3. Experimental Section
3.1. Chemistry
Melting points were obtained on a Büchi Melting Point B-540 apparatus (Büchi Labortechnik, Flawil, Switzerland) and were uncorrected. Mass spectra (MS) were taken in electrospray ionization (ESI) mode on an Agilent 1100 LC-MS (Agilent, Palo Alto, CA, USA). Proton (1H) nuclear magnetic resonance spectroscopy were performed using a Bruker ARX-300, 300 or 400 MHz spectrometers (Bruker Bioscience, Billerica, MA, USA) with tetramethylsilane (TMS) as an internal standard. Thin-layer chromatography (TLC) analysis was carried out on silica gel plates GF254 (Qingdao Haiyang Chemical, Qingdao, China). Unless otherwise noted, all common reagents and solvents were used as obtained from commercial suppliers without further purification.
3.2. General Procedure for Preparation of 2-(4-Aminophenyl)-4-aminoquinazolines (4a–b)
Dioxane (100 mL) was added to a solution of sodium carbonate (3.2 g, 0.03 mol) in water (25 mL). Under argon, 2-chloro-4-aminoquinazoline 3 (0.01 mol), 4-aminophenylboronic acid pinacol ester (2.4 g, 0.011 mol) and Pd(PPh3)2Cl2 (0.7 g, 1 mmol) were added to the mixture successively. After refluxing for 6 h, water (50 mL) was added to the reaction mixture. The mixture was extracted by dichloromethane (50 mL × 3). The organic phase was washed by brine (50 mL × 1), dried, and evaporated to yield 4a–b.
N1-(2-(4-aminophenyl)quinazolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (4a). Yield: 73.5%; MS (ESI) m/z: 308.2 [M + H+]. 2-(4-Aminophenyl)-N-(3-morpholinopropyl)quinazolin-4-amine (4b). Yield: 78.1%; MS (ESI) m/z: 364.5 [M + H+].
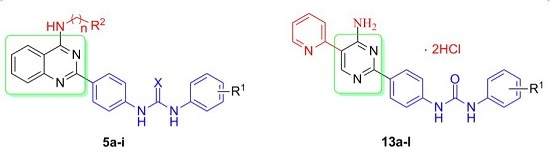
3.3. General Procedure for Preparation of Componds 5a–i
A mixture of intermediate 4 (1 mmmol) and corresponding aromatic isocyanate 6 or isothiocyanate 7 (1.1 mmol) in dry THF (15 mL) was stirred at 30 °C for 6 h and monitored by TLC. The precipitate was collected by filtration, washed with ether, and purified by silica gel chromatography (MeOH:CH2Cl2 = 20:1) to afford target compounds 5a–i.
1-(2-Chloro-6-methylphenyl)-3-(4-(4-((2-(dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)urea (5a). Yield: 62%; m.p.: 144.5–147.0 °C; MS (ESI) m/z: 474.9 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.08 (s, 1H), 8.2.4 (d, J = 8.8 Hz, 2H ), 8.12 (t, J = 5.2, 4.8 Hz, H), 8.04 (d, J = 8.0 Hz, 1H), 7.95 (s, 1H), 7.56 (m, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.29 (m, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 7.05 (m, 1H), 3.17 (q, 2H), 2.47 (t, J = 8.0 Hz, 2H), 2.38 (s, 3H), 2.21 (s, 6H). Anal. Calcd for C26H27ClN6O (%): C, 65.74; H, 5.73; N, 17.69; Found (%): C, 65.71; H, 5.76; N, 17.68.
1-(4-(4-((2-(Dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)-3-(3,4-dimethylphenyl)urea (5b). Yield: 68%; m.p.: 132.0–134.0 °C; MS (ESI) m/z: 455.2 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.04 (s, 1H), 8.39 (t, J = 7.6 Hz, 1H), 8.24 (d, J = 7.2 Hz, 3H), 8.10 (d, J = 8.4 Hz, 1H), 7.55 (m, 3H), 7.46 (d, J = 8.4 Hz, 2H), 7.28 (t, J = 7.2 Hz, 1H), 6.89 (d, J = 7.2 Hz, 1H), 6.60 (d, J = 7.2 Hz, 1H), 3.17 (q, 2H), 2.47 (t, J = 8.0 Hz, 2H), 2.23 (s, 3H), 2.22 (s, 3H), 2.19 (s, 6H). Anal. Calcd for C27H30N6O (%): C, 71.34; H, 6.65; N, 18.49; Found (%): C, 71.36; H, 6.62; N, 18.47.
