
Biogenesis of Triterpene Dimers from Orthoquinones Related to Quinonemethides: Theoretical Study on the Reaction Mechanism

Abstract
:
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Reactivity Global Indexes
3.2. Frontier Orbital Analysis
3.3. Study of the HDA Reactions
3.4. Geometrical and Charge Transfer Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, Q. Natural Diterpene and Triterpene quinone methides: Structures, synthesis, and biological potentials. In Quinone Methides; Rokita, S.E., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; p. 269. [Google Scholar]
- Gunatilaka, A.A.L.; Dhanabalasingham, B.; Karunaratne, V.; Kikuchi, T.; Tezuka, Y. Studies on terpenoids and stereoids. Part 27. Structure of a D:A-friedo-oleanane triterpenoid from Salacia reticulata and revision of the structures of kokoonol and kokzeylanol series of triterpenoids. Tetrahedron 1993, 49, 10397–10404. [Google Scholar] [CrossRef]
- Furbacher, T.R.; Gunatilaka, A.A.L. Catalytic inhibition of topoisomerase IIα by demethylzeylasterone, a 6-oxophenolic triterpenoid from Kokoona zeylanica. J. Nat. Prod. 2001, 64, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Gunatilaka, A.A.L. Triterpenoid Quinonemethides and Related Compounds (Celastroloids). Progress in the Chemistry of Organic Natural Products; Springer: Vienna, Austria, 1996; pp. 2–114. [Google Scholar]
- González, A.G.; Rodríguez, F.M.; Bazzocchi, I.B.; Ravelo, A.G. New terpenoids from Maytenus blepharodes. J. Nat. Prod. 2000, 63, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Shirota, O.; Sekita, S.; Satake, M. Nine New isoxuxuarinse-type triterpene dimers from Maytenus chuchuhuasca. Chem. Biodivers. 2004, 1, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Shirota, O.; Morita, H.; Takeya, K.; Itokawa, H. New geometric and stereoisomeric triterpene dimers from Maytenus chuchuhuasca. Chem. Pharm. Bull. 1998, 46, 102–106. [Google Scholar] [CrossRef]
- Shirota, O.; Sekita, S.; Satake, M. Nine triterpene dimers from Maytenus chuchuhuasca. Helv. Chim. Acta 2004, 87, 1536–1544. [Google Scholar] [CrossRef]
- González, A.G.; Kennedy, M.L.; Rodríguez, F.M.; Bazzocchi, I.L.; Jiménez, I.A.; Ravelo, A.G.; Moujir, L. Absolute configuration of triterpene dimers from Maytenus species (Celastraceae). Tetrahedron 2001, 57, 1283–1287. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Alvarenga, N.L.; Estevez-Braun, A.; Ravelo, A.G.; Bazzocchi, I.L.; Moujir, L. Structure and absolute configuration of triterpene dimers from Maytenus scutioides. Tetrahedron 1996, 52, 9597–9608. [Google Scholar] [CrossRef]
- Shirota, O.; Sekita, S.; Satake, M.; Morita, H.; Takeya, K.; Itokawa, H. Nine regioisomeric and stereoisomeric triterpene dimers from Maytenus chuchuhuasca. Chem. Pharm. Bull. 2004, 52, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Shirota, O.; Morita, H.; Takeya, K.; Itokawa, H. Five new triterpene dimers from Maytenus chuchuhuasca. J. Nat. Prod. 1997, 60, 1100–1104. [Google Scholar] [CrossRef]
- Shirota, O.; Morita, H.; Takeya, K.; Itokawa, H. Revised structures of cangorosins, triterpene dimers from Maytenus ilicifolia. J. Nat. Prod. 1997, 60, 111–115. [Google Scholar] [CrossRef]
- Minami, A.; Oikawa, H. The diels-alderase never ending story. In Biomimetic Organic Synthesis; Poupon, E., Nay, B., Eds.; John Wiley-VCH: Weinheim, Germany, 2011; Volume 2, pp. 753–781. [Google Scholar]
- Shirota, O.; Morita, H.; Takeya, K.; Itokawa, H. Structures of xuxuarines, stereoisomeric triterpene dimers from Maytenus chuchuhuasca. Tetrahedron 1995, 51, 1107–1120. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Alvarenga, N.L.; Bazzocchi, I.L.; Ravelo, A.G.; Moujir, L. Triterpene trimers from Maytenus scutioides: Cycloaddition compounds? J. Nat. Prod. 1999, 62, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, Y.; Wang, L.; Zuo, J.; Zhao, W. Terpenoids from root bark of Celastrus orbiculatus. Phytochemistry 2012, 75, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R. A reexamination of the molecular mechanism of the Diels-Alder reaction between tetrafluoroethene and cyclopentadiene. React. Kinet. Mech. Catal. 2016, 119, 49–57. [Google Scholar] [CrossRef]
