3.2. Syntheses
1,6-Anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-glucopyranose (
3). Compound
3 was synthesized in 7 steps (21% overall yield) from commercially available 1,6-anhydro-β-
d-glucopyranose following the method of Oikawa et al. [
19].
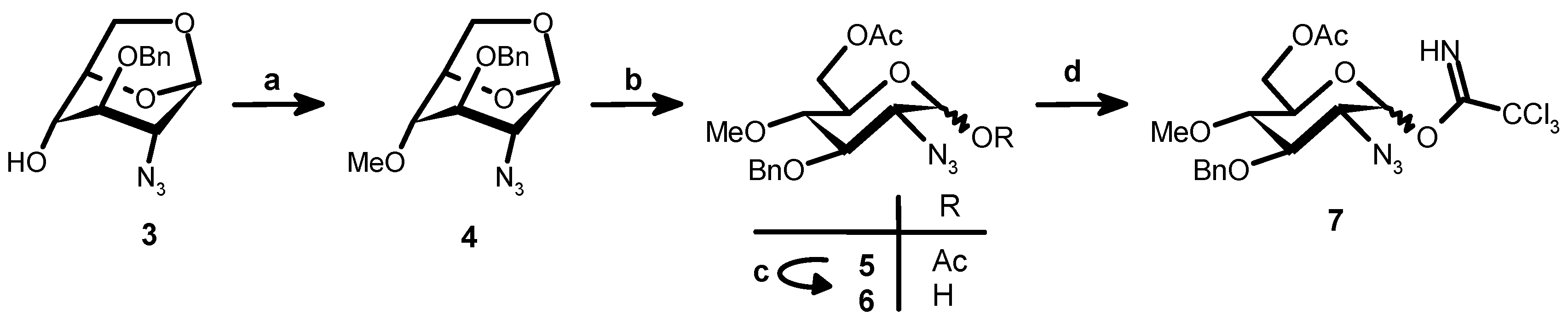
1,6-Anhydro-2-azido-3-O-benzyl-2-deoxy-4-O-methyl-β-d-glucopyranose (4). Methyl iodide (0.5 mL, 8.6 mmol) was added to a cooled (0 °C) solution of 3 (2 g, 7.2 mmol) in dry DMF (32 mL). Sodium hydride 50% in oil (0.5 g, 10.8 mmol) was then introduced in portions and the reaction mixture was stirred at RT for 2 h. After cooling to 0 °C NaH in excess was destroyed by methanol (20 mL). After evaporation under vacuum EtOAc (100 mL) and water (50 mL) were added. The aqueous phase was washed with EtOAc (50 mL) and the combined organic phases were washed with brine (50 mL) and dried (Na2SO4). After evaporation crude 4 (2.3 g) was obtained as syrup and used for the next step without further purification. ESIMS m/z: [M + Na]+: 314.0373, [M + K]+: 330.0077. Calculated for C14H17N3O4 + Na: 314.1117, C14H17N3O4K: 330.0856.
6-O-Acetyl-2-azido-3-O-benzyl-2-deoxy-4-O-methyl-β-d-glucopyranose (6). A mixture of trifluoroacetic acid (5.5 mL, 72 mmol) in acetic anhydride (68 mL, 721 mmol) was added to 4 (2.3 g, 7.2 mmol) and the resulting mixture was stirred overnight at room temperature. After evaporation under vacuum and co-evaporation with toluene the residue was dissolved in CH2Cl2 (100 mL), washed with saturated aqueous NaHCO3 (40 mL) and brine (50 mL × 1). After drying (Na2SO4) and evaporation 5 was obtained (2.93 g, 96.6%) and used for the next step without further purification. ESIMS m/z: [M + Na]+: 416.0497, [M + K]+: 432.0180. Calculated for C18H23N3O7Na: 416.1434, C18H23N3O7K: 432.1173.
Benzylamine (1.4 mL, 12.8 mmol) was added to a solution of 5 (2.7 g, 6.4 mmol) in dry THF after stirring for 3 h at RT the solvent was evaporated under vacuum. Ethyl acetate (100 mL) was added to the residue and the solution was washed with 1 M HCl (20 mL). The aqueous phase was washed with ethyl acetate (50 mL) and the combined organic phases were washed with H2O (20 mL), brine (20 mL) and dried (Na2SO4). After evaporation 6 (3.24 g) was obtained as a brown syrup and used as such in the next step. ESIMS m/z: [M + Na]+: 374.0518, [M + K]+: 390.0207. Calculated for C16H21N3O6Na: 374.1328, C16H21N3O6K: 390.1062.
6-O-Acetyl-2-azido-3-O-benzyl-2-deoxy-4-O-methyl-1-O-trichloroacetimidoyl-d-glucopyranose (7). Cs2CO3 (1.86 g, 5.7 mmol) was added at 0 °C to a solution under nitrogen atmosphere of crude 6 (3.24 g, 6.4 mmol) and Cl3CCN (3.83 mL, 38.2 mmol) in CH2Cl2 (63 mL, dried over molecular sieves). After stirring at RT for 3 h the solution was filtered through Celite and the filter pad was washed with CH2Cl2 (60 mL). The dichloromethane solution was washed with H2O, brine and dried (Na2SO4). After concentration and flash chromatography (98:2–95:5 CH2Cl2–EtOAc) 7 (α:β ratio 1:6) was obtained (1.88 g; 59.9%). ESIMS m/z: [M + Na]+: 516.9294. Calculated for C18H21Cl3N4O6Na: 517.0418.