1-(2-Chloro-6-methylphenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)urea (5c). Yield: 70%; m.p.: 152.5–154.5 °C; MS (ESI) m/z: 531.1 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.20 (s, 1H), 8.38 (d, J = 8.4 Hz, 2H ), 8.28 (t, J = 4.8, 5.2 Hz, H), 8.18 (d, J = 8.4 Hz, 1H), 8.07 (s, 1H), 7.71 (m, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.42 (m, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 7.19 (m, 1H), 3.70 (m, 2H), 3.57 (t, J = 8.4 Hz, 4H), 2.40 (m, 6H), 2.27 (s, 3H), 1.87(m, 2H). Anal. Calcd for C29H31ClN6O2 (%): C, 65.59; H, 5.88; N, 15.83; Found (%): C, 65.61; H, 5.85; N, 15.88.
1-(3,4-Dimethylphenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)urea (5d). Yield: 56%; m.p.: 140.7–143.0 °C; MS (ESI) m/z: 510.9 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.18 (s, 1H), 8.54 (t, J = 7.2 Hz, 1H), 8.37 (d, J = 7.2 Hz, 3H), 8.23 (d, J = 8.0 Hz, 1H), 7.69 (m, 3H), 7.59 (d, J = 8.0 Hz, 2H), 7.41 (t, J = 7.2 Hz, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.73 (d, J = 7.2 Hz, 1H), 3.70 (m, 2H), 3.57 (t, J = 7.2 Hz, 4H), 2.42 (t, J = 7.2 Hz, 2H), 2.37 (t, J = 7.2 Hz, 4H), 2.24 (s, 3H), 2.23 (s, 3H), 1.86 (m, 2H). Anal. Calcd for C30H34N6O2 (%): C, 70.56; H, 6.71; N, 16.46; Found (%): C, 70.55; H, 6.76; N, 16.42.
1-(4-Fluorophenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)urea (5e). Yield: 52%; m.p.: 137.0–139.5 °C; MS (ESI) m/z: 501.2 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.91 (s, 2H), 8.51 (t, J = 5.2 Hz, 1H), 8.39 (d, J = 8.8 Hz, 2H), 8.30 (d, J = 8.4 Hz, 1H), 7.72 (m, 4H), 7.60 (d, J = 8.8 Hz, 2H), 7.43 (m, 1H), 7.36 (d, J = 8.4 Hz, 2H), 4.15 (t, J = 6.8 Hz, 2H ), 3.72 (t, J = 6.4 Hz, 4H), 2.43 (t, J = 6.8 Hz, 2H), 2.38 (t, J = 6.4 Hz, 4H), 1.86 (m, 2H). Anal. Calcd for C28H29FN6O2 (%): C, 67.18; H, 5.84; N, 16.79; Found (%): C, 67.15; H, 5.88; N, 16.75.
1-(4-Chlorophenyl)-3-(4-(4-((2-(dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)thiourea (5f). Yield: 65%; m.p.: 131.5–134.0 °C; MS (ESI) m/z: 477.1 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.75 (s, 2H), 8.31 (d, J = 8.4 Hz, 3H), 8.07 (d, J = 8.1 Hz, 1H), 7.68–7.59 (m, 4H), 7.45 (d, J = 8.4 Hz, 2H), 7.37–7.26 (m, 3H), 3.86–3.77 (q, 2H), 2.73 (t, J = 6.4 Hz, 2H), 2.33 (s, 6H). Anal. Calcd for C25H25ClN6S (%): C, 62.95; H, 5.28; N, 17.62; Found (%): C, 62.94; H, 5.27; N, 17.64.