- Singleton, D.A.; Schulmeier, B.E.; Hang, C.; Thomas, A.A.; Leung, S.W.; Merrigan, S.R. Isotope effects and the distinction between synchronous, asynchronous, and stepwise Diels-Alder reactions. Tetrahedron 2001, 57, 5149–5160. [Google Scholar] [CrossRef]
- Firestones, R.A. Volume of concert and heavy atom effects in Diels-Alder reaction mechanisms. Tetrahedron 1996, 52, 14459–14468. [Google Scholar] [CrossRef]
- Jasinski, R.; Kubik, M.; Krygier, A.L.; Kacka, A.; Dresler, E.; Czubara, A.B. An experimental and theoretical study of the hetero Diels-Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: One-step or zwitterionic, two-step mechanism? React. Kinet. Mech. Catal. 2014, 113, 333–345. [Google Scholar] [CrossRef]
- Jasinski, R. Searching for zwitterionic intermediates in Hetero Diels-Alder reactions between methyl α,p-dinitrocinnamate and vinyl-alkyl ethers. Comput. Theor. Chem. 2014, 1046, 93–98. [Google Scholar] [CrossRef]
- Katayama, K.; Kobayashi, T.; Oikawa, H.; Honna, M.; Ichihara, A. Enzymatic activity and partial purification of solanapyrone synthase: first enzyme catalyzing Diels-Alder reaction. Biochim. Biophys. Acta 1998, 1384, 387–395. [Google Scholar] [CrossRef]
- Pohnert, G. Macrophomate synthase: The first structure of a natural diels-alderase. ChemBioChem 2003, 4, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Vederas, J.C. Biosynthesis of lovastatin and related metabolites formed by fungal iterative PKS enzymes. Biopolymers 2010, 93, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, C.R.W.; Udier-Blagović, M.; Jorgensen, W.L. Macrophomate synthase: QM/MM simulations address the Diels-Alder versus Michael–Aldol reaction mechanism. J. Am. Chem. Soc. 2005, 127, 3577–3588. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.E.; Wijeratne, K.; Corsino, J.; Furlan, M.; Bolzani, V.; Gunatilaka, A.A.L. Biomimetic synthesis of xuxuarines Eα and Eβ: Structure revision of Rzedowskia bistriterpenoids. Bioorg. Med. Chem. 2008, 16, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Pieniazek, S.; Clemente, F.; Houk, K. Sources of error in DFT computations of C–C bond formation thermochemistries: π→σ transformations and error cancellation by DFT methods. Angew. Chem. Int. Ed. 2008, 47, 7746–7749. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the performance of the M05−2X and M06−2X exchange-correlation functionals for noncovalent interactions in biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Brinck, T. Stepwise Diels–Alder: More than just an oddity? A computational mechanistic study. J. Org. Chem. 2012, 77, 6563–6573. [Google Scholar] [CrossRef] [PubMed]
- Tajabadi, J.; Bakavoli, M.; Gholizadeh, M.; Eshghi, H. A mechanistic insight into the effect of piperidine as an organocatalyst on the [3 + 2] cycloaddition reaction of benzalacetone with phenyl azide from a computational study. Org. Biomol. Chem. 2016, 14, 7324–7333. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Quiñones, R.F.; Quesadas-Rojas, M.; Cuevas, G.; Mena-Rejón, G.J. Theoretical study of the Diels-Alder reaction between o-benzoquinone and norbornadiene. IOP Conf. Ser. Mater. Sci. Eng. 2013, 45, 1–4. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Andzelm, J.; Kölmel, C.; Klamt, A. Incorporation of solvent effects into density functional calculations of molecular energies and geometries. J. Chem. Phys. 1995, 103, 9312–9320. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parr, R.G.; von Szentpaly, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comp. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Domingo, L.R.; Gutierrea, M.R.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748–770. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.D.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Lemal, D.M.; Ramanathan, S.; Shellito, J. o-Fluoranil chemistry: Diels-Alder versus hetero-Diels-Alder cycloaddition. J. Org. Chem. 2008, 73, 3392–3396. [Google Scholar] [CrossRef] [PubMed]
