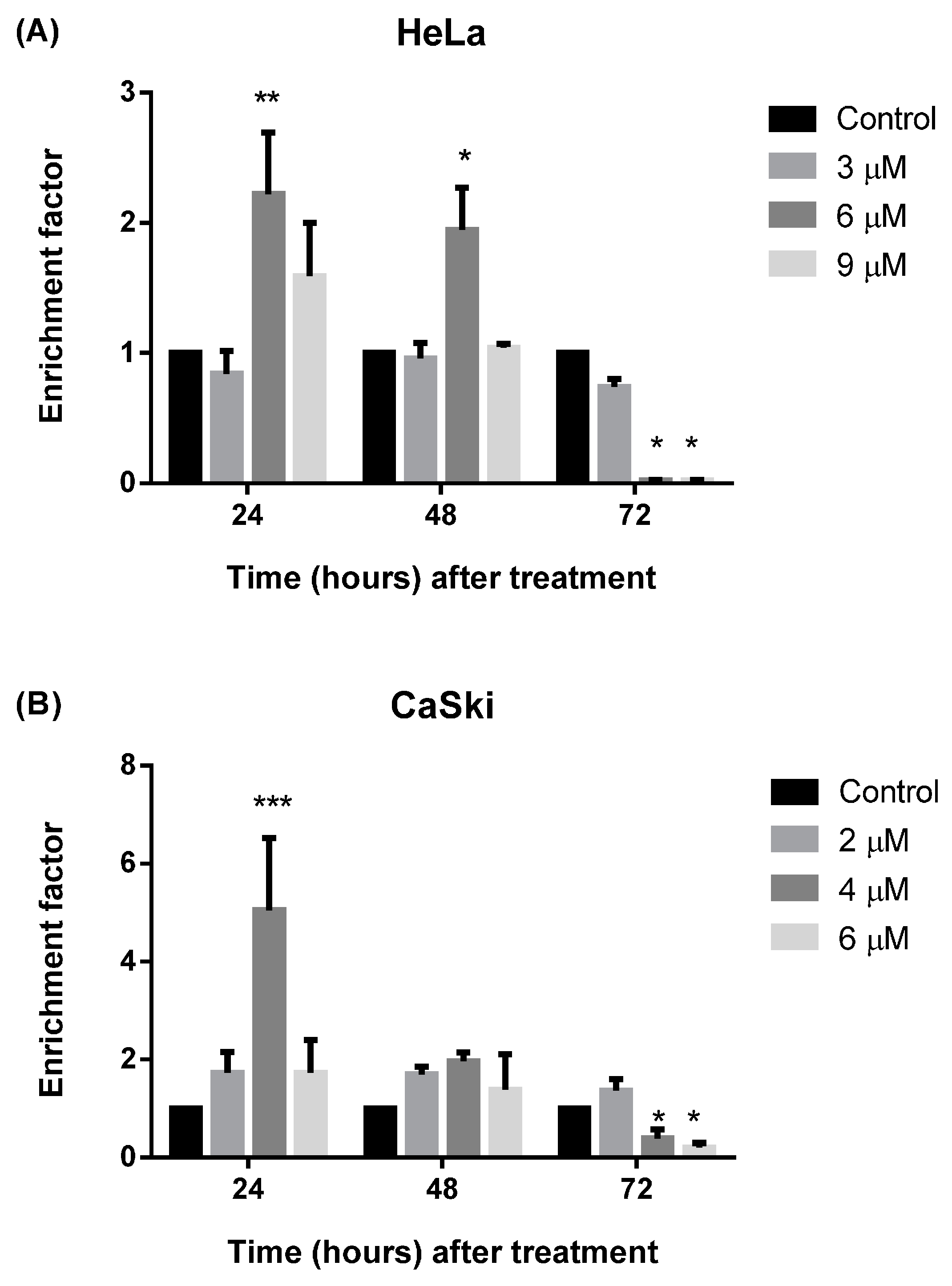
2.2.4. MS17 Induces DNA Fragmentation

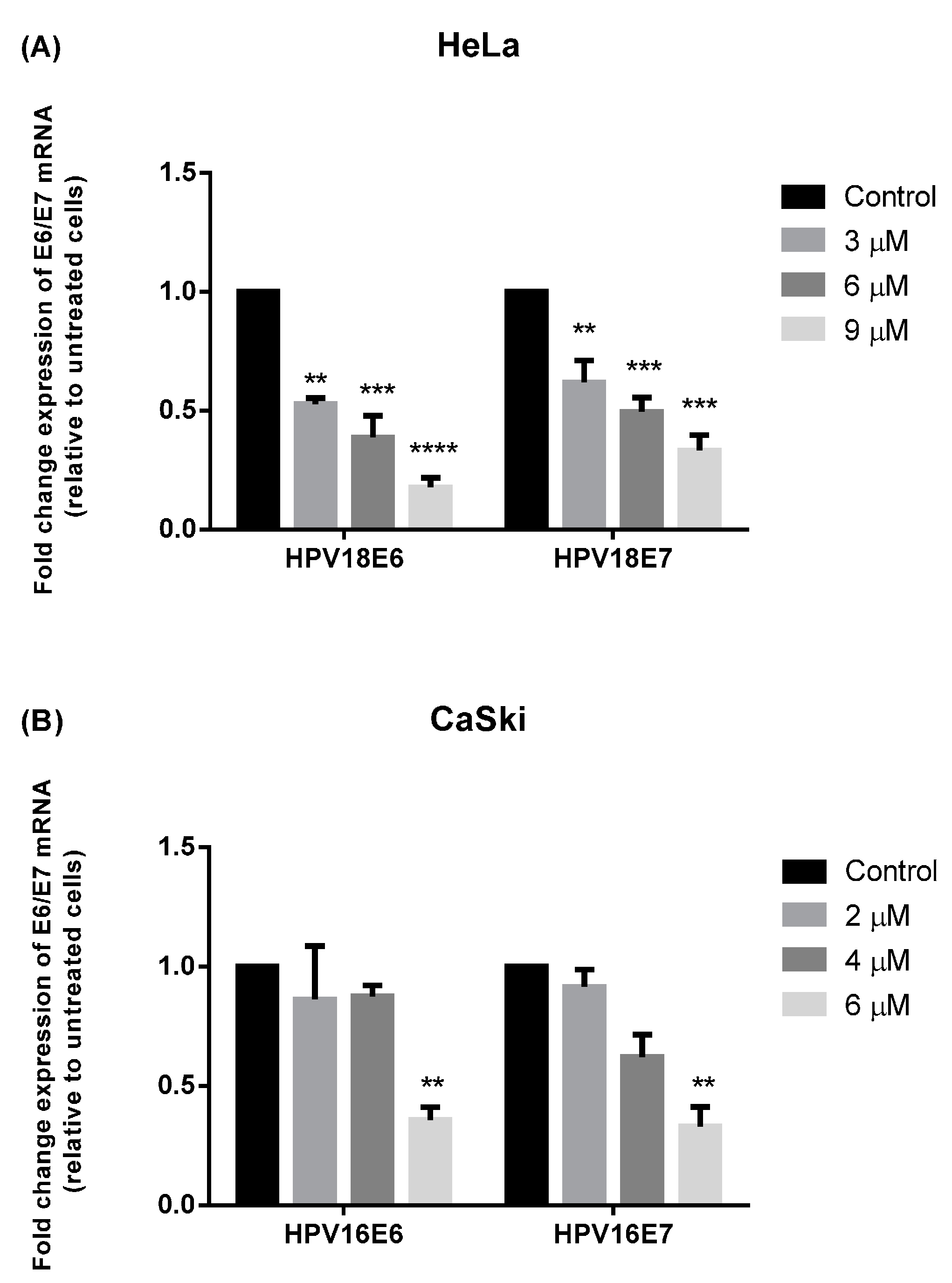
2.3. MS17 Down-Regulates E6 and E7 Viral Oncogene Expression

2.4. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Preparation of Curcumin Analogues
3.3. Cell Viability and Anti-Proliferative Assays
3.4. Induction of Apoptosis with Selected Diarylpentanoids
3.4.1. Acridine Orange-Ethidium Bromide Staining for Morphological Evaluation of Apoptosis
3.4.2. Quantification of Relative Caspase-3 Activity
3.4.3. DNA Fragmentation by Immunochemical Detection of Histone-Complexed Mono- and Oligonucleosomes
3.5. Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-PCR) for Viral Oncogenes E6 and E7
| Target | Forward Primer | Reverse Primer | Fluorescent Hybridisation Probe a |
|---|---|---|---|
| HPV 16E6 | CTGCGACGTGAGGTATATGACTTT | ACATACAGCATATGGATTCCCATCT | 6FAM-CTTTTCGGGATTTATGC-MGB-NFQ [71] |
| HPV 16E7 | CAAGTGTGACTCTACGCTTCGG | GTGGCCCATTAACAGGTCTTCCAA | 6FAM-TGCGTACAAAGCACACACGTAGACATTCGT-TAMRA [71] |
| HPV 18E6 | CTATAGAGGCCAGTGCCATTCG | TTATACTTGTGTTTCTCTGCGTCG | 6FAM-CAACCGAGCACGACAGGAACGACTCCA-TAMRA [70] |
| HPV 18E7 | TAATCATCAACATTTACCAGCCCG | CGTCTGCTGAGCTTTCTACTACTA | 6FAM-CGAGCCGAACCACAACGTCACACAATGTT-TAMRA [70] |
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bruni, L.; Barrionuevo-Rosas, L.; Serrano, B.; Brotons, M.; Albero, G.; Cosano, R.; Muñoz, J.; Bosch, F.X.; de Sanjosé, S.; Castellsagué, X. Human Papillomavirus and Related Diseases in Malaysia. Summary Report 2014–08–22 [Data Accessed]; ICO Information Centre on HPV and Cancer (HPV Information Centre): Barcelona, Spain, 2014. [Google Scholar]
- Bruni, L.; Barrionuevo-Rosas, L.; Serrano, B.; Brotons, M.; Albero, G.; Cosano, R.; Muñoz, J.; Bosch, F.X.; de Sanjosé, S.; Castellsagué, X. Human Papillomavirus and Related Diseases in the World. Summary Report 2014–08–22 [Data Accessed]; ICO Information Centre on HPV and Cancer (HPV Information Centre): Barcelona, Spain, 2014. [Google Scholar]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar]
- Aggarwal, B.B.; Harikumar, K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009, 41, 40–59. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar]
- Mimeault, M.; Batra, S.K. Potential applications of curcumin and its novel synthetic analogs and nanotechnology-based formulations in cancer prevention and therapy. Chin. Med. 2011, 6, 31–49. [Google Scholar] [CrossRef]