1-(4-(4-((2-(Dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)thiourea (5g). Yield: 60%; m.p.: 150.8–153.0 °C; MS (ESI) m/z: 527.2 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.12 (s, 1H), 10.03 (s, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.18 (t, J = 6.8 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.58 (m, 3H), 7.47 (d, J = 8.8 Hz, 2H), 7.31 (m, 3H), 6.96 (d, J = 8.4 Hz, 1H), 3.38 (q, 2H), 2.56 (t, J = 7.2 Hz, 2H), 2.23 (s, 6H). Anal. Calcd for C26H25F3N6OS (%): C, 59.30; H, 4.79; N, 15.96; Found (%): C, 59.32; H, 4.76; N, 15.94.
1-(4-Chlorophenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)thiourea (5h). Yield: 63%; m.p.: 145.5–148.0 °C; MS (ESI) m/z: 533.8 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.90 (s, 2H), 8.42 (d, J = 8.8 Hz, 3H), 8.24 (d, J = 8.4 Hz, 1H), 7.73 (m, 4H), 7.65 (d, J = 8.4 Hz, 2H), 7.44 (m, 1H), 7.38 (d, J = 8.8 Hz, 2H), 3.71 (m, 2H), 3.58 (t, J = 8.4 Hz, 4H), 2.43 (t, J = 7.2 Hz, 2H), 2.38 (t, J = 8.4 Hz, 4H), 1.88 (m, 2H). Anal. Calcd for C28H29ClN6OS (%): C, 63.09; H, 5.48; N, 15.76; Found (%): C, 63.05; H, 5.47; N, 15.79.
1-(4-(4-((3-Morpholinopropyl)amino)quinazolin-2-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)thiourea (5i). Yield: 59%; m.p.: 164.5–166.5 °C; MS (ESI) m/z: 583.9 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.25 (s, 1H), 10.16 (s, 1H), 8.45 (d, J = 8.8 Hz, 2H), 8.35 (t, J = 7.2 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 7.74 (m, 3H), 7.64 (d, J = 8.8 Hz, 2H), 7.48 (m, 3H), 7.11 (d, J = 8.0 Hz, 1H), 3.73 (m, 2H), 3.59 (t, J = 4.4 Hz, 4H), 2.44 (t, J = 7.2 Hz, 2H), 2.39 (t, J = 4.4 Hz, 4H), 1.89 (m, 2H). Anal. Calcd for C29H29F3N6O2S (%): C, 59.78; H, 5.02; N, 14.42; Found (%): C, 59.75; H, 5.01; N, 14.47.
3.4. General Procedure for Preparation of Aromatic Isocyanates (6a–n)
Appropriate aromatic amine (0.02 mol) was added slowly to a stirred solution of BTC (3.0 g, 0.01 mol) in 1,4-dioxane (30 mL) at room temperature. After refluxing for 8-12 h, excess 1,4-dioxane was evaporated. The residue was distilled under reduced pressure to afford compounds 6a–n.
1-Chloro-4-isocyanatobenzene (6a). Purity: 78.2%; b.p.: 110–114 °C (30–40 mmHg).
4-Isocyanato-1,2-dimethylbenzene (6b). Purity: 80.5%; b.p.: 59–65 °C (30–40 mmHg).
1-Chloro-2-isocyanato-3-methylbenzene (6c). Purity: 80.8%; b.p.: 116–118 °C (30–40 mmHg).
1-Fluoro-4-isocyanatobenzene (6d). Purity: 84.2%; b.p.: 94–95 °C (30–40 mmHg).
1-Fluoro-3-isocyanatobenzene (6e). Purity: 83.6%; b.p.: 85–91 °C (30–40 mmHg).
1-Fluoro-2-isocyanatobenzene (6f). Purity: 84.3%; b.p.: 76–80 °C (30–40 mmHg).
1-Chloro-3-isocyanatobenzene (6g). Purity: 80.1%; b.p.: 125–128 °C (30–40 mmHg).
2-Chloro-1-fluoro-4-isocyanatobenzene (6h). Purity: 85.2%; b.p.: 93–98 °C (30–40 mmHg).
1,3-Difluoro-2-isocyanatobenzene (6i). Purity: 80.9%; b.p.: 67–71 °C (30–40 mmHg).
2-Isocyanato-1,3-dimethylbenzene (6j). Purity: 76.9%; b.p.: 67–71 °C (30–40 mmHg).