- Lemal, D.M.; Ramanathan, S. o-Fluoranil: Stereochemistry and mechanism of its Diels-Alder reactions. J. Org. Chem. 2009, 74, 7804–7811. [Google Scholar] [CrossRef] [PubMed]
- Margetic, D.; Johnston, M.R.; Warrener, R.N. High-level computational study of the site-, facial- and stereoselectivities for the Diels-Alder reaction between o-benzoquinone and norbornadiene. Molecules 2000, 5, 1417–1428. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Korotchenko, V.; Stern, C.L.; Zaitsev, V.; Ritzert, J.T. Substituent-induced switch of the role of charge-transfer complexes in the Diels-Alder reactions of o-chloranil and styrenes. J. Org. Chem. 2012, 77, 5971–5981. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Painter, P.P.; Pemberton, R.P.; Wong, B.M.; Ho, K.C.; Tantillo, D.J. The Viability of nitrone–alkene (3 + 2) cycloadditions in alkaloid biosynthesis. J. Org. Chem. 2014, 79, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Tantillo, D.J. Theoretical studies on synthetic and biosynthetic oxidopyrylium-alkene cycloadditions: Pericyclic pathways to intricarene. J. Org. Chem. 2008, 73, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Masel, R.I. Chemical Kinetics and Catalysis; Wiley-Interscience: New York, NY, USA, 2001; p. 21. [Google Scholar]
- Baboul, A.G.; Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-3 theory using density functional geometries and zero-point energies. J. Chem. Phys. 1999, 110, 7650–7657. [Google Scholar] [CrossRef]
- Lau, Y.; Zou, L.; Houk, K.N. Computational methods to calculate accurate activation and reaction energies of 1,3-dipolar cycloadditions of 24 1,3-dipoles. J. Chem. Phys. A 2011, 115, 13906–13920. [Google Scholar]
- Grieco, P.A.; Garner, P.; He, Z. “Micellar” catalysis in the aqueous intermolecular Diels-Alder reaction: Rate acceleration and enhanced selectivity. Tetrahedron Lett. 1983, 24, 1897–1900. [Google Scholar] [CrossRef]
- Rideout, D.; Breslow, R. Hydrophobic acceleration of Diels-Alder reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003; p. 189. [Google Scholar]
- Ose, T.; Watanabe, K.; Mie, T.; Honma, M.; Watanabe, H.; Yao, M.; Oikawa, H.; Tanaka, I. Insight into a natural Diels-Alder reaction from the structure of macrophomate synthase. Nature 2003, 422, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Wannere, C.S.; Paul, A.; Herges, R.; Houk, K.N.; Schaefer, H.F., III; von Ragué Schleyer, P. The existence of secondary orbital interactions. J. Comput. Chem. 2007, 28, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Fujimoto, H. Reexamination of orbital interactions in Diels-Alder reactions. Tetrahedron Lett. 2002, 43, 2055–2057. [Google Scholar] [CrossRef]
- García, J.I.; Mayoral, J.A.; Salvatella, L. The source of the endo rule in the Diels-Alder reaction: Are secondary orbital interactions really necessary? Eur. J. Org. Chem. 2005, 1, 85–90. [Google Scholar] [CrossRef]
- Contini, S.; Leone, S.; Menichetti, S.; Viglianisi, C.; Trimarco, P. [2 + 4] and [4 + 2] cycloadditions of o-thioquinones with 1, 3-dienes: A computational study. J. Org. Chem. 2006, 71, 5507–5514. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Domingo, L.R.; Saéz, J. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.