- Teiten, M.H.; Eifes, S.; Dicato, M.; Diederich, M. Curcumin-the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins 2010, 2, 128–162. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kumar, A.; Aggarwal, M.S.; Shishodia, S. Curcumin derived from turmeric (Curcuma longa): A spice for all seasons. In Phytochemicals in Cancer Chemoprevention; CRC Press: New York, NY, USA, 2005; pp. 349–387. [Google Scholar]
- Divya, C.S.; Pillai, M.R. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Mol. Carcinog. 2006, 45, 320–332. [Google Scholar] [CrossRef]
- Maher, D.M.; Bell, M.C.; O’Donnell, E.A.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Curcumin suppresses human papillomavirus oncoproteins, restores p53, Rb, and PTPN13 proteins and inhibits benzo[a]pyrene-induced upregulation of HPV E7. Mol. Carcinog. 2011, 50, 47–57. [Google Scholar] [CrossRef]
- Singh, M.; Singh, N. Molecular mechanism of curcumin induced cytotoxicity in human cervical carcinoma cells. Mol. Cell. Biochem. 2009, 325, 107–119. [Google Scholar] [CrossRef]
- Ammon, H.P.; Wahl, M.A. Pharmacology of Curcuma longa. Planta Med. 1991, 57, 1–7. [Google Scholar] [CrossRef]
- Chainani-Wu, N. Safety and anti-inflammatory activity of curcumin: A component of tumeric (Curcuma longa). J. Altern. Complement. Med. 2003, 9, 161–168. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar]
- Lao, C.D.; Ruffin, M.T.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10–13. [Google Scholar] [CrossRef]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 2001, 7, 1894–1900. [Google Scholar]
- Garcea, G.; Jones, D.J.; Singh, R.; Dennison, A.R.; Farmer, P.B.; Sharma, R.A.; Steward, W.P.; Gescher, A.J.; Berry, D.P. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br. J. Cancer 2004, 90, 1011–1015. [Google Scholar] [CrossRef]
- Perkins, S.; Verschoyle, R.D.; Hill, K.; Parveen, I.; Threadgill, M.D.; Sharma, R.A.; Williams, M.L.; Steward, W.P.; Gescher, A.J. Chemopreventive efficacy and pharmacokinetics of curcumin in the min/+ mouse, a model of familial adenomatous polyposis. Cancer Epidemiol. Biomark. Prev. 2002, 11, 535–540. [Google Scholar]
- Ravindranath, V.; Chandrasekhara, N. Absorption and tissue distribution of curcumin in rats. Toxicology 1980, 16, 259–265. [Google Scholar] [CrossRef]