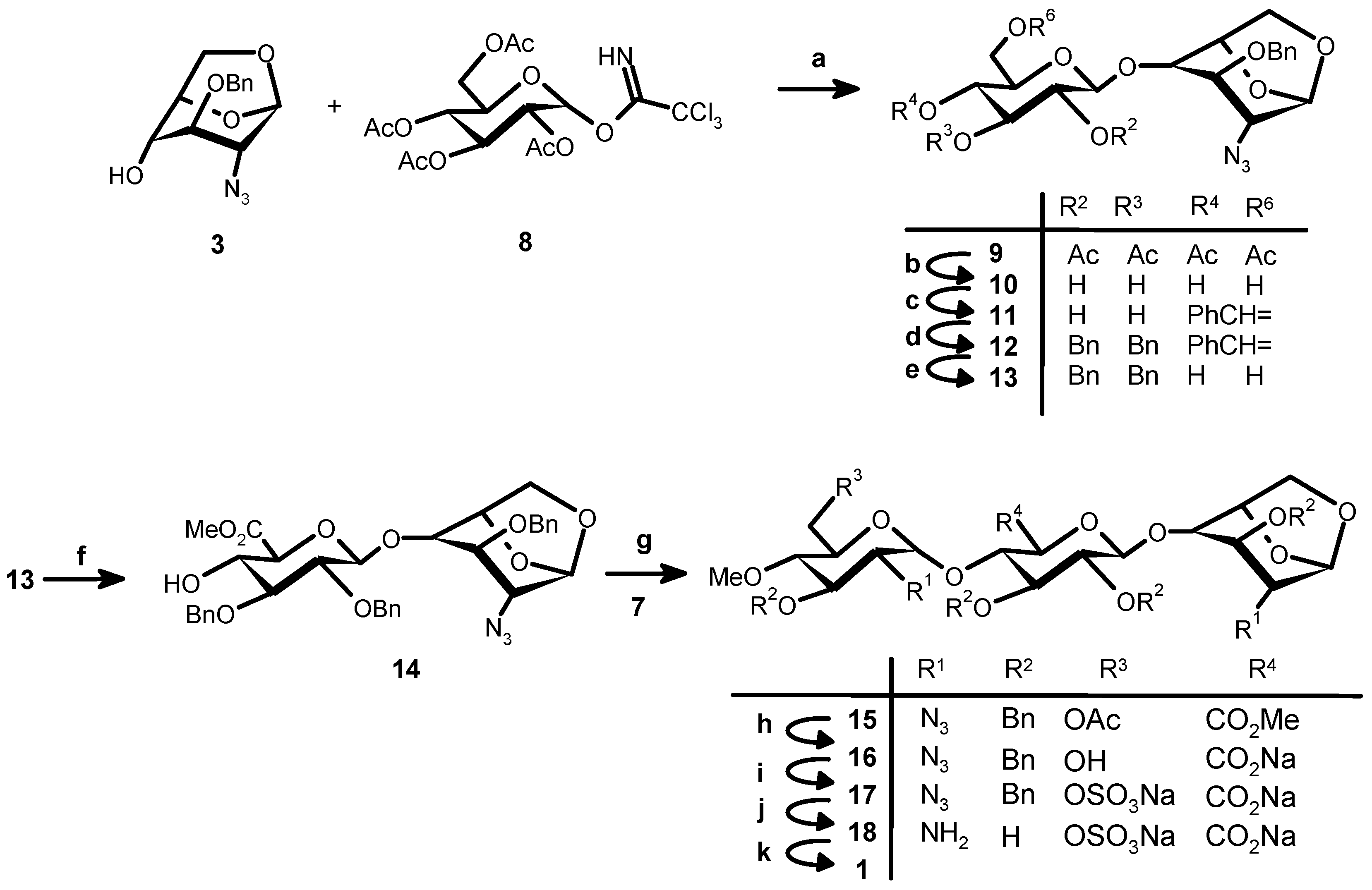
(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-gluco-pyranose (9). A cold (−30 °C) solution of 3 (5 g, 18.21 mmol) and 8 (13.45 g, 27.31 mmol, 1.5 eq) in dry CH2Cl2 (180 mL) containing 4 Å molecular sieves (5 g, previously activated at 400 °C for 4 h), was stirred at RT for 15 min under nitrogen atmosphere. After cooling at −30 °C, a solution of BF3:Et2O (0.75 mL, 5.89 mmol, 0.3 eq) in CH2Cl2 (225 mL) was added dropwise over 30 min. The temperature was slowly raised to room temperature (in about 45 min). After 2 h the reaction mixture was neutralized by TEA. After evaporation under vacuum and flash chromatography (85:15 hexane–EtOAc) 9 was obtained (8.49 g, 76.7%) as a glass. 1H-NMR (500 MHz, CDCl3) δ: 7.36–7.31 (m, 5H, Ph), 5.45 (s, 1H, H-1), 5.21 (t, 1H, J = 9.5 Hz, H-3′), 5.11 (t, 1H, J = 9.5 Hz, H-4′), 5.01 (dd, 1H, J = 9.5 and 8.0 Hz, H-2′), 4.74 (d, 1H, J = 8.0 Hz, H-1′), 4.65 (s, 2H, CH2Ph), 4.58 (d, 1H, J = 5.0 Hz, H-5), 4.20 (dd, 1H, J = 12.5 and 4.5 Hz, H-6′a), 4.14 (m, 1H, H-6′b), 4.09 (d, J = 7.5 Hz, 1H, H-6a), 3.79 (s, 1H, H-3), 3.77 (br s, 2H, H-4 + H-6b), 3.67–3.63 (m, 1H, H-5′), 3.23 (s, 1H, H-2), 2.04 (s, 6H, CH3CO), 2.03 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO). 13C-NMR (CDCl3) δ: 170.47, 170.27, 169.26, 169.16 (4 C=O), 137.33, 128.52, 128.03, 127.65 (Arom. Ph) 100.69 (C-1), 99.36 (C-1′), 77.22 (C-4), 75.96 (C-3), 73.72 (C-5), 72.73 (C-3′), 72.45 (C-5′), 72.14 (CH2Ph), 71.27 (C-2′), 68.21 (C-4′), 65.05 (C-6′), 61.81 (C-6), 59.47 (C-2), 20.54 (CH3CO). ESIMS m/z: [M + Na]+: 630.0705, [M + K]+: 646.0700. Calculated for C27H33N3O13Na: 630.1905, C27H33N3O13K: 646.1644.