1-Isocyanato-2,4-dimethylbenzene (6k). Purity: 77.5%; b.p.: 71–77 °C (30–40 mmHg).
1-Isocyanato-3-(trifluoromethyl)benzene (6l). Purity: 83.2%; b.p.: 105–111 °C (30–40 mmHg).
1-Isocyanato-4-(trifluoromethoxy)benzene (6m). Purity: 81.0%; b.p.: 113–116 °C (30–40 mmHg).
Isocyanatobenzene (6n). Purity: 80.2%; b.p.: 45–50 °C (30–40 mmHg).
3.5. General Procedure for Preparation of Aromatic Isothiocyanates (7a,b)
Appropriate aromatic amine (0.02 mol) and DABCO (6.7 g, 0.06 mol) were dissolved in toluene (25 mL) at room temperature. Under stirring, CS2 (4.6 g, 0.06 mol) was added dropwise to the solution within 20 min. After stirring at room temperature for 12 h, the precipitate was collected by filtration and washed with toluene. The residue was dried and suspended in CHCl3 (25 mL), and BTC (19.6 g, 0.066 mol) in CHCl3 (25 mL) was added dropwise to the mixture slowly in an ice bath. Then the mixture was stirred for 1 h at room temperature and refluxed for 1 h. The mixture was filtered and evaporated to give compounds 7a,b.
1-Chloro-4-isothiocyanatobenzene (7a). Purity: 77.5%.
1-Isothiocyanato-4-(trifluoromethoxy)benzene (7b). Purity: 79.8%.
3.6. Ethyl 4-aminobenzimidate Dihydrochloride (8)
Dry hydrogen chloride was bubbled into the mixture of 4-aminobenzonitrile (15 g, 0.127 mol), 1,4-dioxane (100 mL) and anhydrous ethanol (100 mL) for 6 h in an ice bath. Then the mixture was sealed and stirred at room temperature for 48 h. After completion, the solution was concentrated under reduced pressure and the semisolid residue was diluted with anhydrous ether (200 mL). The suspension was filtered, washed with anhydrous ether and dried to give the title compound 8 as a white solid (29.3 g, 97.7%). MS (ESI) m/z: 164.9 [M + H+].
3.7. 4-Aminobenzimidamide Hydrochloride (9)
Dry ammonia was bubbled into the mixture of compound 8 (29.3 g, 0.123 mol) and anhydrous ethanol (200 mL) for 6 h in an ice bath. Then the mixture was sealed and stirred at room temperature for 24 h. Then the solution was concentrated under a reduced pressure and the semisolid residue was diluted with anhydrous ether (200 mL). The suspension was filtered, washed with anhydrous ether, and dried to give the title compound 9 as a white solid (20.2 g, 95.3%). MS (ESI) m/z: 135.9 [M + H+].
3.8. 3-(Dimethylamino)-2-(pyridin-2-yl)acrylonitrile (10)
A mixture of 2-(pyridin-2-yl)acetonitrile (15.0 g, 0.127 mol), DMF-DMA (30 mL, 0.229 mol) and methanol (36 mL) was stirred at 30 °C for 24 h. The solvent was evaporated, moderate methanol and ethyl acetate was added in. The solution was concentrated under a reduced pressure to remove excess DMF-DMA completely. The residue was poured into petroleum ether (100 mL) and stirred for 6 h. The suspension was separated by filtration and washed with ether to give compound 10 as a dark red solid (18.3 g, 83.2%). MS (ESI) m/z: 174.0 [M + H+].
3.9. 2-(4-Aminophenyl)-5-(pyridin-2-yl)pyrimidin-4-amine (11)
A mixture of compound 9 (20.2 g, 0.118 mol ), compound 10 (17.0 g, 0.098 mol), sodium carbonate (12.5 g, 0.0118 mol), methnol (200 mL) and water (60 mL) was heated to 70 °C for 24 h. The solution was concentrated and added to water (200 mL). The precipitate was collected by filtration, washed with ether, and recrystallized with ethnol to afford compound 11 as a pale white solid (13.9 g, 53.8%). MS (ESI) m/z: 264.1 [M + H+].
