
The C-Terminal O-S Acyl Shift Pathway under Acidic Condition to Propose Peptide-Thioesters

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
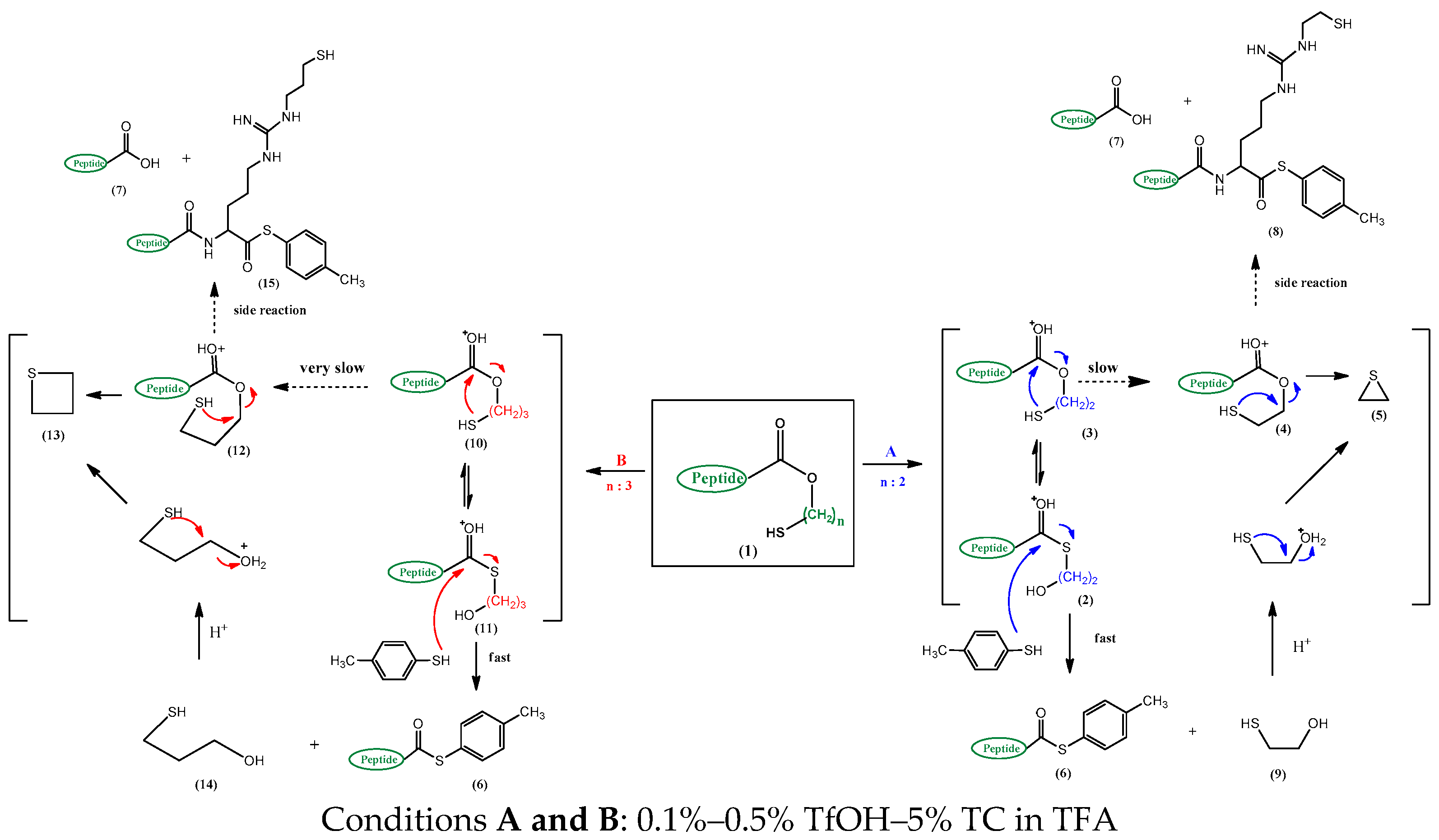
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of Handles
Preparation of the HET and HPT Handles on 2-Chlorotrityl Chloride Resin and Peptide Synthesis
3.2. Tandem Switch Experiment for O-S Acyl Shift
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Bergmann, M.; Brand, E.; Weinmann, F. Rearrangements of peptide-like substances. Derivatives of g-amino-b-hydroxybutyric acid. Physiol. Chem. 1923, 131, 1–17. [Google Scholar] [CrossRef]
- Wieland, T.; Bokelmann, E.; Bauer, L.; Lang, H.U.; Lau, H.; Schafer, W. Über Peptid synthesen. 8. Mitteilung Bildung von S-haltigen Peptiden durch intramolekulare Wanderung von Aminoacylresten. Liebigs Ann. Chem. 1953, 583, 129–149. [Google Scholar] [CrossRef]
- Smith, H.A.; Gorin, G. 2-Methyl-2-thiazoline-4-carboxylic Acid: Formation from N-Acetylcysteine and Hydrolysis. J. Org. Chem. 1961, 26, 820–823. [Google Scholar] [CrossRef]
- Martin, R.B.; Hedrick, R.I.; Parcell, A. Thiazoline and Oxazoline Hydrolyses and Sulfur-Nitrogen and Oxygen-Nitrogen Acyl Transfer Reactions. J. Org. Chem. 1964, 29, 3197–3206. [Google Scholar] [CrossRef]
- Levy, D.; Carpenter, F.H. Insulin methyl ester. Specific cleavage of a peptide chain resulting from a nitrogen to oxygen acyl shift at a threonine residue. Biochemistry 1970, 9, 3215–3222. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, G.; Pinnen, F.; Lucente, G. Cyclization under mild conditions of cysteine containing peptides. Tetrahedron Lett. 1985, 26, 5481–5484. [Google Scholar] [CrossRef]
- Makisumi, S.; Matsuura, S.; Waki, M.; Izumiya, N. Studies of peptide antibiotics. XXV. Synthesis of an immediate precursor of gramicidin S. Bull. Chem. Soc. Jpn. 1971, 44, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Kemp, D.S. The amine capture strategy for peptide bond formation—An outline of progress. Biopolymers 1981, 20, 1793–1804. [Google Scholar] [CrossRef]
- Hojo, H.; Aimoto, S. Polypeptide synthesis using the S-alkyl thioester of a partially protected peptide segment. Synthesis of the DNA-binding domain of c-Myb protein (142–193)-NH2. Bull. Chem. Soc. Jpn. 1991, 64, 111–117. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. A new class of membrane-bound chemokine with a CX3C motif. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Lu, Y.-A.; Liu, C.-F.; Shao, J. Peptide synthesis using unprotected peptides through orthogonal coupling methods. Proc. Natl. Acad. Sci. USA 1995, 92, 12485–12489. [Google Scholar] [CrossRef] [PubMed]
