
A Stereocontrolled Protocol to Highly Functionalized Fluorinated Scaffolds through a Fluoride Opening of Oxiranes



Abstract
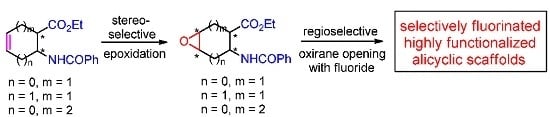
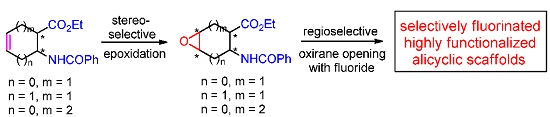
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
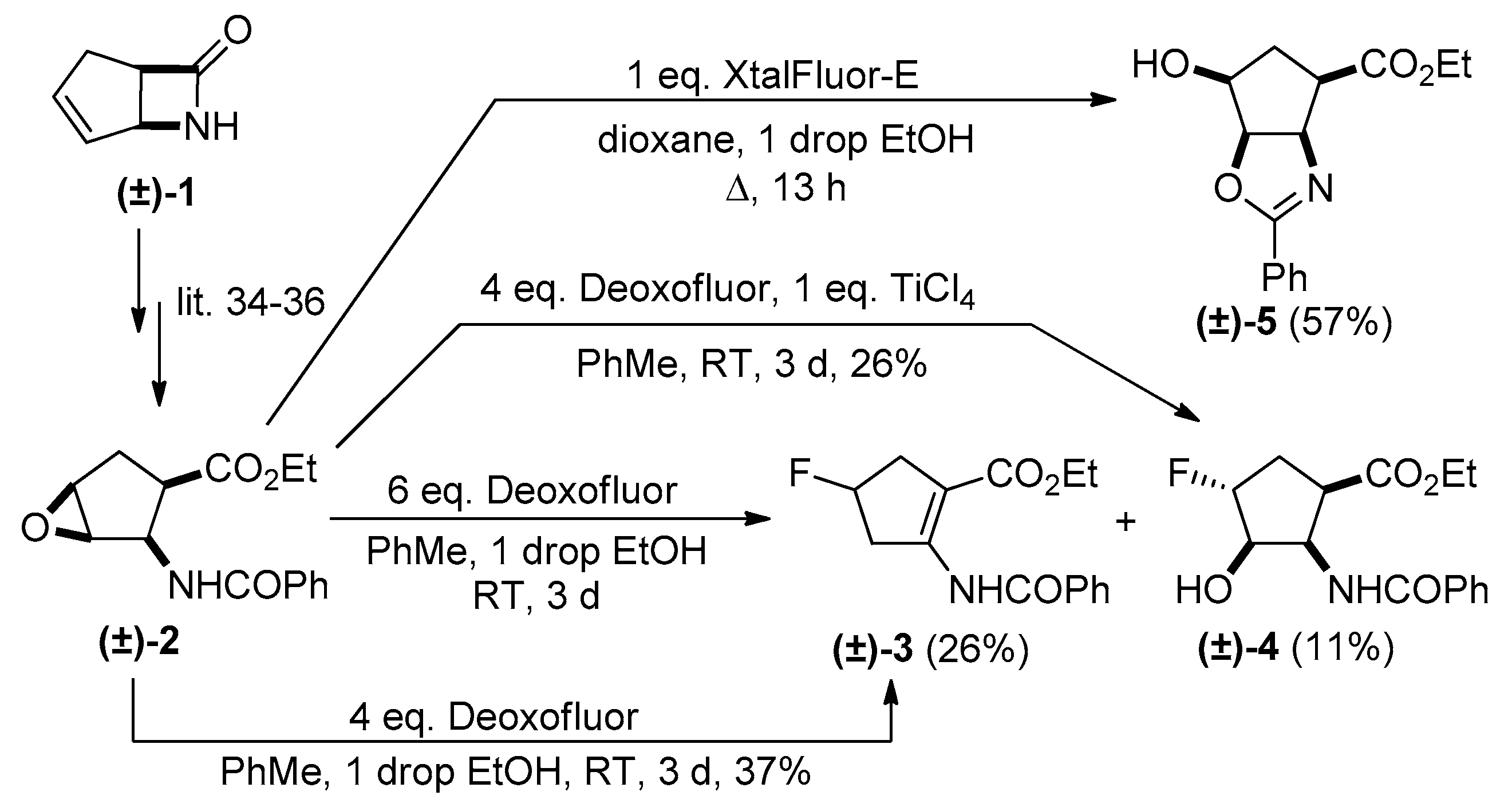
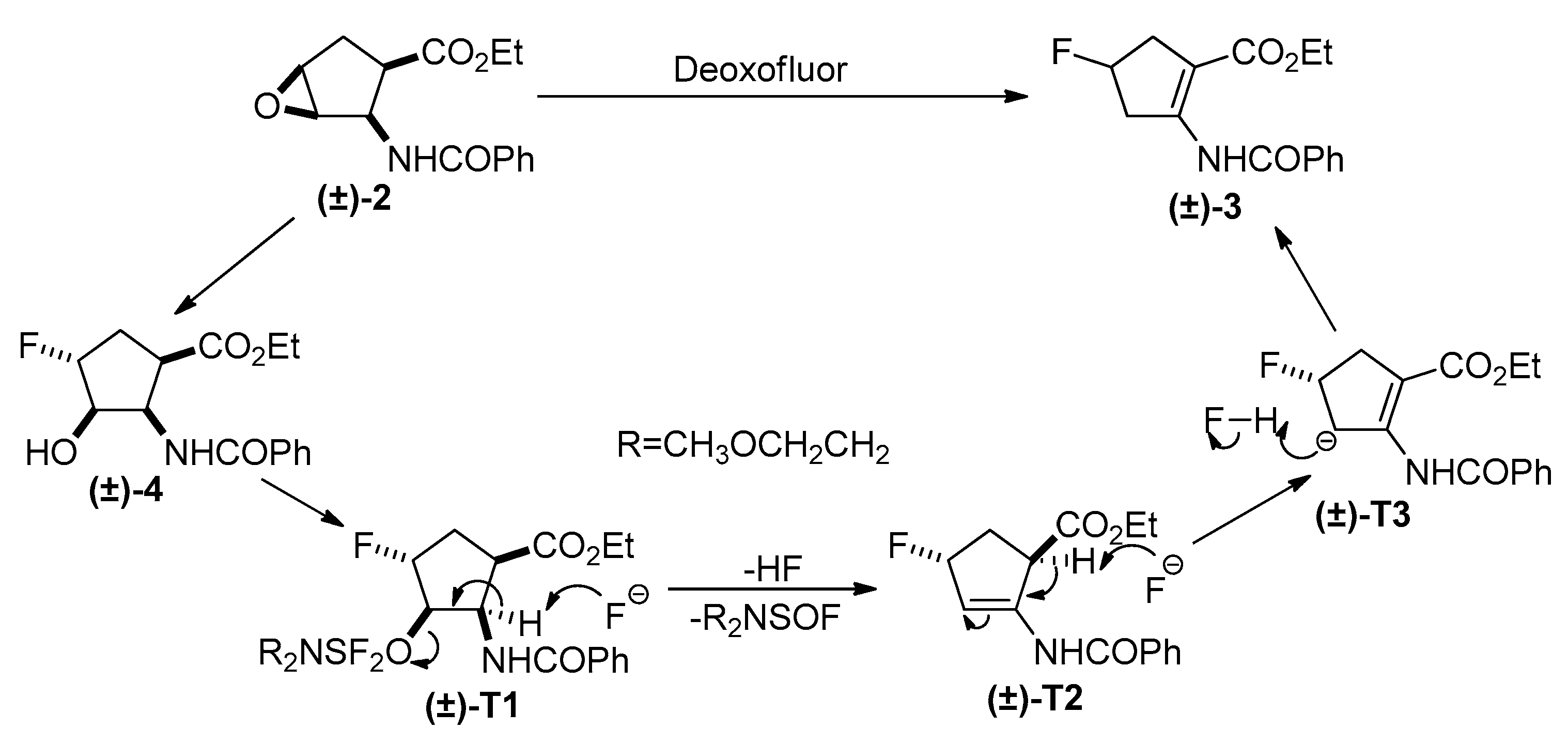
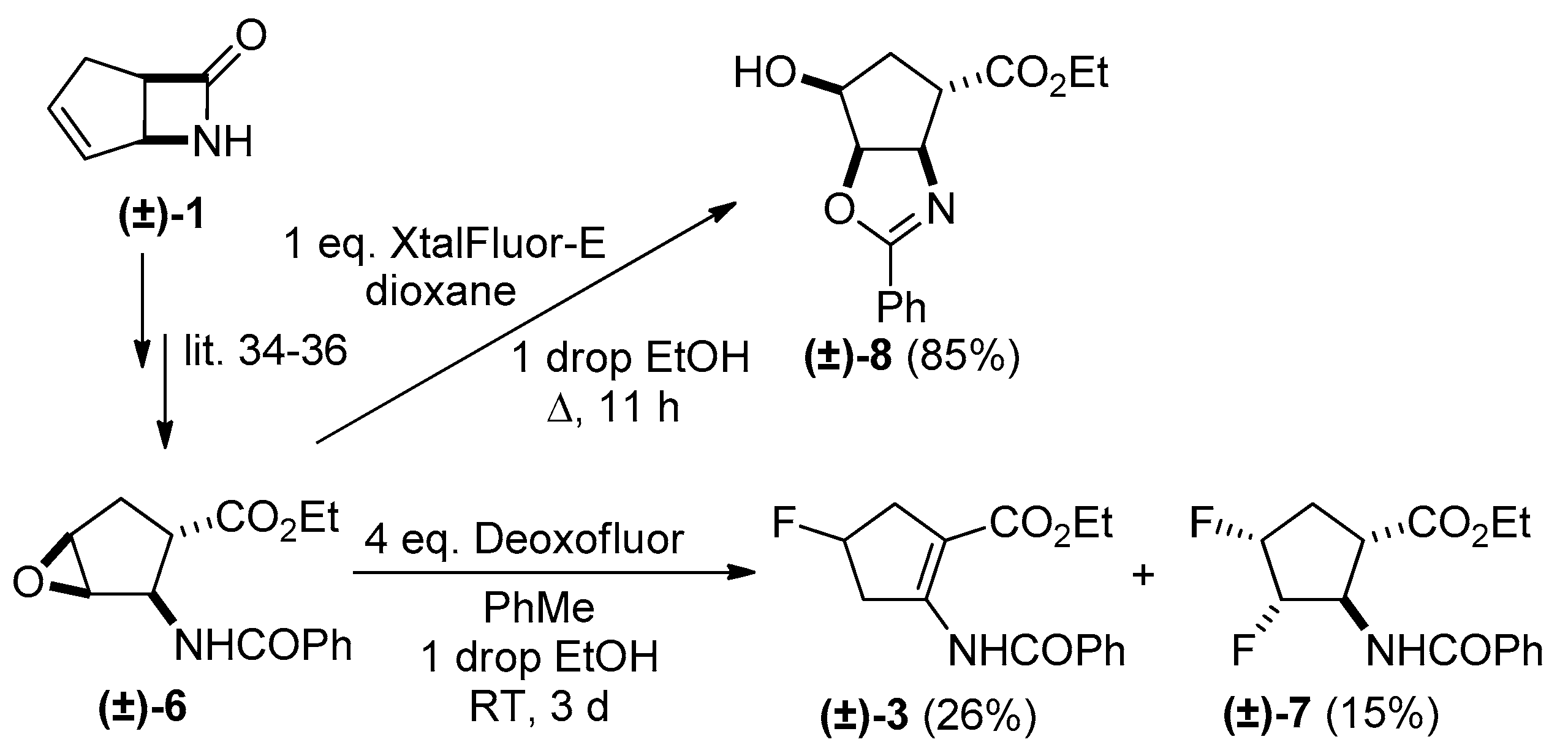
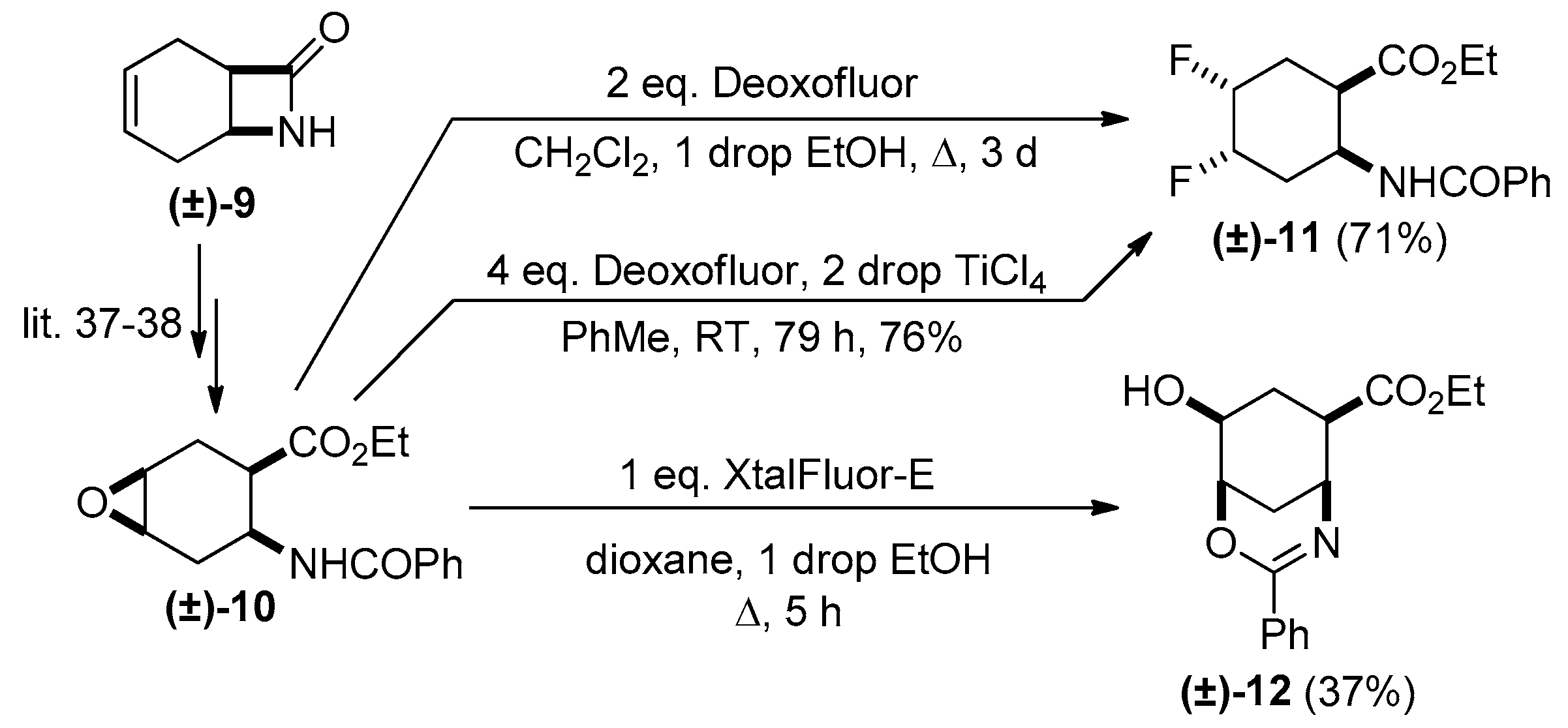
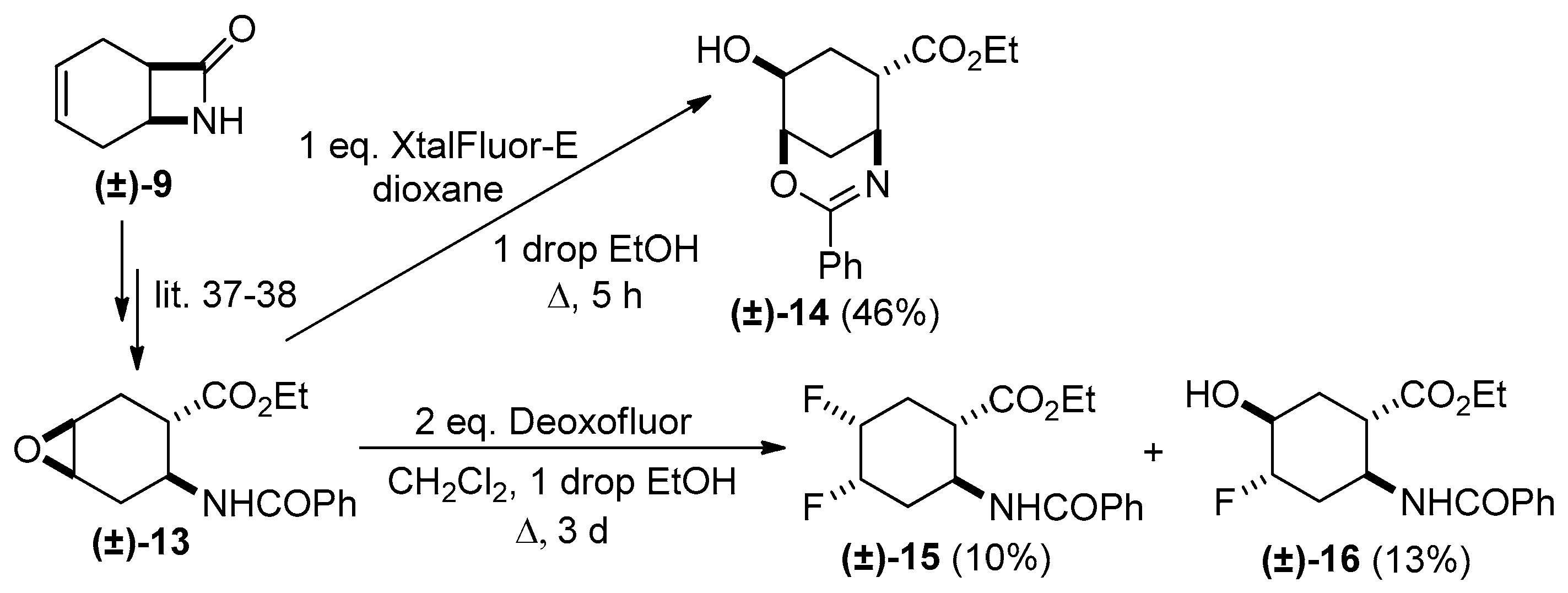
2. Results and Discussion
3. Materials and Methods
Experimental Procedures for the Oxirane Ring Openings
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chawla, R.; Singh, A.K.; Yadav, L.D.S. Organocatalysis in synthesis and reactions of epoxides and aziridines. RSC Adv. 2013, 3, 11385–11403. [Google Scholar] [CrossRef]
- Meninno, S.; Lattanzi, A. Organocatalytic asymmetric reactions of epoxides: Recent progress. Chem. Eur. J. 2016, 22, 3632–3642. [Google Scholar] [CrossRef] [PubMed]
- Krake, S.H.; Bergmeier, S.C. Inter- and intramolecular reactions of epoxides and aziridines with π-nucleophiles. Tetrahedron 2010, 66, 7337–7360. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Q.; Cornwall, R.G.; Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev. 2014, 114, 8199–8256. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Remete, A.M.; Nonn, M.; Fustero, S.; Sillanpaa, R.; Fülöp, F. Substrate-dependent fluorinations of highly functionalized cycloalkanes. Tetrahedron 2016, 72, 781–787. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Fülöp, F. Selective syntheses of novel highly functionalized beta-aminocyclohexanecarboxylic acids. Tetrahedron 2012, 68, 4438–4443. [Google Scholar] [CrossRef]
- Wang, P.A. Organocatalyzed enantioselective desymmetrization of aziridines and epoxides. Beilstein J. Org. Chem. 2013, 9, 1677–1695. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sanchez-Rosello, M.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A. Synthesis of fluorinated beta-amino acids. Synthesis 2011, 3045–3079. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Fustero, S.; Fülöp, F. Selective synthesis of new fluorinated alicyclic β-amino ester stereoisomers. Eur. J.Org. Chem. 2011, 4993–5001. [Google Scholar] [CrossRef]
- Nonn, M.; Kiss, L.; Haukka, M.; Fustero, S.; Fülöp, F. A Novel and selective fluoride opening of aziridines by XtalFluor-E. Synthesis of Fluorinated Diamino Acid Derivatives. Org. Lett. 2015, 17, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Nonn, M.; Forró, E.; Sillanpää, R.; Fustero, S.; Fülöp, F. A selective synthesis of fluorinated cispentacin derivatives. Eur. J. Org. Chem. 2014, 4070–4076. [Google Scholar] [CrossRef]