β-d-Glucopyranosyl-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-glucopyranose (10). A sodium methanolate solution (0.5 M in methanol, 45 mL, 22.5 mmol) was slowly added at 0 °C to a solution of 9 (9.12 g, 15.01 mmol) in THF–methanol (1:1, 98 mL). After 1 h at RT Amberlite® IR-120 (H+-form, previously washed with H2O and MeOH) was added until pH 2. After removal and washing of the resin the solution was concentrated to give 10 as a white solid used as such in the next step. ESIMS m/z: [M + Na]+: 462.5705, [M + K]+: 478.5600. Calculated for C19H25N3O9Na: 462.1488, C19H25N3O9K: 478.1228.
(4,6-O-Benzylidene-l-β-d-glucopyranosyl)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-gluco-pyranose (11). Camphorsulfonic acid (345 mg, 1.5 mmol) followed by benzaldehyde dimethylacetal (45.2 mL, 300 mmol) were added at RT, under a nitrogen atmosphere, to a solution of crude 10 (6.6 g, 15 mmol) in dry DMF (56 mL). After overnight stirring, a saturated aqueous solution of NaHCO3 (30 mL) was added. After dilution with EtOAc (250 mL) the organic phase was washed with brine, dried (Na2SO4), and concentrated. The crude product was purified by flash chromatography (1:1 CH2Cl2–EtOAc) to give 11 as white solid (4.43 g, 56.3% from 9). 1H-NMR (500 MHz, CDCl3) δ: 7.51–7.32 (m, 10H, aroma), 5.55 (s, 1H, acetal), 5.53 (s, 1H, H-1), 4.71 (d, J = 5.5 Hz, 1H, H-5), 4.67, 4.60 (AB system, J = 12 Hz, 2H, CH2Ph), 4.53 (d, J = 7.5 Hz, 1H, H-1′), 4.27–4.24 (m, 1H, H-6′), 4.17 (d, J = 7.0 Hz, 1H, H-6), 3.83 (d, J = 5.5 Hz, 1H, H-3), 3.82 (s, 1H, H-4), 3.81 (m, 1H, H-6), 3.81 (m, 1H, H-3′), 4.25–3.76(m, 1H, H-6′), 3.61–3.59 (m, 1H, H-2′), 3.59–3.56 (m, 1H, H-4′), 3.43–3.38 (m, 1H, H-5′), 3.22 (s, 1H, H-2). 13C-NMR (CDCl3) δ: 137.3–127.6 (Bn), 109.2 (CH Acetal), 102.2 (C-1′), 102.1 (C-1), 80.5 (C-4′), 77.3 (C-3), 75.6 (C-4), 74.3 (C-2′), 72.8 (CH2Ph), 68.7 (C-6′), 66.8 (C-5′), 65.2 (C-6), 59.2 (C-2). ESIMS m/z: [M + Na]+: 550.0657, [M + K]+: 566.0370 calculated for C26H29N3O9Na: 550.1796, C26H29N3O9 K: 566.1535.