3.10. General Procedure for Preparation of Compounds 12a–l and 13a–l
A mixture of intermediate 11 (1 mmmol) and corresponding aromatic isocyanate 6 (1.1 mmol) in dry THF (15 mL) was stirred at 30 °C for 6 h and monitored by TLC. The precipitate was collected by filtration, washed with ether, and dried to afford compounds 12a–l without further purification. At room temperature, 20%–30% hydrochloride ethanol solution (1 mL) was added dropwise to a stirred solution of compound 12 dissolved in CHCl3 (10 mL). The suspension was stirred for 1 h, filtered and dried to give the corresponding compounds 13a–l.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-phenylurea dihydrochloride (13a). Yield: 79%; m.p.: 303.0–306.0 °C; MS (ESI) m/z: 383.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.41 (s, 1H), 9.11 (s, 1H), 8.93 (s, 2H), 8.77 (s, 1H), 8.68 (d, J = 4.4 Hz, 1H), 8.32 (d, J = 8.4 Hz, 2H), 8.10 (d, J = 8.0 Hz, 1H), 7.94 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.4 Hz), 7.38–7.32 (m, 1H), 7.30 (t, J = 7.7 Hz, 2H), 7.00 (t, J = 6.9 Hz). Anal. Calcd for C22H20Cl2N6O (%): C, 58.03; H, 4.43; N, 18.46; Found (%): C, 58.07; H, 4.46; N, 18.42.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2,4-dimethylphenyl)urea dihydrochloride (13b). Yield: 65%; m.p.: 286.0–288.5 °C; MS (ESI) m/z: 411.3 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.77 (s, 1H), 8.89 (s, 1H), 8.74 (d, J = 4.6 Hz, 1H), 8.25(d, J = 8.6 Hz, 2H), 8.22 (d, J = 7.8 Hz, 1H), 8.05 (t, J = 7.9 Hz, 1H), 7.72(d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.1 Hz, 1H), 7.52 (t, J = 4.8 Hz, 1H), 7.02 (s, 1H), 6.98 (d, J = 8.2 Hz, 2H), 2.24 (s, 6H). Anal. Calcd for C24H24Cl2N6O (%): C, 59.63; H, 5.00; N, 17.39; Found (%): C, 59.60; H, 5.06; N, 17.34.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(6-chloro-2-methylphenyl)urea dihydrochloride (13c). Yield: 72%; m.p.: 252.0–254.0 °C; MS (ESI) m/z: 431.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.78 (s, 1H), 8.85 (s, 1H), 8.74 (d, J = 4.2 Hz, 1H), 8.56 (s, 1H), 8.26 (d, J = 8.7 Hz, 2H), 8.22 (d, J = 8.1 Hz, 1H), 8.04 (t, J = 6.7 Hz, 1H), 7.72 (d, J = 8.9 Hz, 2H), 7.54–7.50 (m, 1H), 7.36 (d, J = 5.4 Hz, 1H), 7.27–7.18 (m, 2H), 2.28 (s, 3H). Anal. Calcd for C23H21Cl3N6O (%): C, 54.83; H, 4.20; N, 16.68; Found (%): C, 54.84; H, 4.23; N, 16.65.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2,6-dimethylphenyl)urea dihydrochloride (13d). Yield: 76%; m.p.: 271.0–274.0 °C; MS (ESI) m/z: 411.2 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.83 (s, 1H), 8.85 (s, 1H), 8.74(d, J = 4.2 Hz, 1H), 8.34 (s, 1H), 8.23 (d, J = 9.3 Hz, 2H), 8.21 (d, J = 8.1 Hz, 1H), 8.05(t, J = 7.8 Hz, 1H), 7.72 (d, J = 9.0 Hz, 2H), 7.54–7.50 (m, 1H), 7.08 (s, 3H), 2.23 (s, 6H). Anal. Calcd for C24H24Cl2N6O (%): C, 59.63; H, 5.00; N, 17.39; Found (%): C, 59.67; H, 5.02; N, 17.36.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2,6-difluorophenyl)urea dihydrochloride (13e). Yield: 68%; m.p.: 279.5–281.0 °C; MS (ESI) m/z: 419.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.78 (s, 1H), 8.88 (s, 1H), 8.73 (d, J = 4.7 Hz, 1H), 8.53 (s, 1H), 8.40 (s, 2H), 8.24 (d, J = 8.8 Hz, 2H), 8.20 (d, J = 9.3 Hz, 1H), 8.04 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 8.6 Hz, 2H), 7.54–7.49 (m, 1H), 7.32–7.25 (m, 3H). Anal. Calcd for C22H18Cl2F2N6O (%): C, 53.78; H, 3.69; N, 17.10; Found (%): C, 53.75; H, 3.72; N, 17.08.