
Excited-State Dynamics of Melamine and Its Lysine Derivative Investigated by Femtosecond Transient Absorption Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
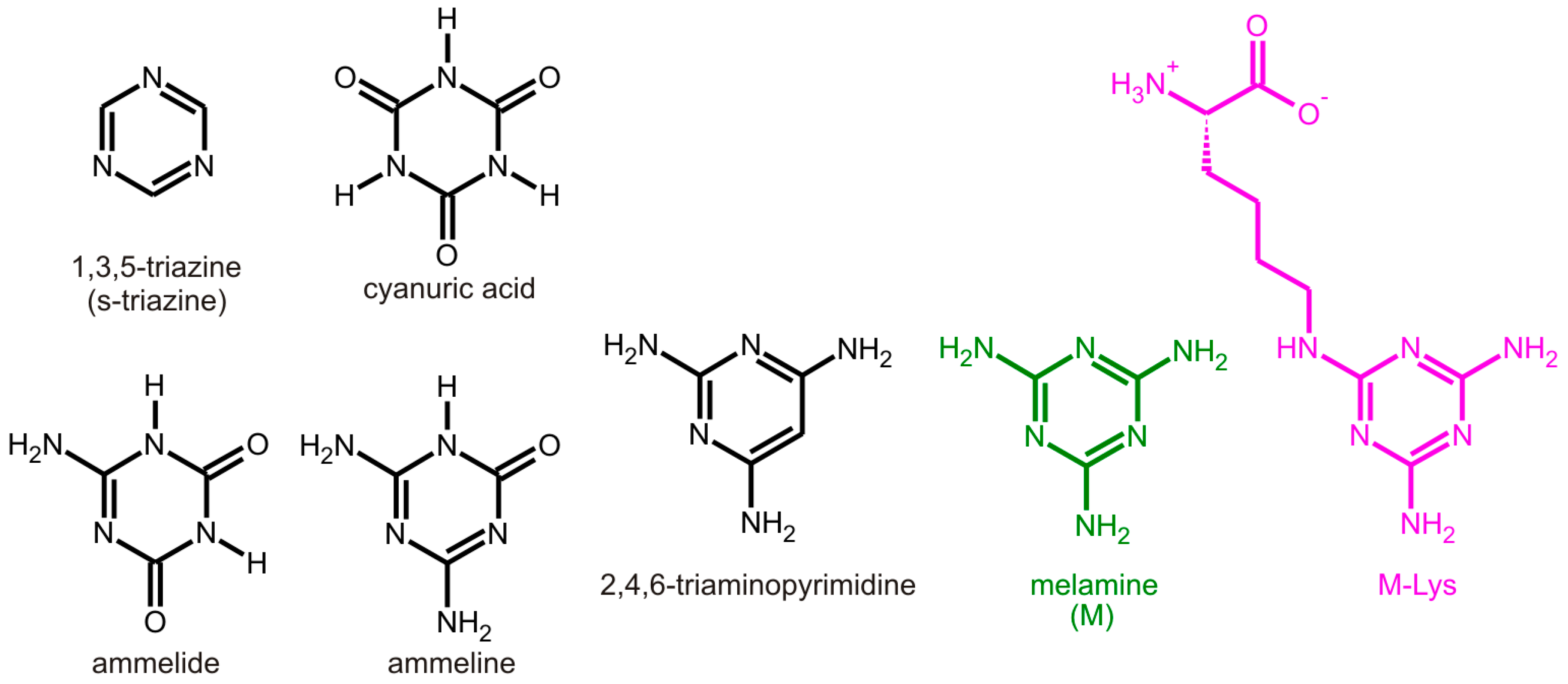
:1. Introduction
2. Results and Discussion
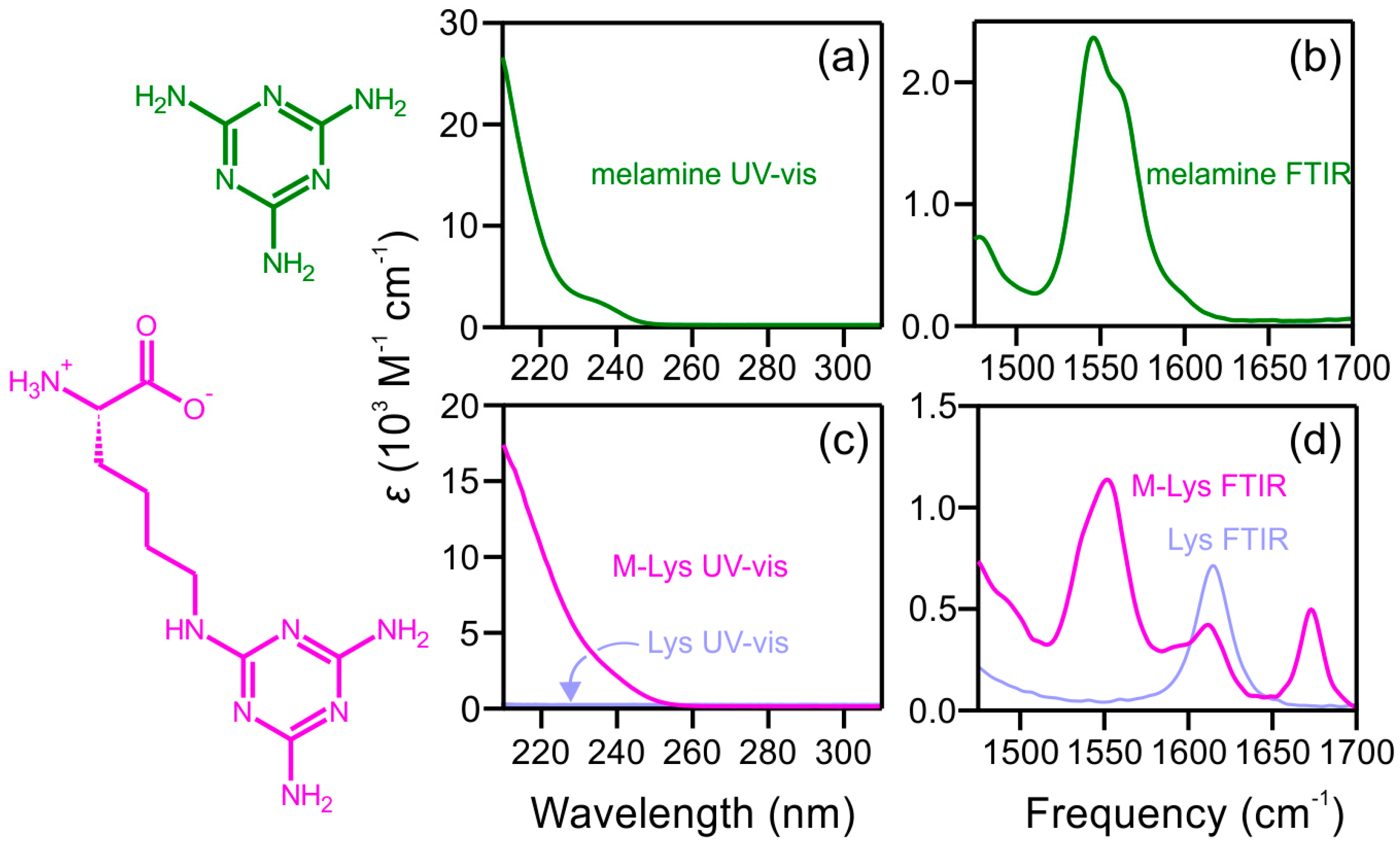
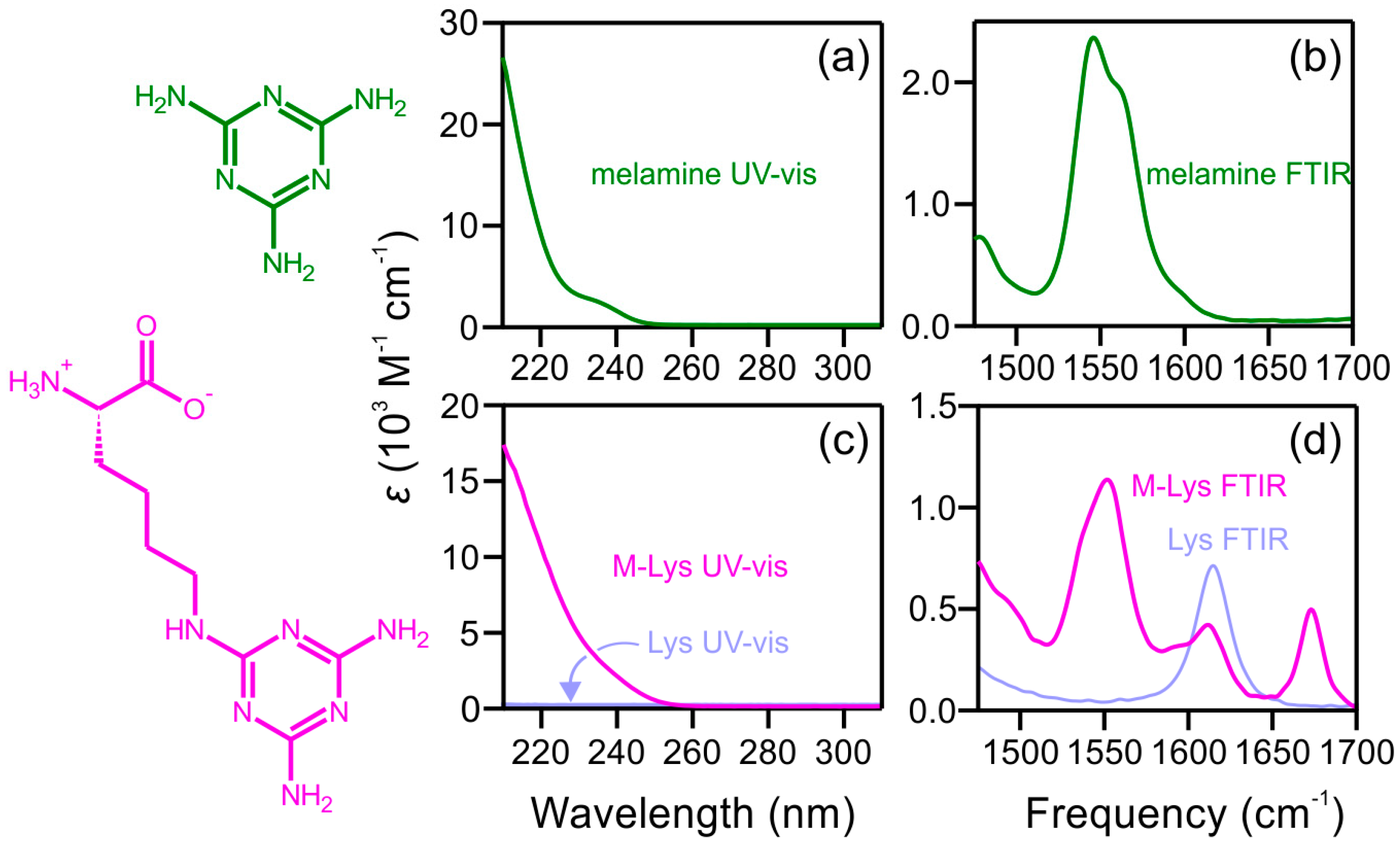
2.1. Steady-State Spectra
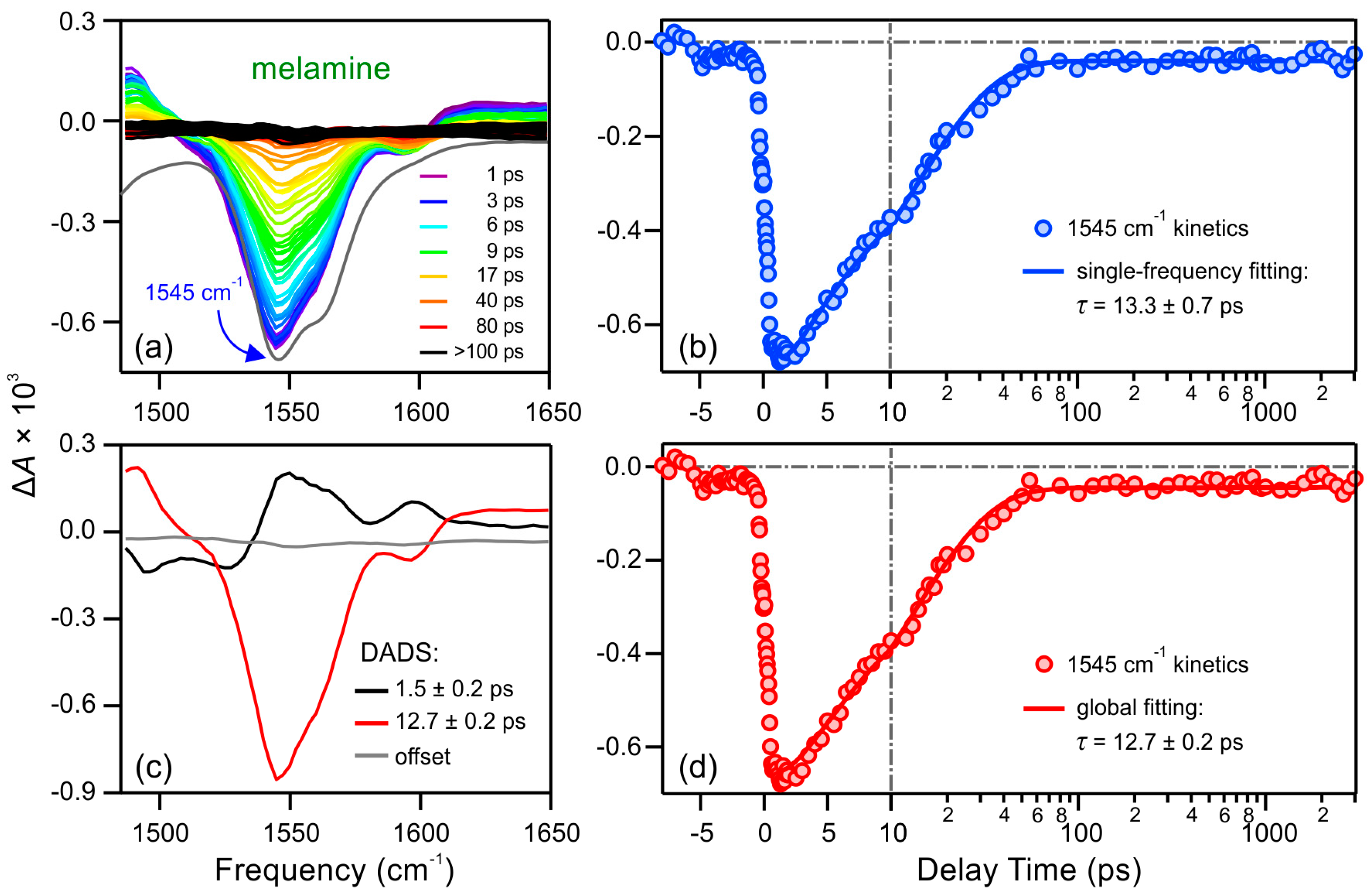
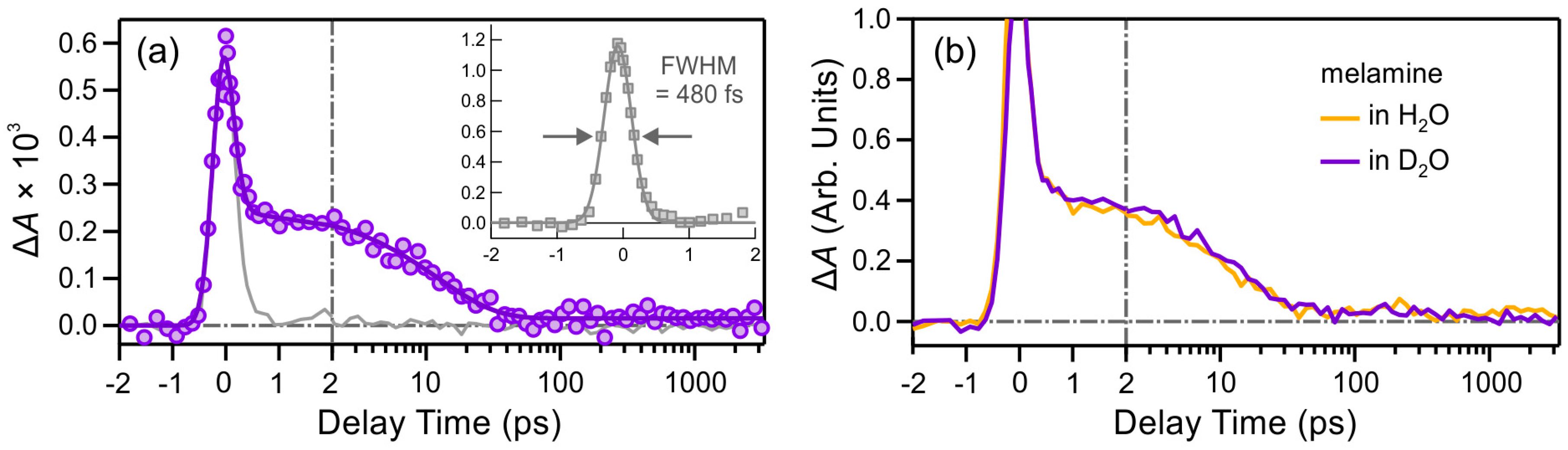
2.2. Excited-State Dynamics
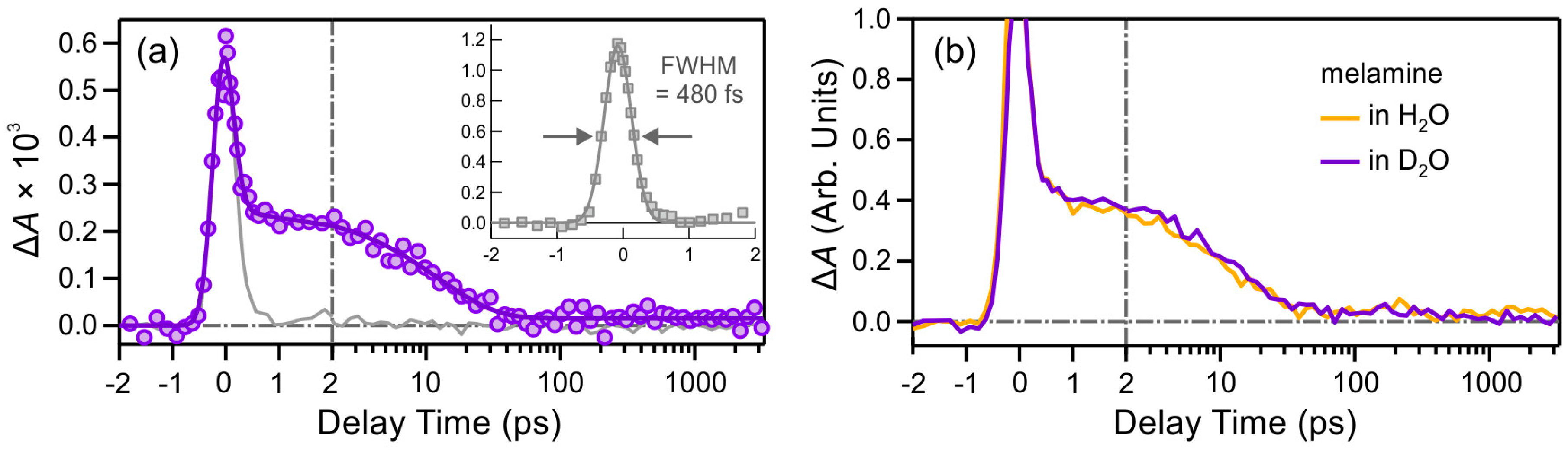
2.2.1. Melamine (M)
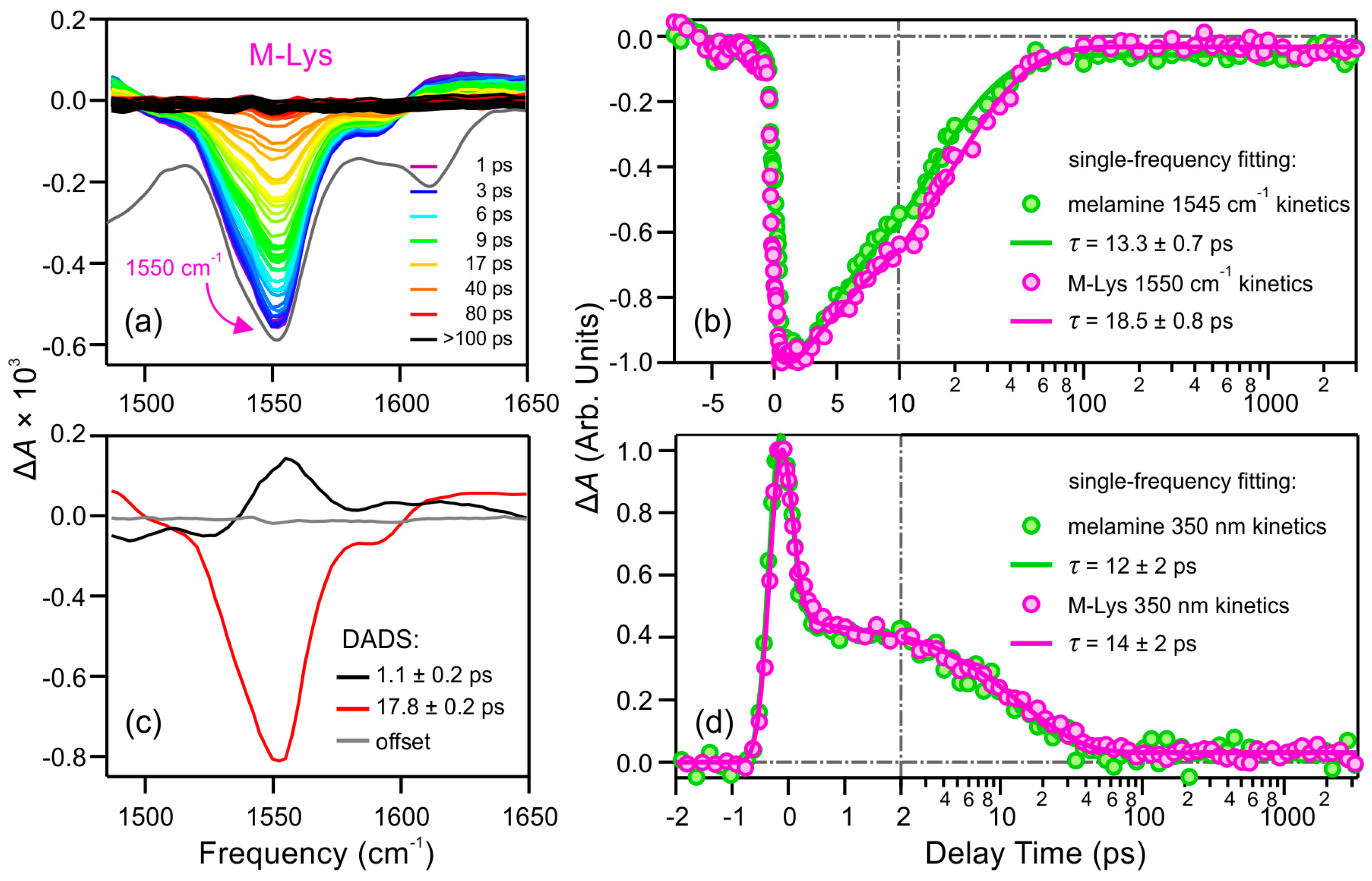
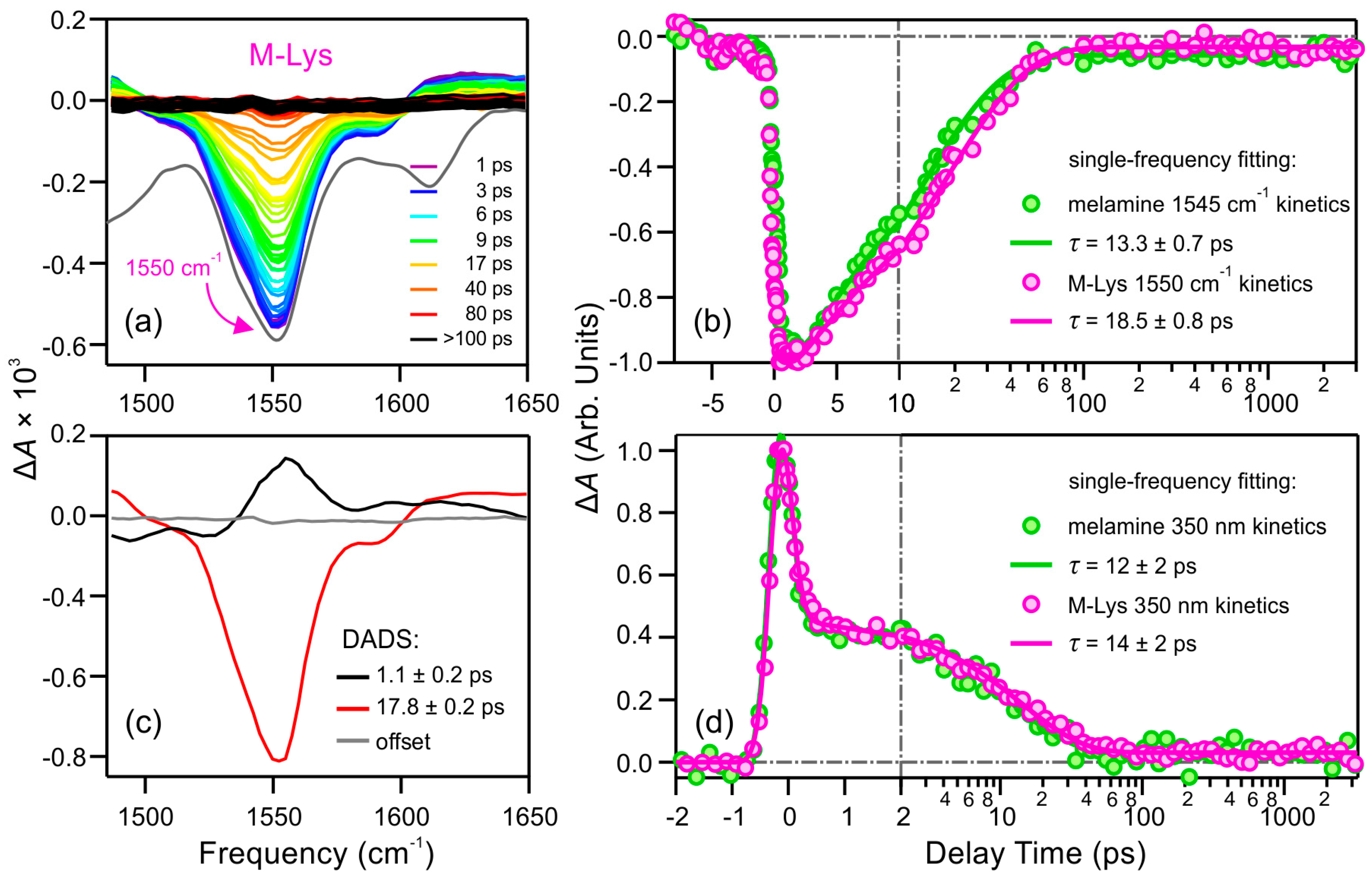
2.2.2. Lysine Derivative of Melamine (M-Lys)
3. Materials and Methods
3.1. TRIR Spectroscopy
3.2. UV-Pump/UV-Probe Transient Absorption (TA) Spectroscopy
3.3. Synthesis of the Lysine Derivative of Melamine
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Menor-Salván, C.; Ruiz-Bermejo, D.M.; Guzmán, M.I.; Osuna-Esteban, S.; Veintemillas-Verdaguer, S. Synthesis of pyrimidines and triazines in ice: Implications for the prebiotic chemistry of nucleobases. Chem. Eur. J. 2009, 15, 4411–4418. [Google Scholar] [CrossRef] [PubMed]
- Seto, C.T.; Whitesides, G.M. Self-assembly based on the cyanuric acid melamine lattice. J. Am. Chem. Soc. 1990, 112, 6409–6411. [Google Scholar] [CrossRef]