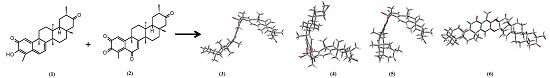{kind=link}
{kind=link}
{kind=link}
{kind=link}
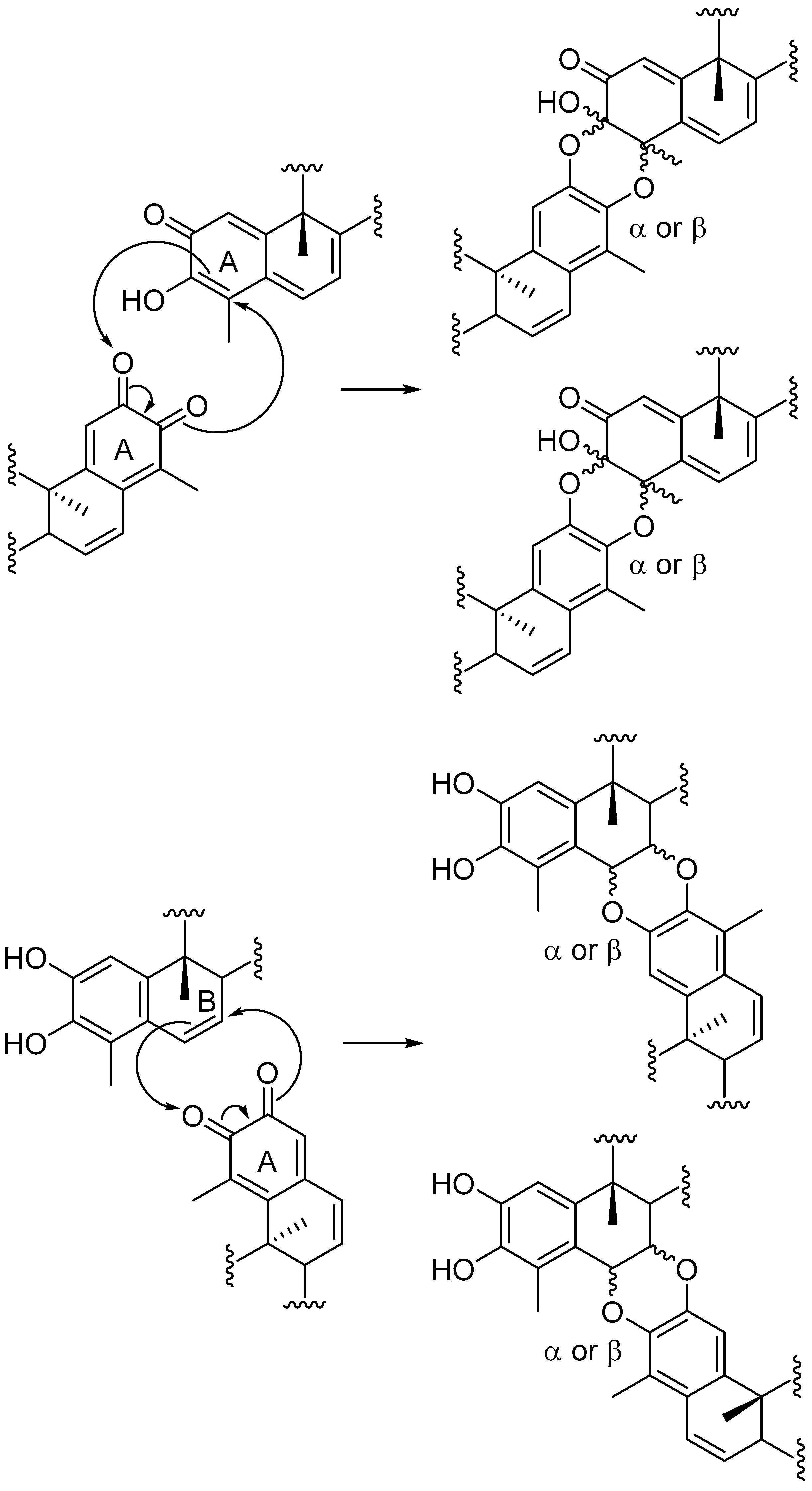{kind=link}
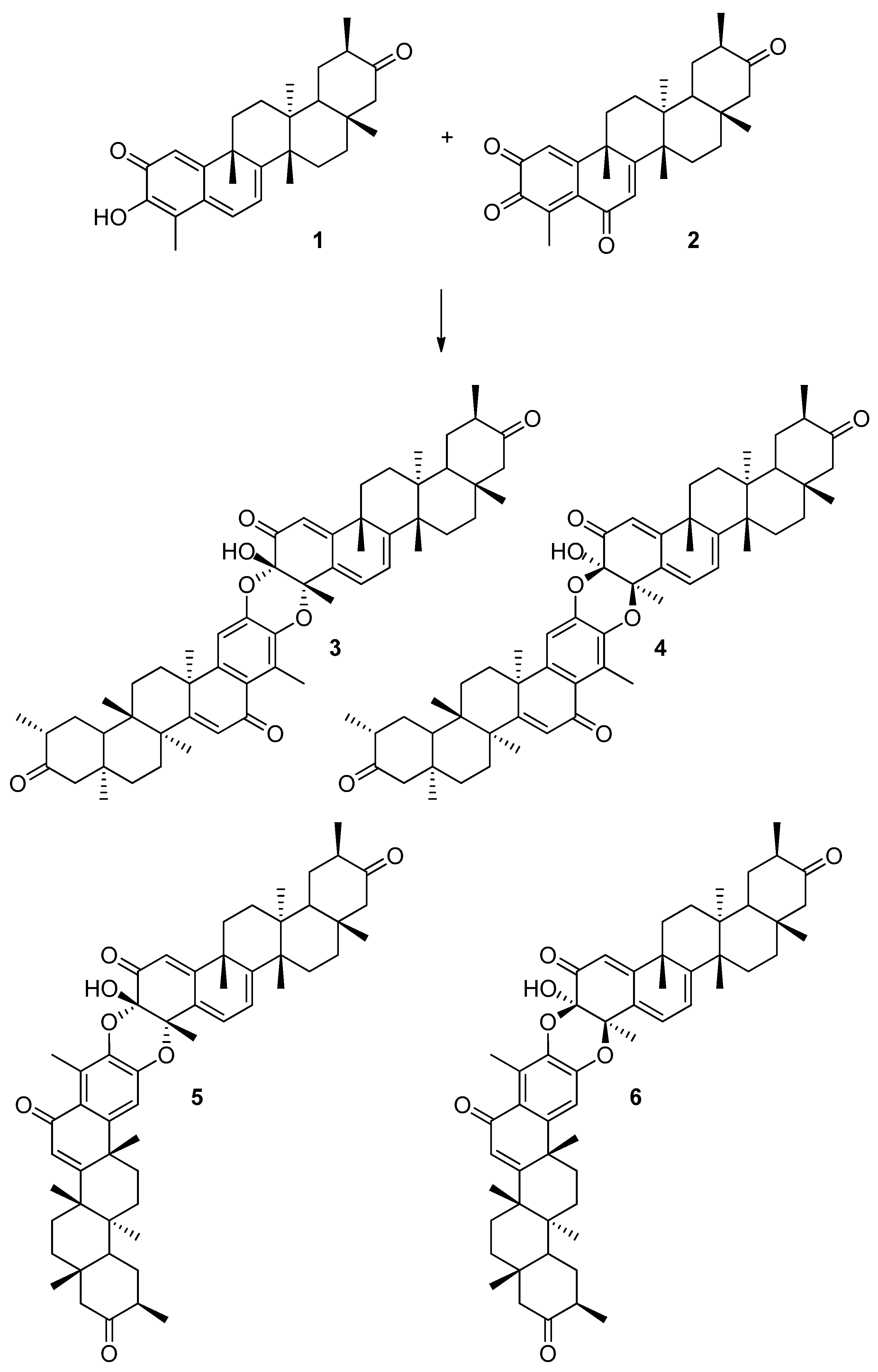{kind=link}
{kind=link}
| Molecule | µ (eV) | ω (eV) | N (eV) |
|---|---|---|---|
| tingenone 1 | −4.218 | 1.409 | −6.742 |
| oq-6-oxotingenol 2 | −5.415 | 2.084 | −8.330 |
| isopristimerol 7 | −3.020 | 0.551 | −6.460 |
| oq-isopristimerol 8 | −4.816 | 1.707 | −7.590 |
| Diene | Dienophile | ΔNED a | ΔIED b |
|---|---|---|---|
| oq-6-oxotingenol 2 | tingenone 1 | 6.626 | 4.100 |
| oq-isopristimerol 8 | isopristimerol 7 | 8.019 | 4.326 |
| Molecule | ΔGrxn | ΔHrxn | TΔSrxn | endo | exo | ||||
|---|---|---|---|---|---|---|---|---|---|
| ΔGact | ΔHact | TΔSact | ΔGact | ΔHact | TΔSact | ||||
| Xuxuarine Aα 3 | −27.1 | −44.4 | −17.3 | 27.6 | 8.8 | −18.8 | 30.6 | 14.1 | −16.4 |
| Xuxuarine Aβ 4 | −24.3 | −42.1 | −17.7 | 28.1 | 11.2 | −16.8 | 30.6 | 13.8 | −16.8 |
| Isoxuxuarine Aα 5 | −27.5 | −43.7 | −16.1 | 25.5 | 5.2 | −20.2 | 30.9 | 14.6 | −16.2 |
| Isoxuxuarine Aβ 6 | −25.8 | −42.1 | −16.3 | 29.7 | 11.9 | −17.8 | 29.3 | 13.3 | −16.0 |