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(3-chloro-4-fluorophenyl)urea dihydrochloride (13f). Yield: 80%; m.p.: 289.5–291.5 °C; MS (ESI) m/z: 435.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.69 (s, 1H), 9.55 (s, 1H), 8.88 (s, 1H), 8.74 (d, J = 4.4 Hz, 1H), 8.25 (d, J = 8.8 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.05 (t, J = 7.8 Hz, 1H), 7.85–7.82 (m, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.54–7.49 (m, 1H), 7.40–7.34 (m, 2H). Anal. Calcd for C22H18Cl3FN6O (%): C, 52.04; H, 3.57; N, 16.55; Found (%): C, 52.05; H, 3.52; N, 16.56.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(3-chlorophenyl)urea dihydrochloride (13g). Yield: 71%; m.p.: 270.0–272.5 °C; MS (ESI) m/z: 417.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.89 (s, 1H), 9.71 (s, 1H), 8.86 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.25 (d, J = 8.7 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.05 (t, J = 7.9 Hz, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.72 (s, 1H), 7.55–7.51 (m, 1H), 7.33–7.30 (m, 2H), 7.04 (d, J = 5.8 Hz, 1H). Anal. Calcd for C22H19Cl3N6O (%): C, 53.95; H, 3.91; N, 17.16; Found (%): C, 53.94; H, 3.92; N, 17.14.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea dihydrochloride (13h). Yield: 65%; m.p.: 267.0–269.5 °C; MS (ESI) m/z: 451.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.78 (s, 1H), 9.74 (s, 1H), 8.89 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.26 (d, J = 8.8 Hz, 2H), 8.21 (m, 2H), 8.05 (s, 2H), 7.74 (d, J = 8.8 Hz, 2H), 7.61–7.50 (m, 4H), 7.35 (d, J = 7.6 Hz, 1H). Anal. Calcd for C23H19Cl2F3N6O (%): C, 52.79; H, 3.66; N, 16.06; Found (%): C, 52.75; H, 3.69; N, 16.05.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)urea dihydrochloride (13i). Yield: 78%; m.p.: 308.5–310.5 °C; MS (ESI) m/z: 467.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.50 (s, 1H), 9.35 (s, 1H), 8.89 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.24 (d, J = 8.7 Hz, 2H), 8.19 (d, J = 8.4 Hz, 1H), 8.04 (t, J = 6.7 Hz, 1H), 7.71 (d, J = 8.7 Hz, 2H), 7.59 (d, 2H), 7.60–7.51 (m, 1H), 7.32 (d, J = 8.1 Hz, 2H). Anal. Calcd for C23H19Cl2F3N6O2 (%): C, 51.22; H, 3.55; N, 15.58; Found (%): C, 51.24; H, 3.59; N, 15.54.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(4-fluorophenyl)urea dihydrochloride (13j). Yield: 75%; m.p.: 264.0–267.0 °C; MS (ESI) m/z: 401.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.80 (s, 1H), 9.47 (s, 1H), 8.87 (s, 1H), 8.75 (d, J = 4.6 Hz, 1H), 8.26 (d, J = 8.7 Hz, 2H), 8.22 (d, J = 7.8 Hz, 1H), 8.07 (t, J = 7.9 Hz, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.56–7.52 (m, 2H), 7.49 (t, J = 4.8 Hz, 1H), 7.15 (t, J = 8.8 Hz, 2H). Anal. Calcd for C22H19Cl2FN6O (%): C, 55.82; H, 4.05; N, 17.76; Found (%): C, 55.84; H, 4.01; N, 17.79.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(3-fluorophenyl)urea dihydrochloride (13k). Yield: 67%; m.p.: 228.5–231.0 °C; MS (ESI) m/z: 401.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.89 (s, 1H), 9.73 (s, 1H), 8.86 (s, 1H), 8.74 (d, J = 4.5 Hz), 8.26 (d, J = 8.9 Hz, 2H), 8.22 (d, J = 8.4 Hz, 1H), 8.05 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 8.9 Hz, 2H), 7.55 (s, 1H), 7.53–7.50 (m, 1H), 7.37–7.29 (m, 1H), 7.14 (d, J = 8.1 Hz, 1H), 6.82 (t, J = 8.4 Hz, 1H). Anal. Calcd for C22H19Cl2FN6O (%): C, 55.82; H, 4.05; N, 17.76; Found (%): C, 55.81; H, 4.06; N, 17.72.