- Mittapalli, G.K.; Reddy, K.R.; Xiong, H.; Munoz, O.; Han, B.; de Riccardis, F.; Krishnamurthy, R.; Eschenmoser, A. Mapping the landscape of potentially primordial informational oligomers: Oligodipeptides and oligodipeptoids tagged with triazines as recognition elements. Angew. Chem. Int. Ed. 2007, 46, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Pratumyot, Y.; Piao, X.; Bong, D. Discrete assembly of synthetic peptide-DNA triplex structures from polyvalent melamine-thymine bifacial recognition. J. Am. Chem. Soc. 2012, 134, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.; Xia, X.; Bong, D. Bifacial peptide nucleic acid directs cooperative folding and assembly of binary, ternary, and quaternary DNA complexes. Biochemistry 2013, 52, 6313–6323. [Google Scholar] [CrossRef] [PubMed]
- Hysell, M.; Siegel, J.S.; Tor, Y. Synthesis and stability of exocyclic triazine nucleosides. Org. Biomol. Chem. 2005, 3, 2946–2952. [Google Scholar] [CrossRef] [PubMed]
- Beckstead, A.A.; Zhang, Y.; De Vries, M.S.; Kohler, B. Life in the light: Nucleic acid photoproperties as a legacy of chemical evolution. Phys. Chem. Chem. Phys. 2016, 18, 24228–24238. [Google Scholar] [CrossRef] [PubMed]