| Cangorosin A 9 | −23.1 | −39.2 | −16.1 | 36.0 | 16.7 | −19.3 | 38.7 | 23.1 | −15.6 |
| Cangorosin Aβ 10 | −20.9 | −37.4 | −16.5 | 40.0 | 25.0 | −15.0 | 37.8 | 21.9 | −16.0 |
| Isocangorosin A 11 | −23.4 | −39.2 | −15.9 | 36.8 | 18.7 | −18.1 | 38.5 | 23.3 | −15.2 |
| Isocangorosin Aβ 12 | −21.0 | −37.6 | −16.6 | 41.4 | 22.0 | −19.3 | 36.2 | 21.4 | −14.9 |
| Molecule | exo TS | endo TS | Products a | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AD | CT b | D1 | D2 | AD | CT b | D1 | D2 | D1 | D2 | |
| Xuxuarine Aα 3 | 0.19 | 0.397 | 1.92 | 2.20 | 0.06 | 0.357 | 2.02 | 2.11 | 1.42 | 1.43 |
| Xuxuarine Aβ 4 | 0.21 | 0.404 | 1.90 | 2.22 | 0.07 | 0.366 | 2.01 | 2.12 | 1.42 | 1.43 |
| Isoxuxuarine Aα 5 | 0.10 | 0.382 | 1.98 | 2.12 | 0.15 | 0.418 | 2.16 | 1.94 | 1.42 | 1.42 |
| Isoxuxuarine Aβ 6 | 0.09 | 0.381 | 1.98 | 2.11 | 0.07 | 0.378 | 2.11 | 2.02 | 1.42 | 1.42 |
| Cangorosin A 9 | 0.16 | 0.319 | 1.92 | 2.16 | 0.02 | 0.307 | 2.01 | 2.05 | 1.42 | 1.43 |
| Cangorosin Aβ 10 | 0.16 | 0.297 | 1.97 | 2.20 | 0.03 | 0.317 | 2.03 | 2.07 | 1.42 | 1.42 |