(2,3-di-O-Benzyl-4,6-O-benzylidenel-β-d-glucopyranosyl)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-glucopyranose (12). Benzyl bromide (2.4 mL, 20.31 mmol) was added under nitrogen atmosphere to a solution of 11 (4.12 g, 7.81 mmol) in dry DMF (27 mL). After cooling (0 °C) NaH (1.50 g, 50% dispersion in oil, 31.24 mmol) was added in portions. After stirring at RT for 2 h the solution was cooled to 0 °C and NaH in excess was destroyed by slow addition of methanol (25 mL). After evaporation in vacuo EtOAc (150 mL) and H2O (50 mL) were added. The aqueous phase was extracted by EtOAc (50 mL) and the combined organic solutions were washed with brine (50 mL) dried (Na2SO4) and concentrated to dryness to give crude 12 (6.3 g) as a syrup used as such in the next step. ESIMS m/z: [M + Na]+: 730.1302, [M + K]+: 746.0999. Calculated for C40H41N3O9Na: 730.2735, C40H41N3O9K: 746.8676.
(2,3-di-O-Benzyl-β-d-glucopyranosyl)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-gluco-pyranose (13). An aqueous TFA solution (70%, 12 mL) was added to a solution of 12 (6.3 g, 7.81 mmol) in CH2Cl2 (270 mL). After stirring at RT for 4 h, aqueous NaHCO3 (saturated solution, 200 mL) was added. The aqueous phase was washed with EtOAc (50 mL × 1). Combined organic phases were washed once by brine, dried (Na2SO4), filtered and concentrated to give 13 (6.11 g) as colorless syrup.
ESIMS m/z: [M + Na]+: 642.1171, [M + K]+: 658.0868 calculated for C33H37N3O9Na: 642.2422, C33H37N3O9K: 658.2161.
(Methyl 2,3-di-O-benzyl-β-d-glucopyranosyluronate)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-glucopyranose (14). TEMPO (122 mg, 0.781 mmol) and 1,3-dibromo-5,5-dimethyl hydantin (Dibromantin, 4.47 g, 15.62 mmol) were added to a solution of crude 13 (~7.81 mmol) in CH3CN (260 mL and 1% NaHCO3 aqueous solution (260 mL) and stirred at RT for 2.5 h. pH = 8. 1 M Na2S2O3 aqueous solution was added until the yellow colour was disappeared. At 0 °C 1 N H2SO4 solution was added to pH = 2. 150 mL of EtOAc was added. After two phases were separated, aqueous phase was extracted by EtOAc (120 mL × 2). Combined organic phases were dried over Na2SO4, filtered and concentrated to give crude uronic acid (7 g) directly for methylation. The above crude uronic acid (7 g,) was dissolved in dry DMF (76 mL). Solid NaHCO3 (6.56 g, 78.10 mmol, 10 eq) and iodomethane (4.86 mL, 78.10 mmol, 10 eq) were added under a nitrogen atmosphere and stirred at RT overnight. The reaction mixture was diluted with EtOAc (250 mL) and H2O (120 mL). After the two phases were separated, the aqueous phase was extracted with EtOAc (100 mL × 1). The combined organic phases were washed with 1 M Na2S2O3 (80 mL × 1) and brine (80 mL × 1). The crude product (6.16 g) was submitted to flash chromatography (hexane–EtOAc 6:4) to give compound 14 (4.13 g, yield: 81.8% of 4 steps) as a crystalline glass. 1H-NMR (500 MHz, CDCl3) δ: 7.39–7.28 (m, 15H, 3Ph), 5.52 (s, 1H, H-1), 5.01, 4.74 (AB system, J = 11.0 Hz, 2H, CH2Ph), 4.91, 4.83 (AB system, J = 11.5 Hz, 2H, CH2Ph), 4.67, 4.64 (AB system, J = 11.5 Hz, 2H, CH2Ph), 4.60 (d, J = 5.0 Hz, 1H, H-5), 4.57 (d, J = 7.5 Hz, 1H, H-1′), 4.05 (d, J = 7.0 Hz, 1H, H-6), 3.95 (t, J = 1.5 Hz, 1H, H-3), 3.90 (t, J = 9.5 Hz, 1H, H-4′), 3.79–3.75 (m, 1H, H-6), 3.78 (s, 1H, H-4), 3.78 (t, J = 10.0 Hz, 1H, H-5′), 3.76 (s, 3H, CH3O), 3.54–3.52 (m, 1H, H-2′), 3.51–3.49 (m, 1H, H-3′), 3.21 (s, 1H, H-2). 13C-NMR (CDCl3) δ: 169.39 (CO), 138.44,138.22,137.60 (Ph), 128.52, 128.49, 128.42, 128.38, 127.92, 127.62 (Ph), 103.91 (C-1′), 100.71 (C-1), 83.02 (C-3′), 80.78 (C-2′), 77.85 (C-3), 75.42 (C-4), 75.23, 74.42 (CH2Ph), 74.15 (C-5), 72.63 (C-5′), 71.56 (C-4′), 64.97 (C-6), 59.84 (C-2), 52.66 (CH3O). ESIMS m/z: [M + Na]+: 670.1001, [M + K]+: 686.068. Calculated for C34H37N3O10 + Na: 670.2371, C34H37N3O10K: 686.2111.