- Ravindranath, V.; Chandrasekhara, N. Metabolism of curcumin—Studies with [3H]curcumin. Toxicology 1981, 22, 337–344. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharmacol. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Terlikowska, K.M.; Witkowska, A.M.; Zujko, M.E.; Dobrzycka, B.; Terlikowski, S.J. Potential application of curcumin and its analogues in the treatment strategy of patients with primary epithelial ovarian cancer. Int. J. Mol. Sci. 2014, 15, 21703–21722. [Google Scholar] [CrossRef]
- Zhou, Q.M.; Chen, Q.L.; Du, J.; Wang, X.F.; Lu, Y.Y.; Zhang, H.; Su, S.B. Synergistic effect of combinatorial treatment with curcumin and mitomycin C on the induction of apoptosis of breast cancer cells: A cDNA microarray analysis. Int. J. Mol. Sci. 2014, 15, 16284–16301. [Google Scholar] [CrossRef]
- Zhou, D.H.; Wang, X.; Yang, M.; Shi, X.; Huang, W.; Feng, Q. Combination of Low Concentration of (−)-Epigallocatechin Gallate (EGCG) and Curcumin Strongly Suppresses the Growth of Non-Small Cell Lung Cancer in Vitro and in Vivo through Causing Cell Cycle Arrest. Int. J. Mol. Sci. 2013, 14, 12023–12036. [Google Scholar] [CrossRef]
- Ohori, H.; Yamakoshi, H.; Tomizawa, M.; Shibuya, M.; Kakudo, Y.; Takahashi, A.; Takahashi, S.; Kato, S.; Suzuki, T.; Ishioka, C.; et al. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol. Cancer Ther. 2006, 5, 2563–2571. [Google Scholar] [CrossRef]
- Yamakoshi, H.; Ohori, H.; Kudo, C.; Sato, A.; Kanoh, N.; Ishioka, C.; Shibata, H.; Iwabuchi, Y. Structure-activity relationship of C5-curcuminoids and synthesis of their molecular probes thereof. Bioorg. Med. Chem. 2010, 18, 1083–1092. [Google Scholar] [CrossRef]
- Cen, L.; Hutzen, B.; Ball, S.; DeAngelis, S.; Chen, C.L.; Fuchs, J.R.; Li, C.; Li, P-K.; Lin, J. New structural analogues of curcumin exhibit potent growth suppressive activity in human colorectal carcinoma cells. BMC Cancer 2009, 9. [Google Scholar] [CrossRef]
- Fajardo, A.M.; MacKenzie, D.A.; Ji, M.; Deck, L.M.; Vander Jagt, D.L.; Thompson, T.A.; Bisoffi, M. The curcumin analog ca27 down-regulates androgen receptor through an oxidative stress mediated mechanism in human prostate cancer cells. Prostate 2012, 72, 612–625. [Google Scholar] [CrossRef]