- Raz, R.; Rademann, J. Fmoc-Based Synthesis of Peptide Thioesters for Native Chemical Ligation Employing a tert-Butyl Thiol Linker. Org. Lett. 2011, 13, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “Traceless” Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef] [PubMed]
- Mühlberg, M.; Jaradat, D.M.M.; Kleineweischede, R.; Papp, I.; Dechtrirat, D.; Muth, S.; Broncel, M.; Hackenberger, C.P.R. Acidic and basic deprotection strategies of borane-protected phosphinothioesters for the traceless Staudinger ligation. Bioorg. Med. Chem. 2010, 18, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Dheur, J.; Ollivier, N.; Melnyk, O. Synthesis of Thiazolidine Thioester Peptides and Acceleration of Native Chemical Ligation. Org. Lett. 2011, 13, 1560–1563. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-F.; Tam, J.P. Chemical Ligation Approach to Form a Peptide Bond between Unprotected Peptide Segments. Concept and Model Study. J. Am. Chem. Soc. 1994, 116, 4149–4153. [Google Scholar] [CrossRef]
- Cohen, A.S.; Dubikovskaya, E.A.; Rush, J.S.; Bertozzi, C.R. Real-Time Bioluminescence Imaging of Glycans on Live Cells. J. Am. Chem. Soc. 2010, 132, 8563–8565. [Google Scholar] [CrossRef] [PubMed]
- Nisic, F.; Bernardi, A. Stereoselective synthesis of N-galactofuranosyl amides. Carbohydr. Res. 2011, 346, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Mauger, A.B.; Stuart, O.A. Slow O, N-acyl shift in an actinomycin-related peptide lactone. Int. J. Pept. Protein Res. 1987, 30, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Kent, S.B.H. Synthesis of Native Proteins by Chemical Ligation. Annu. Rev. Biochem. 2000, 69, 923–960. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Xu, J.; Eom, K.D. Methods and strategies of peptide ligation. Biopolymers 2001, 60, 194–205. [Google Scholar] [CrossRef]
- Muir, T.W. Semisynthesis of Proteins by Expressed Protein Ligation. Annu. Rev. Biochem. 2003, 72, 249–289. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.L.; Soellner, M.B.; Raines, R.T. Chemical Synthesis of Proteins. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Mende, F.; Seitz, O. 9-Fluorenylmethoxycarbonyl-Based Solid-Phase Synthesis of Peptide α-Thioesters. Angew. Chem. Int. Ed. 2011, 50, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Taichi, M.; Hemu, X.; Qiu, Y.; Tam, J.P. A Thioethylalkylamido (TEA) Thioester Surrogate in the Synthesis of a Cyclic Peptide via a Tandem Acyl Shift. Org. Lett. 2013, 15, 2620–2623. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.S.; Chen, X.; Thang, S.; Chang, H.N.; Wang, F.L.; Zuo, C. A New Method for Synthesis of Peptide Thioesters via Irreversible N-to-S Acyl Transfer. Org. Lett. 2014, 16, 4908–4911. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Sasaki, T.; Kaiser, E.T. Peptide segment synthesis catalyzed by the semisynthetic enzyme thiolsubtilisin. J. Am. Chem. Soc. 1987, 109, 3808–3810. [Google Scholar] [CrossRef]