(6-O-Acetyl-2-azido-3-O-benzyl-2-deoxy-4-O-methyl-α-d-glucopyranosyl)-O-(1→4)-(methyl 2,3-di-O benzyl-β-d-glucopyranosyluronate)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-glucopyranose (15). Compound 14 (1.679 g, 2.44 mmol), 7 (1.45 g, 2.928 mmol, 1.3 eq) and 4 Å molecular sieves (1.6 g) were stirred in dry DCM at RT for 1 h under nitrogen atmosphere. After cooling (−20 °C), TMSOTf (0.106 mL, 0.2 eq respect to donor) in DCM (16 mL) was added dropwise over 30 min. A precipitate formed and dissolved again. Additional compound 7 (0.19 g, 0.3 eq) and TMSOTf (0.025 mL) were added and after 15 min at −20 °C the mixture was allowed to warm up and maintained at RT for 1 h. TEA was added till pH 7. After filtration and evaporation purification was performed by flash chromatography (CH2Cl2–EtOAc, 95:5 to 92:8) to give 15 (1.387 g, 58%) and its beta anomer (0.465 g, 19.5%). 1H-NMR (500 MHz, CDCl3) (α anomer) δ: 7.4–7.26 (m, 20H, arom. Ph), 5.54 (d, J = 3.75 Hz, 1H, H-1′′), 5.50 (s, 1H, H-1), 5.04, 4.70 (AB system, J = 10.8 Hz, 2H, CH2Ph), 5.03, 4.82 (AB system, J = 10.8 Hz, 2H, CH2Ph), 4.85 (s, 2H, CH2Ph), 4.63, 4.60 (AB system, J = 10.8 Hz, 2H, CH2Ph), 4.61 (d, J = 7.7 Hz, 1H, H-1′), 4.59 (s, 1H, H-5), 4.26 (br s, 2H, H-6′′a + H-6′′b), 4.12 (t, J = 9.1 Hz, 1H, H-4′), 4.07 (d, J = 7.4 Hz, H-6a), 3.92 (d, J = 9.6 Hz, 1H, H-5′), 3.88 (br s, 1H, H-3′′), 3.79–3.74 (m, 4H, H-3 + H-4 + H-3′ + H-6b), 3.74 (s, 3H, Me ester), 3.63 (t, J = 7.7 Hz, 1H, H-2′), 3.50 (s, 3H, OMe), 3.48 (br s, 1H, H-5′′), 3.22–3.19 (m, 3H, H-2 + H-2′ + H-4′′), 2.10 (s, 3H, CH3CO). 13C-NMR (CDCl3) (α anomer) δ: 170.7 (C=O, acetyl), 168.3 (C=O, Me ester), 138.1–137.4 (Bn), 128.1–127.2 (Bn), 103.3 (C-1′), 100.7 (C-1), 97.5 (C-1′′), 83.8 (C-3′), 81.4 (C-2′), 80.0 (C-4′′), 79.5 (C-4), 77.5 (C-3′′), 76.7 (C-3), 75.1 (C-5), 74.3 (C-4′), 74.2 (C-5′), 75.3–74.2 (CH2, Bn), 72.7 (CH2, Bn), 69.7 (C-5′′), 65.0 (C-6), 63.0 (C-2′′), 62.3 (C-6′′), 60.8 (OMe), 59.9 (C-2), 52.7 (CH3, Me ester), 20.8 (CH3CO). ESIMS m/z: [M + Na]+: 1003.1659, [M + K]+: 1019.1354 calculated for C50H56N6O15Na: 1003.3696, C50H56N6O15K: 1019.3435. 1H-NMR (500 MHz, CDCl3) (β anomer) δ: 7.37–7.24 (m, 20H, 4Ph), 5.49 (s, 1H, H-1), 4.98, 4.72 (AB system, J = 11.4 Hz, 2H, CH2Ph), 4.91, 4.66 (AB system, J = 10.7 Hz, 2H, CH2Ph), 4.83, 4.78 (AB system, J = 11.3 Hz, 2H, CH2Ph), 4.64, 4.60 (AB system, J = 11.3 Hz, 2H, CH2Ph), 4.57 (d, J = 7.5 Hz, 1H, H-1′), 4.57 (d, J = 7.5 Hz, 1H, H-5), 4.42 (d, J = 7.6 Hz, H-1′′), 4.20 (m, 1H, H-4′), 4.16 8d, J = 12.5 Hz, 1H, H-6′′a), 4.12 (dd, J = 12.5 and 4.6 Hz, 1H, H-6′′b), 4.05 (d, J = 7.9 Hz, 1H, H-6a), 3.94 (d, J = 9.7 Hz, 1H, H-6a), 3.94 (d, J = 9.7 Hz, 1H, H-5′), 3.88 (br s, 1H, H-3), 3.80 (s, 3H, Me ester), 3.75–3.72 (m, 2H, H-6b + H-4), 3.61 (t, J = 9.3 Hz, 1H, H-3′), 3.55 (t, J = 7.9 Hz, 1H, H-2′), 3.47 (s, 3H, OMe), 3.30–3.25 (m, 3H, H-5′′ + H-3′′ + H-2′′), 3.20–3.18 (m, 2H, H-2 + H-4′′), 1.92 (s, 3H, CH3CO). 13C-NMR (CDCl3) (β anomer) δ: 170.7 (CO, acetyl), 168.2 (CO, Me ester), 129.0–126.2 (Bn), 103.8 (C-1′), 101.6 (C-1′′), 100.6 (C-1), 82.4 (C-3′′), 81.8 (C-3′), 80.8 (C-2′), 79.8 (C-4′′), 78.4 (C-4′), 77.6 (C-3), 76.8 (C-4), 75.5 (CH2, Bn), 75.3 (CH2, Bn), 74.8 (CH2, Bn), 74.4 (C-5′), 73.1 (C-5′′), 72.6 (CH2, Bn), 66.4 (C-2′′), 65.0 (C-6), 62.8 (C-6′′), 60.8 (OMe), 59.9 (C-2), 57.0 (CH3, Me ester), 20.5 (CH3CO).