- Wu, J. Review of recent advances in nucleophilic C–F bond-forming reactions at SP3 centers. Tetrahedron Lett. 2014, 55, 4289–4294. [Google Scholar] [CrossRef]
- Ma, J.A.; Cahard, D. Asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 2008, 108, PR1–PR43. [Google Scholar] [CrossRef] [PubMed]
- Champagne, P.A.; Desroches, J.; Hamel, J.D.; Vandamme, M.; Paquin, J.F. Monofluorination of organic compounds: 10 years of innovation. Chem. Rev. 2015, 115, 9073–9174. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed]
- Husstedt, W.S.; Wiehle, S.; Stillig, C.; Bergander, C.; Grimme, S.; Haufe, G. Synthesis and preferred conformations of all regio- and diastereoisomeric methyl 2,3-fluorohydroxyalkanoates. Eur. J. Org. Chem. 2011, 355–363. [Google Scholar] [CrossRef]
- O’Hagan, D. Organofluorine chemistry: Synthesis and conformation of vicinal fluoromethylene motifs. J. Org. Chem. 2012, 77, 3689–3699. [Google Scholar] [CrossRef] [PubMed]
- Hunter, L.; Jolliffe, K.A.; Jordan, M.J.T.; Jensen, P.; Macquart, R.B. Synthesis and conformational analysis of alfa,beta-difluoro-gamma-amino acid derivatives. Chem. Eur. J. 2011, 17, 2340–2343. [Google Scholar] [CrossRef] [PubMed]
- Bykova, T.; Al-Maharik, N.; Slawin, A.M.Z.; O’Hagan, D. Multicomponent reactions of methyl substituted all-cis tetrafluorocyclohexane aldehydes. Org. Biomol. Chem. 2016, 14, 1117–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile, J.M.; Mayoral, J.A.; Salvatella, L. Theoretical study on the BF3-catalyzed meinwald rearrangement reaction. J. Org. Chem. 2014, 79, 5993–5999. [Google Scholar] [CrossRef] [PubMed]