- Cockell, C.S.; Horneck, G. The history of the UV radiation climate of the earth—Theoretical and space-based observations. Photochem. Photobiol. 2001, 73, 447–451. [Google Scholar] [CrossRef]
- Cnossen, I.; Sanz-Forcada, J.; Favata, F.; Witasse, O.; Zegers, T.; Arnold, N.F. Habitat of early life: Solar X-ray and UV radiation at Earth’s surface 4–3.5 billion years ago. J. Geophys. Res. Planets 2007, 112, 1–19. [Google Scholar] [CrossRef]
- Pecourt, J.M.L.; Peon, J.; Kohler, B. Ultrafast internal conversion of electronically excited RNA and DNA nucleosides in water. J. Am. Chem. Soc. 2000, 122, 9348–9349. [Google Scholar] [CrossRef]
- Pecourt, J.M.L.; Peon, J.; Kohler, B. DNA excited-state dynamics: Ultrafast internal conversion and vibrational cooling in a series of nucleosides. J. Am. Chem. Soc. 2001, 123, 10370–10378. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 2004, 104, 1977–2019. [Google Scholar] [CrossRef] [PubMed]
- Brister, M.M.; Pollum, M.; Crespo-Hernández, C.E. Photochemical etiology of promising ancestors of the RNA nucleobases. Phys. Chem. Chem. Phys. 2016, 18, 20097–20103. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, G.K.; Osornio, Y.M.; Guerrero, M.A.; Reddy, K.R.; Krishnamurthy, R.; Eschenmoser, A. Mapping the landscape of potentially primordial informational oligomers: Oligodipeptides tagged with 2,4-disubstituted 5-aminopyrimidines as recognition elements. Angew. Chem. Int. Ed. 2007, 46, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.K.; Woodberry, N.T.; Costa, G.W. The dissociation constants of melamine and certain of its compounds. J. Am. Chem. Soc. 1947, 69, 599–603. [Google Scholar] [CrossRef]
- Jang, Y.H.; Hwang, S.; Chang, S.B.; Ku, J.; Chung, D.S. Acid dissociation constants of melamine derivatives from density functional theory calculations. J. Phys. Chem. A 2009, 113, 13036–13040. [Google Scholar] [CrossRef] [PubMed]
- Kolb, V.M.; Dworkin, J.P.; Miller, S.L. Alternative bases in the RNA world—The prebiotic synthesis of urazole and its ribosides. J. Mol. Evol. 1994, 38, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hirt, R.C.; Salley, D.J. Ultraviolet absorption spectra of derivatives of symmetric triazine. I. Amino triazines. J. Chem. Phys. 1953, 21, 1181–1184. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Paoloni, L. The electronic structure of melamine. Trans. Faraday Soc. 1957, 53, 261–271. [Google Scholar] [CrossRef]
- Costa, G.W.; Hirt, R.C.; Salley, D.J. Near ultraviolet absorption spectra of melamine and some related compounds. J. Chem. Phys. 1950, 18, 434–437. [Google Scholar] [CrossRef]