(2-Azido-3-O-benzyl-2-deoxy-4-O-methyl-α-d-glucopyranosyl)-O-(1→4)-(2,3-di-O-benzyl-β-d-gluco-pyranosyluronate)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-glucopyranose (16). Lithium hydroxide (0.7 M in H2O, 37.3 mL) was added was slowly added to a cooled (−5 °C) solution of 15 (1.35 g, 1.3759 mmol) in THF–MeOH (1:1, 240 mL) containing H2O2 (41.8 mL). After overnight stirring at RT the mixture was acidified to pH 2 by 1 M HCl. The product was extracted by DCM (200 ml and 80 mL) washed with 5% aqueous Na2S2O3 and brine and dried (Na2SO4) to give 13 (1.12 g, 82%) after evaporation. ESIMS m/z: [M − H]+: 923.3761 . Calculated for C47H51N6O14: 923.3458.
(2-Azido-3-O-benzyl-2-deoxy-4-O-methyl-6-O-sodiumsulfonato-α-d-glucopyranosyl)-O-(1→4)-(2,3-di-O-benzyl-β-d-sodium glucopyranosyluronate)-O-(1→4)-1,6-anhydro-2-azido-3-O-benzyl-2-deoxy-β-d-gluco-pyrano se (17). SO3:Py complex (854 mg, 5.37 mmol) was added to a solution of 16 (0.993 g, 1.07 mmol) in dry pyridine (34 mL) and the reaction mixture was heated under nitrogen atmosphere at 55 °C for 2.0 h. After evaporation the crude product was purified by flash chromatography (DCM–MeOH, 97:3) to give 17 (825 mg, 76%). 1H-NMR (500 MHz, CDCl3) δ: 7.37–7.24 (m, 20H, Ph), 5.48 (s, 1H, H-1), 5.43 (d, 1H, J = 3.85 Hz, H-1′′), 5.03, 4.66 (AB system, 2H, J = 10.8 Hz, CH2Ph), 5.03, 4.80 (AB system, 2H, J = 10.8 Hz, CH2Ph), 4.85 (2H, CH2Ph), 4.59 (s, 2H, CH2Ph), 4.57 (s, 1H, H-1′), 4.56 (m, 1H, H-5), 4.20 (dd, 1H, J = 2.1 and 11.0 Hz, H-6′′a), 4.17 (m, 2H, H-6′′b + H-6a), 4.00 (t, 1H, J = 9.0 Hz, H-4′′), 3.89 (d, 1H, J = 7.4 Hz, H-6a), 3.83 (s, 1H, H-5′), 3.80–3.71 (m, 5H, H-3 + H-3′ + H-3′′ + H-4 + H-2′), 3.56 (t, 1H, J = 8.45 Hz, H-5′′), 3.52 (s, 3H, OMe), 3.19–3.13 (m, 3H, H-2′′ + H-2 + H-4’). 13C-NMR (CDCl3): δ: 169.4(C=O), 138.1–137.5 (Ph), 128.1–127.2 (Ph), 103.2 (C-1′), 100.5 (C-1), 97.5 (C-1′′), 83.8 (C-3′), 81.4 (C-2′) 80.0 (C-4′), 79.4 (C-3′), 78.2 (C-3), 76.4 (C-4), 76.0 (C-4′′), 76.1 (CH2, Bn), 75.8 (CH2, Bn), 74.9 (C-5), 75.2 (C-5′), 73.2 (CH2, Bn), 72.5 (C-5′′), 65.1 (C-6), 63.6 (C-2′′), 62.1 (C-6′′), 61.5 (OMe), 60.5 (C-2). ESIMS: [M − H]+: 1003.3061. Calculated for C47H51N6O17S: 1003.3026.