- Cresswell, A.J.; Davies, S.G.; Lee, J.A.; Roberts, P.M.; Russell, A.J.; Thomson, J.E.; Tyte, M. β-Fluoroamphetamines via the stereoselective synthesis of benzylic fluorides. J. Org. Lett. 2010, 12, 2936–2939. [Google Scholar] [CrossRef] [PubMed]
- Cresswell, A.J.; Davies, S.G.; Lee, J.A.; Morris, M.J.; Roberts, P.M.; Thomson, J.E. Ring-opening hydrofluorination of 2,3- and 3,4-epoxy amines by HBF43OEt2: Application to the asymmetric synthesis of (S,S)-3-deoxy-3-fluorosafingol. J. Org. Chem. 2011, 76, 4617–4627. [Google Scholar] [CrossRef] [PubMed]
- Cresswell, A.J.; Davies, S.G.; Lee, J.A.; Morris, M.J.; Roberts, P.M.; Thomson, J.E. Diastereodivergent hydroxyfluorination of cyclic and acyclic allylic amines: Synthesis of 4-deoxy-4-fluorophytosphingosines. J. Org. Chem. 2012, 77, 7262–7281. [Google Scholar] [CrossRef] [PubMed]
- Kondapi, V.P.K.; Soueidan, O.M.; Hosseini, S.N.; Jabari, N.; West, F.G. Efficient and easy access to optically pure tetrasubstituted tetrahydrofurans via stereoselective opening of C2-symmetric epoxide and aziridine rings. Eur. J. Org. Chem. 2016, 2016, 1367–1379. [Google Scholar] [CrossRef]