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2-fluorophenyl)urea dihydrochloride (13l). Yield: 73%; m.p.: 258.5–261.5 °C; MS (ESI) m/z: 401.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 10.01 (s, 1H), 8.94 (s, 1H), 8.87 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.27 (d, J = 9.0 Hz, 2H), 8.22 (d, J = 8.1 Hz, 1H), 8.16–8.03 (m, 1H), 7.74 (d, J = 8.7 Hz, 2H), 7.55–7.50 (m, 1H), 7.29–7.03 (m, 3H). Anal. Calcd for C22H19Cl2FN6O (%): C, 55.82; H, 4.05; N, 17.76; Found (%): C, 55.86; H, 4.03; N, 17.75.
3.11. Evaluation of the Biological Activity
The antitumor activity of compounds 5a–i and 13a–l was evaluated with HT-29, H-460, A549, and MDA-MB-231 by the MTT method in vitro, with sorafenib as positive control. The cancer cells were cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS).
Approximately 4 × 103 cells, suspended in MEM medium, were plated onto each well of a 96-well plate and incubated in 5% CO2 at 37 °C for 24 h. The test compounds were added to the culture medium at the indicated final concentrations and the cell cultures were continued for 72 h. Fresh MTT was added to each well at a final concentration of 5 μg/mL and incubated with cells at 37 °C for 4 h. The formazan crystals were dissolved in 100 μL DMSO per each well, and the absorbency at 492 nm (for the absorbance of MTT formazan) and 630 nm (for the reference wavelength) was measured with the ELISA reader. All of the compounds were tested twice in each of the cell lines. The results expressed as IC50 (inhibitory concentration of 50%) were the averages of two determinations and were calculated by using the Bacus Laboratories Incorporated Slide Scanner (Bliss) software.
3.12. EGFR and VEGFR2/KDR Kinases Assay In Vitro
The target compound
5a was tested for its activity against EGFR and VEGFR2/KDR kinases through the mobility shift assay. All kinase assays were performed in 96-well plates in a 50 μL reaction volume. The kinase buffer contains 50 mM HEPES, pH 7.5, 10 mM MgCl
2, 0.0015% Brij-35 and 2 mM DTT. The stop buffer contains 100 mM HEPES, pH 7.5, 0.015% Brij-35, 0.2% Coating Reagent #3, and 50 mM ethylene diamine tetraacetic acid (EDTA). Compounds were diluted to 500 μM by 100% DMSO, then 10 μL of the compounds were transfered to a new 96-well plate as the intermediate plate, and 90 μL kinase buffer was added to each well. Five microliters of each well of the intermediate plate was transferred to a 384-well plate. The following amounts of enzyme and substrate were used per well: kinase base buffer, FAM-labeled peptide, ATP, and enzyme solution. Wells containing the substrate, enzyme, and DMSO without compound were used as the DMSO control. Wells containing just the substrate without enzyme were used as the low control. The compounds were incubated at room temperature for 10 min. Ten microliters of peptide solution was added to each well and incubated at 28 °C for a specified period of time and the reaction stopped by 25 μL of stop buffer. Finally, data was collected using the Caliper program, which converted conversion values to inhibition values.
where “max” stands for DMSO control; “min” stands for low control.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