- Jackson, D.Y.; Bumier, J.; Quan, C.; Stanley, M.; Tom, J.; Wells, J. A Designed Peptide Ligase for Total Synthesis of Ribonuclease A with Unnatural Catalytic Residues. Science 1994, 266, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Kane, P.M.; Yamashiro, C.T.; Wolczyk, D.F.; Neff, N.; Goebl, M.; Stevens, T.H. Protein Splicing Coverts the Yeast TEPI Gene Product to the 69 kD Subunit of the Vacuolar H+—Adenosine Triphosphatase. Science 1990, 250, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Frutos, S.; Goger, M.; Giovani, B.; Cowburn, D.; Muir, T.W. Branched intermediate formation stimulates peptide bond cleavage in protein splicing. Nat. Chem. Biol. 2010, 6, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, D.; Hilvert, D. Facile, Facile, Fmoc-Compatible Solid-Phase Synthesis of Peptide C-Terminal Thioesters. Org. Lett. 2000, 2, 2439–2442. [Google Scholar] [CrossRef] [PubMed]
- Sewing, A.; Hilvert, D. Fmoc-Compatible Solid-Phase Peptide Synthesis of Long C-Terminal Peptide Thioesters. Angew. Chem. Int. Ed. 2001, 40, 3395–3396. [Google Scholar] [CrossRef]
- Botti, P.; Villain, M.; Manganiello, S.; Gaertner, H. Native Chemical Ligation through in Situ O to S Acyl Shift. Org. Lett. 2004, 6, 4861–4864. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Warren, J.D.; Chen, J.; Wu, B.; Wan, Q.; Danishefsky, S.J. Studies Related to the Relative Thermodynamic Stability of C-Terminal Peptidyl Esters of O-Hydroxy Thiophenol: Emergence of a Doable Strategy for Non-Cysteine Ligation Applicable to the Chemical Synthesis of Glycopeptides. J. Am. Chem. Soc. 2006, 128, 7460–7462. [Google Scholar] [CrossRef] [PubMed]
- Tofteng, A.P.; Jensen, K.J.; Hoeg-Jensen, T. Peptide dithiodiethanol esters for in situ generation of thioesters for use in native ligation. Tetrahedron Lett. 2007, 48, 2105–2107. [Google Scholar] [CrossRef]
- Chen, G.; Wan, Q.; Tan, Z.; Kan, C.; Hua, Z.; Ranganathan, K.; Danishefsky, S.J. Development of Efficient Methods for Accomplishing Cysteine-Free Peptide and Glycopeptide Coupling. Angew. Chem. Int. Ed. 2007, 46, 7383–7387. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Chen, J.; Yuan, Y.; Danishefsky, S.J. Oxo-ester Mediated Native Chemical Ligation: Concept and Applications. J. Am. Chem. Soc. 2008, 130, 15814–15816. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Shang, S.; Halkina, T.; Yuan, Y.; Danishefsky, S.J. Toward Homogeneous Erythropoietin: Non-NCL-Based Chemical Synthesis of the Gln78−Arg166 Glycopeptide Domain. J. Am. Chem. Soc. 2009, 131, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Trzupek, J.D.; Wu, B.; Wan, Q.; Chen, G.; Tan, Z.; Yuan, Y.; Danishefsky, S.J. Toward Homogeneous Erythropoietin: Chemical Synthesis of the Ala1−Gly28 Glycopeptide Domain by “Alanine” Ligation. J. Am. Chem. Soc. 2009, 131, 5438–5443. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Xi, W.-X.; Wang, F.-L.; Li, J.; Guo, Q.-X. Fmoc-SPPS chemistry compatible approach for the generation of (glyco)peptide aryl thioesters. Tetrahedron Lett. 2011, 52, 2655–2660. [Google Scholar] [CrossRef]
- Danger, G.; Michaunt, A.; Bucci, M.; Boiteau, L.; Canal, J.; Plasson, R.; Pascal, R. 5(4H)-Oxazolones as Intermediates in the Carbodiimide- and Cyanamide-Promoted Peptide Activations in Aqueous Solution. Angew. Chem. Int. Ed. 2013, 52, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Metrano, A.J.; Miller, S.J. Peptide-Catalyzed Conversion of Racemic Oxazol-5(4H)-ones into Enantiomerically Enriched α-Amino Acid Derivatives. J. Org. Chem. 2014, 79, 1542–1544. [Google Scholar] [CrossRef] [PubMed]
- Eom, K.D.; Tam, J.P. Acid-Catalyzed Tandem Thiol Switch for Preparing Peptide Thioesters from Mercaptoethyl Esters. Org. Lett. 2011, 13, 2610–2613. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.; McGahren, W.J. Mechanistic studies of peptide oxazolone racemization. Tetrahedron 1967, 23, 2031–2050. [Google Scholar] [CrossRef]
- Lu, W.; Qasim, M.A.; Kent, S.B.H. Comparative Total Syntheses of Turkey Ovomucoid Third Domain by Both Stepwise Solid Phase Peptide Synthesis and Native Chemical Ligation. J. Am. Chem. Soc. 1996, 118, 8518–8523. [Google Scholar] [CrossRef]
- Hackeng, T.M.; Griffin, J.H.; Dawson, P.E. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. USA 1999, 96, 10068–10073. [Google Scholar] [CrossRef] [PubMed]
- Langenhan, J.M.; Guzei, I.A.; Gellman, S.H. Parallel Sheet Secondary Structure in β-Peptides. Angew. Chem. 2003, 115, 2504–2507. [Google Scholar]
- Tsuboi, M.; Shimanouchi, T.; Mizushima, S.-I. Near Infrared Spectra of Compounds with Two Peptide Bonds and the Configuration of a Polypeptide Chain. VII. On the Extended Forms of Polypeptide Chains. J. Am. Chem. Soc. 1959, 81, 1406–1411. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available.




© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.M. The C-Terminal O-S Acyl Shift Pathway under Acidic Condition to Propose Peptide-Thioesters. Molecules 2016, 21, 1559. https://doi.org/10.3390/molecules21111559
Kim BM. The C-Terminal O-S Acyl Shift Pathway under Acidic Condition to Propose Peptide-Thioesters. Molecules. 2016; 21(11):1559. https://doi.org/10.3390/molecules21111559
Chicago/Turabian StyleKim, Bo Mi. 2016. "The C-Terminal O-S Acyl Shift Pathway under Acidic Condition to Propose Peptide-Thioesters" Molecules 21, no. 11: 1559. https://doi.org/10.3390/molecules21111559