- Yan, N.; Fang, Z.; Liu, Q.Q.; Guo, X.H.; Hu, X.G. Conformation-induced regioselective and divergent opening of epoxides by fluoride: Facile access to hydroxylated fluoro-piperidines. Org. Biomol. Chem. 2016, 14, 3469–3475. [Google Scholar] [CrossRef] [PubMed]
- Bruns, S.; Haufe, G. Enantioselective introduction of fluoride into organic compounds. First asymmetric ring opening of epoxides by hydrofluorinating reagents. J. Fluor. Chem. 2000, 104, 247–254. [Google Scholar] [CrossRef]
- Haufe, G.; Bruns, S.; Runge, M. Enantioselective ring opening of epoxides by HF-reagents. Asymmetric synthesis of fluorolactones. J. Fluor. Chem. 2001, 112, 55–61. [Google Scholar] [CrossRef]
- Althaus, M.; Togni, A.; Mezzetti, A. Asymmetric oxidative alfa-fluorination of 2-alkylphenylacetaldehydes with AgHF2 and ruthenium/PNNP catalysts. J. Fluor. Chem. 2009, 130, 702–707. [Google Scholar] [CrossRef]
- Kalow, J.A.; Doyle, A.G. Enantioselective ring opening of epoxides by fluoride anion promoted by a cooperative dual-catalyst system. J. Am. Chem. Soc. 2010, 132, 3268–3269. [Google Scholar] [CrossRef] [PubMed]
- Kalow, J.A.; Doyle, A.G. Mechanistic investigations of cooperative catalysis in the enantioselective fluorination of epoxides. J. Am. Chem. Soc. 2011, 133, 16001–16012. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Fülöp, F. Synthesis of carbocyclic and heterocyclic β-aminocarboxylic acids. Chem. Rev. 2014, 114, 1116–1169. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Forró, E.; Sillanpää, R.; Fülöp, F. Diastereo- and enantioselective synthesis of orthogonally protected 2,4-diaminocyclopentanecarboxylates: A flip from β-amino- to β,γ-diaminocarboxylates. J. Org. Chem. 2007, 72, 8786–8790. [Google Scholar] [CrossRef] [PubMed]
- Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Synthesis of novel isoxazoline-fused cyclic beta-amino esters by regio- and stereoselective 1,3-dipolar cycloaddition. Tetrahedron 2011, 67, 4079–4085. [Google Scholar] [CrossRef]
- Cherepanova, M.; Kiss, L.; Forró, E.; Fülöp, F. A De Novo Stereocontrolled Approach to syn- and anti-Disubstituted Acyclic β-2,3-Amino Acid Enantiomers. Eur. J. Org. Chem. 2014, 403–409. [Google Scholar] [CrossRef]
- L’Heureux, A.; Beaulieu, F.; Bennett, C.; Bill, D.R.; Clayton, S.; LaFlamme, F.; Mirmehrabi, M.; Tadayon, S.; Tovell, D.; Couturier, M. Aminodifluorosulfinium Salts: Selective Fluorination Reagents with Enhanced Thermal Stability and Ease of Handling. J. Org. Chem. 2010, 75, 3401–3411. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.S.; Pez, G.P.; Pesaresi, R.J.; Prozonic, F.M.; Cheng, H. Bis(2-methoxyethyl)aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability. J. Org. Chem. 1999, 71, 7048–7054. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Martinek, T.A.; Bernáth, G.; de Kimpe, N.; Fülöp, F. Stereoselective synthesis of hydroxylated beta-aminocyclohexanecarboxylic acids. Tetrahedron 2008, 64, 5036–5043. [Google Scholar] [CrossRef]
- Cherepanova, M.; Kiss, L.; Fülöp, F. Stereocontrolled transformation of cyclohexene beta-amino esters into syn- or anti-difunctionalized acyclic beta2,3-amino acid derivatives. Tetrahedron 2014, 70, 2515–2522. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available in mg amounts from the authors.





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remete, A.M.; Nonn, M.; Fustero, S.; Fülöp, F.; Kiss, L. A Stereocontrolled Protocol to Highly Functionalized Fluorinated Scaffolds through a Fluoride Opening of Oxiranes. Molecules 2016, 21, 1493. https://doi.org/10.3390/molecules21111493
Remete AM, Nonn M, Fustero S, Fülöp F, Kiss L. A Stereocontrolled Protocol to Highly Functionalized Fluorinated Scaffolds through a Fluoride Opening of Oxiranes. Molecules. 2016; 21(11):1493. https://doi.org/10.3390/molecules21111493
Chicago/Turabian StyleRemete, Attila Márió, Melinda Nonn, Santos Fustero, Ferenc Fülöp, and Loránd Kiss. 2016. "A Stereocontrolled Protocol to Highly Functionalized Fluorinated Scaffolds through a Fluoride Opening of Oxiranes" Molecules 21, no. 11: 1493. https://doi.org/10.3390/molecules21111493