- Oliva, J.M.; Azenha, E.M.D.G.; Burrows, H.D.; Coimbra, R.; de Melo, J.S.S.; Canle, M.L.; Fernández, M.I.; Santaballa, J.A.; Serrano-Andrés, L. On the low-lying excited states of sym-triazine-based herbicides. ChemPhysChem 2005, 6, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Mozhayskiy, V.A.; Krylov, A.I. Jahn-Teller distortions in the electronically excited states of sym-triazine. Mol. Phys. 2009, 107, 929–938. [Google Scholar] [CrossRef]
- Wang, Y.L.; Mebel, A.M.; Wu, C.J.; Chen, Y.T.; Lin, C.E.; Jiang, J.C. IR spectroscopy and theoretical vibrational calculation of the melamine molecule. J. Chem. Soc. Faraday Trans. 1997, 93, 3445–3451. [Google Scholar] [CrossRef]
- Sawodny, W.; Niedenzu, K.; Dawson, J.W. Vibrational spectrum and assignment of normal vibrations of melamine. J. Chem. Phys. 1966, 45, 3155–3156. [Google Scholar] [CrossRef]
- Liquier, J.; Taillandier, E. Infrared Spectroscopy of Nucleic Acids; Wiley-Liss: New York, NY, USA, 1996. [Google Scholar]
- Banyay, M.; Sarkar, M.; Gräslund, A. A library of IR bands of nucleic acids in solution. Biophys. Chem. 2003, 104, 477–488. [Google Scholar] [CrossRef]