- Friedman, L.; Lin, L.; Ball, S.; Bekaii-Saab, T.; Fuchs, J.; Li, P.K.; Li, C.; Lin, J. Curcumin analogues exhibit enhanced growth suppressive activity in human pancreatic cancer cells. Anticancer Drugs 2009, 20, 444–449. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Du, Y.; Thomas, S.L.; Zhao, J.; Sun, S.Y.; Khuri, F.R.; Wang, C.Y.; Shoji, M.; Sun, A.; Snyder, J.P.; et al. Inhibition of IkappaB kinase-nuclear factor-kappaB signaling pathway by 3,5-bis(2-flurobenzylidene)piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol. Pharmacol. 2008, 74, 654–661. [Google Scholar] [CrossRef]
- Kudo, C.; Yamakoshi, H.; Sato, H.; Ohori, H.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Synthesis of 86 species of 1,5-diaryl-3-oxo-1,4-pentadienes analogs of curcumin can yield a good lead in vivo. BMC Pharmacol. 2011, 11. [Google Scholar] [CrossRef]
- Olivera, A.; Moore, T.W.; Hu, F.; Brown, A.P.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Yoon, Y.; Shim, H.; Marcus, A.I.; et al. Inhibition of the NF-kappaB signaling pathway by the curcumin analog, 3,5-Bis(2-pyridinylmethylidene)-4-piperidone (EF31): Anti-inflammatory and anti-cancer properties. Int. Immunopharmacol. 2012, 12, 368–377. [Google Scholar] [CrossRef]
- Selvendiran, K.; Ahmed, S.; Dayton, A.; Kuppusamy, M.L.; Tazi, M.; Bratasz, A.; Tong, L.; Rivera, B.K.; Kalai, T.; Hideg, K.; et al. Safe and targeted anticancer efficacy of a novel class of antioxidant-conjugated difluorodiarylidenyl piperidones: Differential cytotoxicity in healthy and cancer cells. Free Radic. Biol. Med. 2010, 48, 1228–1235. [Google Scholar] [CrossRef]
- Selvendiran, K.; Tong, L.; Bratasz, A.; Kuppusamy, M.L.; Ahmed, S.; Ravi, Y.; Trigg, N.J.; Rivera, B.K.; Kalai, T.; Hideg, K.; et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol. Cancer Ther. 2010, 9, 1169–1179. [Google Scholar] [CrossRef]
- Subramaniam, D.; May, R.; Sureban, S.M.; Lee, K.B.; George, R.; Kuppusamy, P.; Ramanujam, R.P.; Hideg, K.; Dieckgraefe, B.K.; Houchen, C.W.; et al. Diphenyl difluoroketone: A curcumin derivative with potent in vivo anticancer activity. Cancer Res. 2008, 68, 1962–1969. [Google Scholar] [CrossRef]
- Tan, X.; Sidell, N.; Mancini, A.; Huang, R.P.; Shenming, W.; Horowitz, I.R.; Liotta, D.C.; Taylor, R.N.; Wieser, F. Multiple anticancer activities of EF24, a novel curcumin analog, on human ovarian carcinoma cells. Reprod. Sci. 2010, 17, 931–940. [Google Scholar] [CrossRef]
- Thomas, S.L.; Zhao, J.; Li, Z.; Lou, B.; Du, Y.; Purcell, J.; Snyder, J.P.; Khuri, F.R.; Liotta, D.; Fu, H. Activation of the p38 pathway by a novel monoketone curcumin analog, EF24, suggests a potential combination strategy. Biochem. Pharmacol. 2010, 80, 1309–1316. [Google Scholar] [CrossRef]