(2-Amino-2-deoxy-4-O-methyl-6-O-sodium sulfonato-α-d-glucopyranosyl)-O-(1→4)-(2,3-di-O-benzyl-β-d-sodium glucopyranosyluronate)-O-(1→4)-1,6-anhydro-2-amino-3-O-benzyl-2-deoxy-β-d-glucopyranose (18). Compound 17 (760 mg, 0.7562 mmol) dissolved in t-BuOH–H2O (1:1) (76 mL) was stirred under H2 in the presence of 10% Pd/C catalyst (760 mg) for 60 h. Filtration through Celite and evaporation of the solvent gave 18 (450 mg, yield 100%). 1H-NMR (500 MHz, D2O) δ: 5.45 (s, 1H, H-1), 5.44 (s, 1H, H-1′′), 4.78 (d, J = 5.5 Hz, 1H, H-5), 4.62 (d, d = 8.0 Hz, 1H, H-1′), 4.29 (dd, J = 2.2 and 11.0 Hz, 1H, H-6′′a), 4.16 (dd, J = 2.2 and 11.0 Hz, 1H, H-6b), 4.18 (d, J = 7.2 Hz, 1H, H-6′′b), 3.94 (t, J = 1.5 Hz, 1H, H-3), 3.90 (dt, J = 2.1 and 10.1 Hz, 1H, H-5′′), 3.82-3.73 (m, 5H, H-6b + H-3′ + H-4 + H-4′ + H-5′), 3.68 (t, J = 9.8 Hz, 1H, H-3′′), 3.58 (s, 3H, OMe ), 3.41 (dd, J = 7.8 and 9.2 Hz, 1H, H-2′), 3.28 (t, J = 9.7 Hz, 1H, H-4′′), 2.87 (dd, J = 4.0 and 10.2 Hz, 1H, H-2′′), 2.82 (s, 1H, H-2). 13C-NMR (D2O): δ: 179.0 (COOH), 104.9 (C-1), 104.5 (C-1′), 101.2 (C-1′′), 81.4 (C-4′′), 80.7 (C-5′), 79.4 (C-4 + C-4′), 78.8 (C-3′), 77.2 (C-5), 76.0 (C-2′), 75.2 (C-3′′), 73.8 (C-3), 73.1 (C-5′′), 69.1 (C-6′′), 67.8 (C-6), 62.8 (OMe), 57.7 (H-2′′), 55.8 (H-2). ESIMS m/z: [M − H]+: 591.1395. Calculated for C19H31N2O17S: 591.1337.
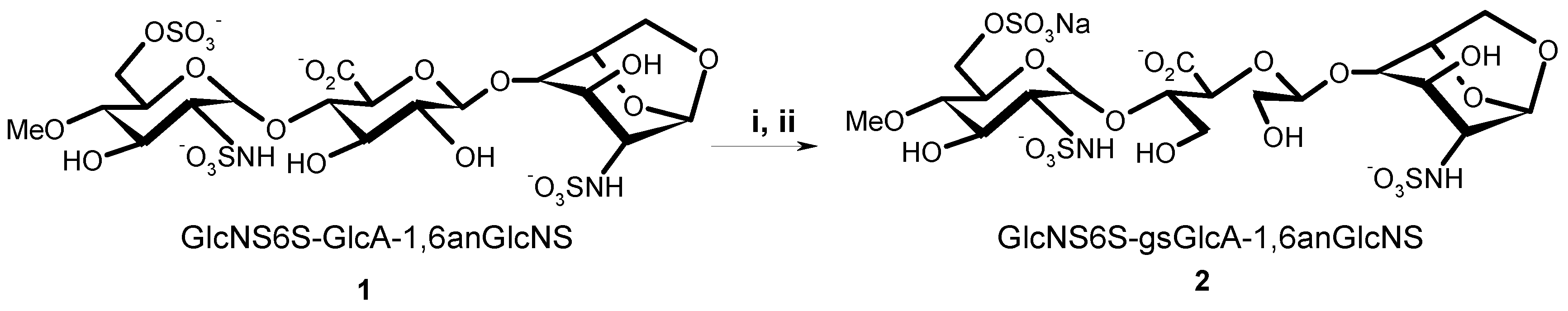
(2-Deoxy-4-O-methyl-2-sodium sulfonatamido-6-O-sodium sulfonato-α-d-glucopyranosyl-O-(1→4)-(sodium β-d-glucopyranosyluronate)-O-(1→4)-1,6-anhydro-2-deoxy-2-sodium sulfonatamido-β-d-glucopyranose (1). Aqueous sodium hydroxide (2 M) was added till pH 8.5 to a solution of 15 (100 mg, 0.169 mmol) in H2O (15 mL). Sulphur trioxide-pyridine complex (970 mg, 6.08 mmol) was then added at RT in five portions in a 1.5 h interval while the pH maintained to 8–9 by addition of 2 M NaOH. After stirring at RT overnight the solution was concentrated to 7 mL and layered on top of a Sephadex G10 column (2.5 cm × 80 cm) eluted by aqueous 0.2 M NaCl. The fractions were collected and concentrated. Compound 1 (81 mg, 57%) was obtained after desalting on Sephadex G 10 and lyophilisation. 1H-NMR (500 MHz, D2O) δ: 5.63 (s, 1H, H-1), 5.61 (d, J = 3.75 Hz, 1H, H-1′′), 4.78 (d, J = 5.4 Hz, 1H, H-5), 4.62 (d, J = 7.9 Hz, 1H, H-1′), 4.30 (dd, J = 2.1 and 11.0 Hz, 1H, H-6′′a), 4.18 (m, 1H, H-6′′b), 4.18 (d, J = 11.0, 1H, H-6a), 3.95 (H-3), 3.88 (H-5′′), 3.86 (H-3′), 3.83 (H-5′), 3.82 (H-4), 3.78 (H-4′), 3.80 (m, 1H, H-6b), 3.67 (t, J = 9.7 Hz, 1H, H-3′′), 3.58 (s, 3H, OMe), 3.44 (t, J = 9.7 Hz, 1H, H-2’), 3.36 (t, J = 7.9 Hz, 1H, H-4′′), 3.28 (dd, J = 3.8 and 10.5 Hz, 1H, H-2′′), 3.21 (s, 1H, H-2). 