- Schreier, W.J.; Schrader, T.E.; Koller, F.O.; Gilch, P.; Crespo-Hernández, C.E.; Swaminathan, V.N.; Carell, T.; Zinth, W.; Kohler, B. Thymine dimerization in DNA is an ultrafast photoreaction. Science 2007, 315, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Kuimova, M.K.; Dyer, J.; George, M.W.; Grills, D.C.; Kelly, J.M.; Matousek, P.; Parker, A.W.; Sun, X.Z.; Towrie, M.; Whelan, A.M. Monitoring the effect of ultrafast deactivation of the electronic excited states of DNA bases and polynucleotides following 267 nm laser excitation using picosecond time-resolved infrared spectroscopy. Chem. Commun. 2005, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, J.; Kohler, B. Hydrogen bond donors accelerate vibrational cooling of hot purine derivatives in heavy water. J. Phys. Chem. A 2013, 117, 6771–6780. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Kohler, B. Influence of secondary structure on electronic energy relaxation in adenine homopolymers. J. Phys. Chem. B 2004, 108, 11182–11188. [Google Scholar] [CrossRef]
- Zhang, Y.; Dood, J.; Beckstead, A.; Chen, J.; Li, X.-B.; Burrows, C.J.; Lu, Z.; Matsika, S.; Kohler, B. Ultrafast excited-state dynamics and vibrational cooling of 8-oxo-7,8-dihydro-2′-deoxyguanosine in D2O. J. Phys. Chem. A 2013, 117, 12851–12857. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.M.; Marroux, H.J.B.; Grubb, M.P.; Ashfold, M.N.R.; Orr-Ewing, A.J. On the participation of photoinduced N-H bond fission in aqueous adenine at 266 and 220 nm: A combined ultrafast transient electronic and vibrational absorption spectroscopy study. J. Phys. Chem. A 2014, 118, 11211–11225. [Google Scholar] [CrossRef] [PubMed]
- Hare, P.M.; Crespo-Hernández, C.E.; Kohler, B. Internal conversion to the electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution. Proc. Natl. Acad. Sci. USA 2007, 104, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Liu, Y.; Li, L.; Zhang, F. Studying on the steady-state and time-resolved fluorescence characteristics of melamine. Spectrochim. Acta Part A 2010, 75, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- West, B.A.; Womick, J.M.; Moran, A.M. Interplay between vibrational energy transfer and excited state deactivation in DNA components. J. Phys. Chem. A 2013, 117, 5865–5874. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, B.D.; Switzer, C. An alternative nucleobase code: Characterization of purine-purine DNA double helices bearing guanine-isoguanine and diaminopurine 7-deaza-xanthine base pairs. ChemBioChem 2008, 9, 2779–2783. [Google Scholar] [CrossRef] [PubMed]
- Röttger, K.; Siewertsen, R.; Temps, F. Ultrafast electronic deactivation dynamics of the rare natural nucleobase hypoxanthine. Chem. Phys. Lett. 2012, 536, 140–146. [Google Scholar] [CrossRef]
- Chen, J.; Thazhathveetil, A.K.; Lewis, F.D.; Kohler, B. Ultrafast excited-state dynamics in hexaethyleneglycol-linked DNA homoduplexes made of A·T base pairs. J. Am. Chem. Soc. 2013, 135, 10290–10293. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Beckstead, A.A.; Hu, Y.; Piao, X.; Bong, D.; Kohler, B. Excited-State Dynamics of Melamine and Its Lysine Derivative Investigated by Femtosecond Transient Absorption Spectroscopy. Molecules 2016, 21, 1645. https://doi.org/10.3390/molecules21121645
Zhang Y, Beckstead AA, Hu Y, Piao X, Bong D, Kohler B. Excited-State Dynamics of Melamine and Its Lysine Derivative Investigated by Femtosecond Transient Absorption Spectroscopy. Molecules. 2016; 21(12):1645. https://doi.org/10.3390/molecules21121645
Chicago/Turabian StyleZhang, Yuyuan, Ashley A. Beckstead, Yuesong Hu, Xijun Piao, Dennis Bong, and Bern Kohler. 2016. "Excited-State Dynamics of Melamine and Its Lysine Derivative Investigated by Femtosecond Transient Absorption Spectroscopy" Molecules 21, no. 12: 1645. https://doi.org/10.3390/molecules21121645