- Zhu, S.; Moore, T.W.; Lin, X.; Morii, N.; Mancini, A.; Howard, R.B.; Culver, D.; Arrendale, R.F.; Reddy, P.; Evers, T.J.; et al. Synthetic curcumin analog EF31 inhibits the growth of head and neck squamous cell carcinoma xenografts. Integr. Biol. 2012, 4, 633–640. [Google Scholar] [CrossRef]
- Citalingam, K.; Abas, F.; Lajis, N.H.; Othman, I.; Naidu, R. Anti-proliferative effect and induction of apoptosis in androgen-independent human prostate cancer cells by 1,5-bis(2-hydroxyphenyl)-1,4-pentadiene-3-one. Molecules 2015, 20, 3406–3430. [Google Scholar] [CrossRef]
- Popiolkiewicz, J.; Polkowski, K.; Skierski, J.S.; Mazurek, A.P. In vitro toxicity evaluation in the development of new anticancer drugs-genistein glycosides. Cancer Lett. 2005, 229, 67–75. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Amarante-Mendes, G.P.; Finucane, D.; Brunner, T.; Bossy-Wetzel, E.; Green, D.R. Acridine Orange/Ethidium Bromide (AO/EB) Staining to Detect Apoptosis. CSH Protoc. 2006, 3. [Google Scholar] [CrossRef]
- Squier, M.K.; Cohen, J.J. Standard quantitative assays for apoptosis. Mol. Biotechnol. 2001, 19, 305–312. [Google Scholar] [CrossRef]
- Mosley, C.A.; Liotta, D.C.; Snyder, J.P. Highly active anticancer curcumin analogues. Adv. Exp. Med. Biol. 2007, 595, 77–103. [Google Scholar]
- Lin, L.; Hutzen, B.; Ball, S.; Foust, E.; Sobo, M.; Deangelis, S.; Pandit, B.; Friedman, L.; Li, C.; Li, P.K.; et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 2009, 100, 1719–1727. [Google Scholar] [CrossRef]
- Eischen, C.M.; Kottke, T.J.; Martins, L.M.; Basi, G.S.; Tung, J.S.; Earnshaw, W.C.; Leibson, P.J.; Kaufmann, S.H. Comparison of apoptosis in wild-type and Fas-resistant cells: Chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand interactions. Blood 1997, 90, 935–943. [Google Scholar]
- Holm, B.; Jensen, P.B.; Sehested, M.; Hansen, H.H. In vivo inhibition of etoposide-mediated apoptosis, toxicity, and antitumor effect by the topoisomerase II-uncoupling anthracycline aclarubicin. Cancer Chemother. Pharmacol. 1994, 34, 503–508. [Google Scholar] [CrossRef]
- Ferreira, C.G.; Epping, M.; Kruyt, F.A.E.; Giaccone, G. Apoptosis: Target of Cancer Therapy. Clin. Cancer Res. 2002, 8, 2024–2034. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Nunez, G.; Benedict, M.A.; Hu, Y.; Inohara, N. Caspases: The proteases of the apoptotic pathway. Oncogene 1998, 17, 3237–3245. [Google Scholar] [CrossRef]