13C-NMR (D2O): 178.6 (COOH), 104.5 (C-1), 103.0 (C-1′), 100.4 (C-1′′), 81.5 (C-4′′), 79.8 (C-3′), 79.6 (C-4′), 79.3 (C-4), 79.1 (C-5′), 76.4 (C-5), 75.4 (C-2′), 73.8 (C-3′′), 73.2 (C-3), 71.5 (C-5′′), 69.2 (C-6′′), 68.1 (C-6), 68.2 (OMe), 60.9 (C-2′′), 58.6 (C-2). LC-MS: [M – H]+: 751.0561. Calculated for C19H31N2O23S3: 751.0474.
(2-Deoxy-4-O-methyl-2-sodium sulfonatamido-6-O-sodium sulfonato-α-d-glucopyranosyl)-O-(1→4)-(glycol-split sodium β-d-glucopyranosyluronate)-O-(1→4)-1,6-anhydro-2-deoxy-2-sodium_sulfonatamido-β-d-glucopyranose. Or: sodium(2S,3S)-4-hydroxy-2-((R)-2-hydroxy-1-(((1R,2S,3R,4R,5R)-3-hydroxy-4-(sulfonatoamino)-6,8-dioxabicyclo[3.2.1]octan-2-yl)oxy)ethoxy)-3-(((2R,3R,4R,5S,6R)-4-hydroxy-5-methoxy-3-(sulfonatoamino)-6-((sulfonatooxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)butanoate (2). An aqueous solution of sodium metaperiodate (0.2 M, 2.1 mL) was added at RT to a solution of 1 (55 mg, 0.0731 mmol) in H2O (2.1 mL). After 5 h, ethylene glycol (0.24 mL, 4.4 mmol) was added and the solution was stirred at RT for 1 h (N.B.: light must be avoided during periodate oxidation). Sodium borohydride (100 mg, 2.633 mmol) was then added in portions at 0 °C. After 16 h, HOAc (0.175 mL) was added at 0 °C to pH 7. After concentration the salt were removed using a Sephadex G10 column eluted by 10% EtOH–H2O. Lyophilisation gave 2 (35.2 mg, 70.4%). 1H-NMR (500 MHz, D2O) δ: 5.59 (s, 1H, H-1), 5.34 (d, J = 3.65 Hz, 1H, H-1′′), 4.86 (t, J = 4.59 Hz, 1H, H-1′), 4.69 (d, J = 5.45 Hz, 1H, H-5), 4.29 (dd, J = 6.0 and 11.0 Hz, 1H, H-6′′a), 4.21 (m, 1H, H-6′′b), 4.16 (d, J = 6.0 Hz, 1H, H-4), 4.15 (m, 1H, H-6a), 3.97 (d, J = 8.0 Hz, 1H, H-5′′), 3.94 (m, 1H, H-4′), 3.94 (m, 1H, H-3), 3.92 (m, 1H, H-3′a), 3.90 (m, 1H, H-5′), 3.73 (m, 1H, H-6b), 3.80 (m, 1H, H-3′b), 3.68 (m, 1H, 1H, H-3′′), 3.66 (m, 2H, H-2′a, H2′b), 3.58 (s, 3H, OMe), 3.35 (t, J = 9.75 Hz, 1H, H-4′′), 3.29 (dd, J = 3.65 and 10.45 Hz, 1H, H-2′′), 3.20 (s, 1H, H-2). 13C-NMR (D2O) δ: 179.58 (COOH), 104.2 (C-1), 103.9 (C-1′), 98.4 (C-1′′), 81.8 (C-4′′), 80.8 (C-4′), 80.5 (C-4), 77.8 (C-5′), 76. 5 (C-5), 73.8 (C-3′′), 73.4 (C-3), 71.8 (C-5′′), 69.2 (C-6′′), 67.9 (C-6), 64.6 (C-2′), 62.6, (OCH3), 61.8 (C-3′), 60.8 (C-2′′), 58.3 (C-2). LC-MS: [M − H]+: 753.0884. Calculated for C19H31N2O23S3: 753.0708.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