| Isocangorosin A 11 | 0.06 | 0.310 | 1.98 | 2.08 | 0.16 | 0.329 | 2.11 | 1.89 | 1.42 | 1.43 |
| Isocangorosin Aβ 12 | 0.19 | 0.300 | 1.96 | 2.23 | 0.03 | 0.308 | 2.07 | 2.02 | 1.42 | 1.42 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quesadas-Rojas, M.; Mena-Rejón, G.J.; Cáceres-Castillo, D.; Cuevas, G.; Quijano-Quiñones, R.F. Biogenesis of Triterpene Dimers from Orthoquinones Related to Quinonemethides: Theoretical Study on the Reaction Mechanism. Molecules 2016, 21, 1551. https://doi.org/10.3390/molecules21111551
Quesadas-Rojas M, Mena-Rejón GJ, Cáceres-Castillo D, Cuevas G, Quijano-Quiñones RF. Biogenesis of Triterpene Dimers from Orthoquinones Related to Quinonemethides: Theoretical Study on the Reaction Mechanism. Molecules. 2016; 21(11):1551. https://doi.org/10.3390/molecules21111551
Chicago/Turabian StyleQuesadas-Rojas, Mariana, Gonzalo J. Mena-Rejón, David Cáceres-Castillo, Gabriel Cuevas, and Ramiro F. Quijano-Quiñones. 2016. "Biogenesis of Triterpene Dimers from Orthoquinones Related to Quinonemethides: Theoretical Study on the Reaction Mechanism" Molecules 21, no. 11: 1551. https://doi.org/10.3390/molecules21111551