- Vitale, I.; Antoccia, A.; Crateri, P.; Leone, S.; Arancia, G.; Tanzarella, C. Caspase-independent apoptosis is activated by diazepam-induced mitotic failure in HeLa cells, but not in human primary fibroblasts. Apoptosis 2005, 10, 909–920. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef]
- Munger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Von Knebel Doeberitz, M.; Rittmuller, C.; Aengeneyndt, F.; Jansen-Durr, P.; Spitkovsky, D. Reversible repression of papillomavirus oncogene expression in cervical carcinoma cells: Consequences for the phenotype and E6-p53 and E7-pRB interactions. J. Virol. 1994, 68, 2811–2821. [Google Scholar]
- Von Knebel Doeberitz, M.; Rittmuller, C.; zur Hausen, H.; Durst, M. Inhibition of tumorigenicity of cervical cancer cells in nude mice by HPV E6-E7 anti-sense RNA. Int. J. Cancer 1992, 51, 831–834. [Google Scholar] [CrossRef]
- Zerfass, K.; Schulze, A.; Spitkovsky, D.; Friedman, V.; Henglein, B.; Jansen-Durr, P. Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J. Virol. 1995, 69, 6389–6399. [Google Scholar]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Durr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef]
- Singh, M.; Singh, N. Curcumin counteracts the proliferative effect of estradiol and induces apoptosis in cervical cancer cells. Mol. Cell. Biochem. 2011, 347, 1–11. [Google Scholar] [CrossRef]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pandey, A.; Tyagi, A.; Vishnoi, K.; Basir, S.F.; Das, B.C.; Bharti, A.C. Functional regulatory role of STAT3 in HPV16-mediated cervical carcinogenesis. PLoS ONE 2013, 8, e67849. [Google Scholar] [CrossRef]
- Kewitz, S.; Volkmer, I.; Staege, M.S. Curcuma Contra Cancer? Curcumin and Hodgkin’s Lymphoma. Cancer Growth Metastasis 2013, 6, 35–52. [Google Scholar]
- Lee, K.H.; Ab Aziz, F.H.; Syahida, A.; Abas, F.; Shaari, K.; Israf, D.A.; Lajis, N.H. Synthesis and biological evaluation of curcumin-like diarylpentanoid analogues for anti-inflammatory, antioxidant and anti-tyrosinase activities. Eur. J. Med. Chem. 2009, 44, 3195–3200. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Chew, A.J.; Zainal Abidin, N.; Abdul Wahab, N. Anti-proliferation and anti-migration activities of ten selected Zingiberaceae species against MDA-MB-231 cells. J. Med. Plants Res. 2012, 6, 3711–3723. [Google Scholar]
- Brady, H.J.M. Apoptosis Methods and Protocols; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Cattani, P.; Siddu, A.; D’Onghia, S.; Marchetti, S.; Santangelo, R.; Vellone, V.G.; Zannoni, G.F.; Fadda, G. RNA (E6 and E7) assays versus DNA (E6 and E7) assays for risk evaluation for women infected with human papillomavirus. J. Clin. Microbiol. 2009, 47, 2136–2141. [Google Scholar] [CrossRef]
- Schache, A.G.; Liloglou, T.; Risk, J.M.; Filia, A.; Jones, T.M.; Sheard, J.; Woolgar, J.A.; Helliwell, T.R.; Triantafyllou, A.; Robinson, M.; et al. Evaluation of human papilloma virus diagnostic testing in oropharyngeal squamous cell carcinoma: Sensitivity, specificity, and prognostic discrimination. Clin. Cancer Res. 2011, 17, 6262–6271. [Google Scholar] [CrossRef]
- Samples Availability: Samples of all the compounds used here are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulraj, F.; Abas, F.; Lajis, N.H.; Othman, I.; Hassan, S.S.; Naidu, R. The Curcumin Analogue 1,5-Bis(2-hydroxyphenyl)-1,4-pentadiene-3-one Induces Apoptosis and Downregulates E6 and E7 Oncogene Expression in HPV16 and HPV18-Infected Cervical Cancer Cells. Molecules 2015, 20, 11830-11860. https://doi.org/10.3390/molecules200711830
Paulraj F, Abas F, Lajis NH, Othman I, Hassan SS, Naidu R. The Curcumin Analogue 1,5-Bis(2-hydroxyphenyl)-1,4-pentadiene-3-one Induces Apoptosis and Downregulates E6 and E7 Oncogene Expression in HPV16 and HPV18-Infected Cervical Cancer Cells. Molecules. 2015; 20(7):11830-11860. https://doi.org/10.3390/molecules200711830
Chicago/Turabian StylePaulraj, Felicia, Faridah Abas, Nordin H. Lajis, Iekhsan Othman, Sharifah Syed Hassan, and Rakesh Naidu. 2015. "The Curcumin Analogue 1,5-Bis(2-hydroxyphenyl)-1,4-pentadiene-3-one Induces Apoptosis and Downregulates E6 and E7 Oncogene Expression in HPV16 and HPV18-Infected Cervical Cancer Cells" Molecules 20, no. 7: 11830-11860. https://doi.org/10.3390/molecules200711830
APA StylePaulraj, F., Abas, F., Lajis, N. H., Othman, I., Hassan, S. S., & Naidu, R. (2015). The Curcumin Analogue 1,5-Bis(2-hydroxyphenyl)-1,4-pentadiene-3-one Induces Apoptosis and Downregulates E6 and E7 Oncogene Expression in HPV16 and HPV18-Infected Cervical Cancer Cells. Molecules, 20(7), 11830-11860. https://doi.org/10.3390/molecules200711830