
Assessment of the Potential Energy Hypersurfaces in Thymine within Multiconfigurational Theory: CASSCF vs. CASPT2


Abstract
:1. Introduction
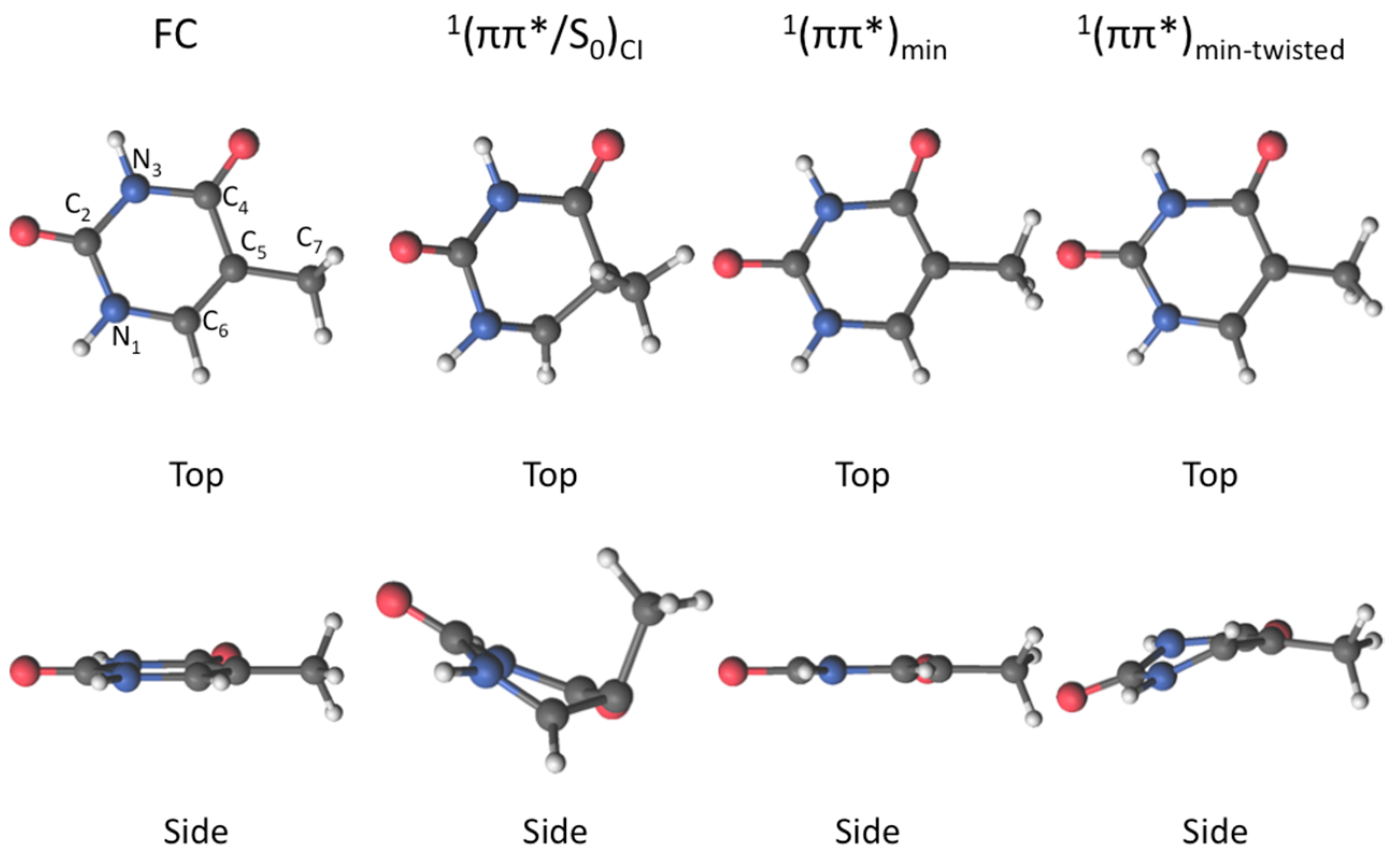
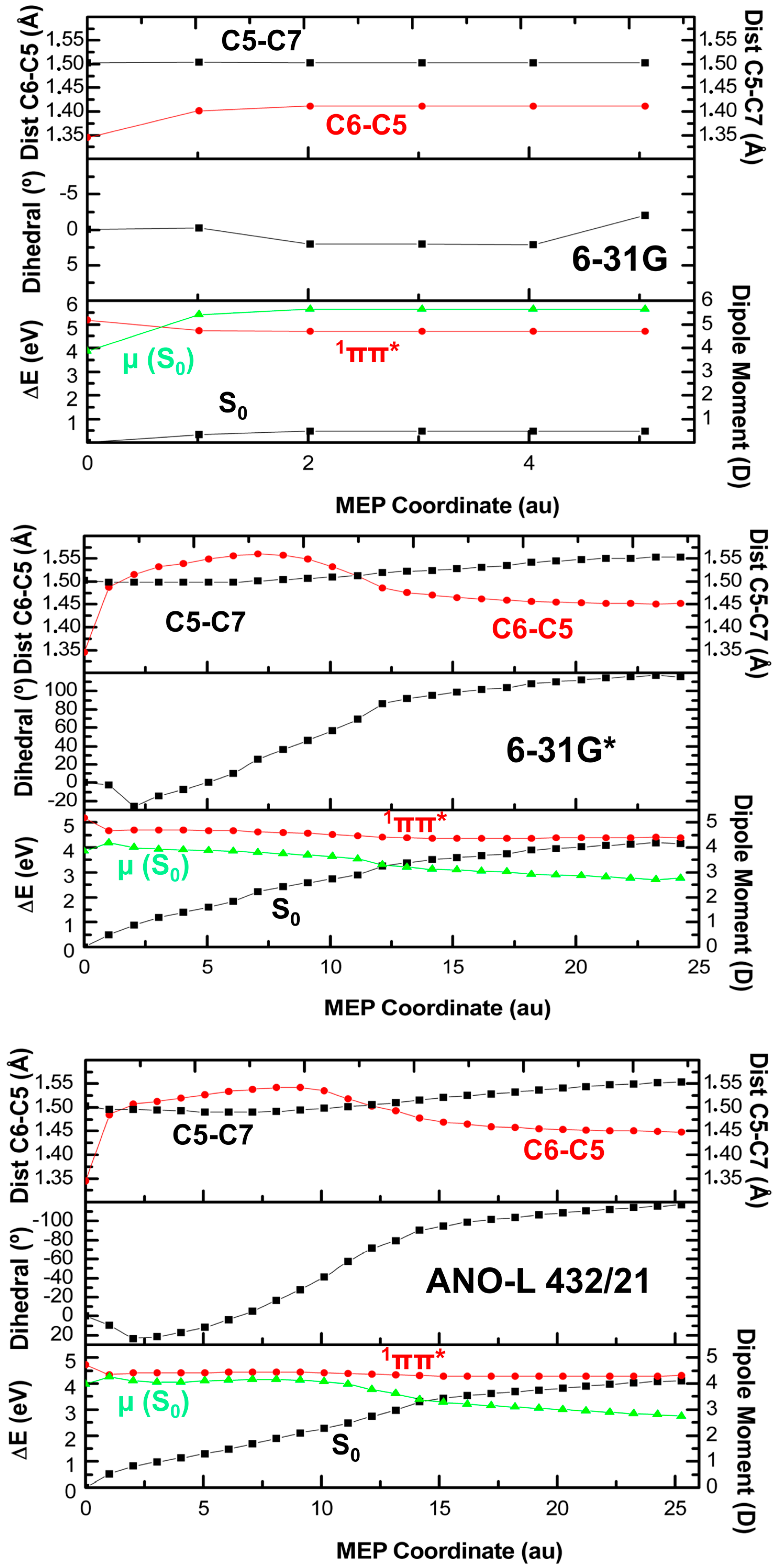
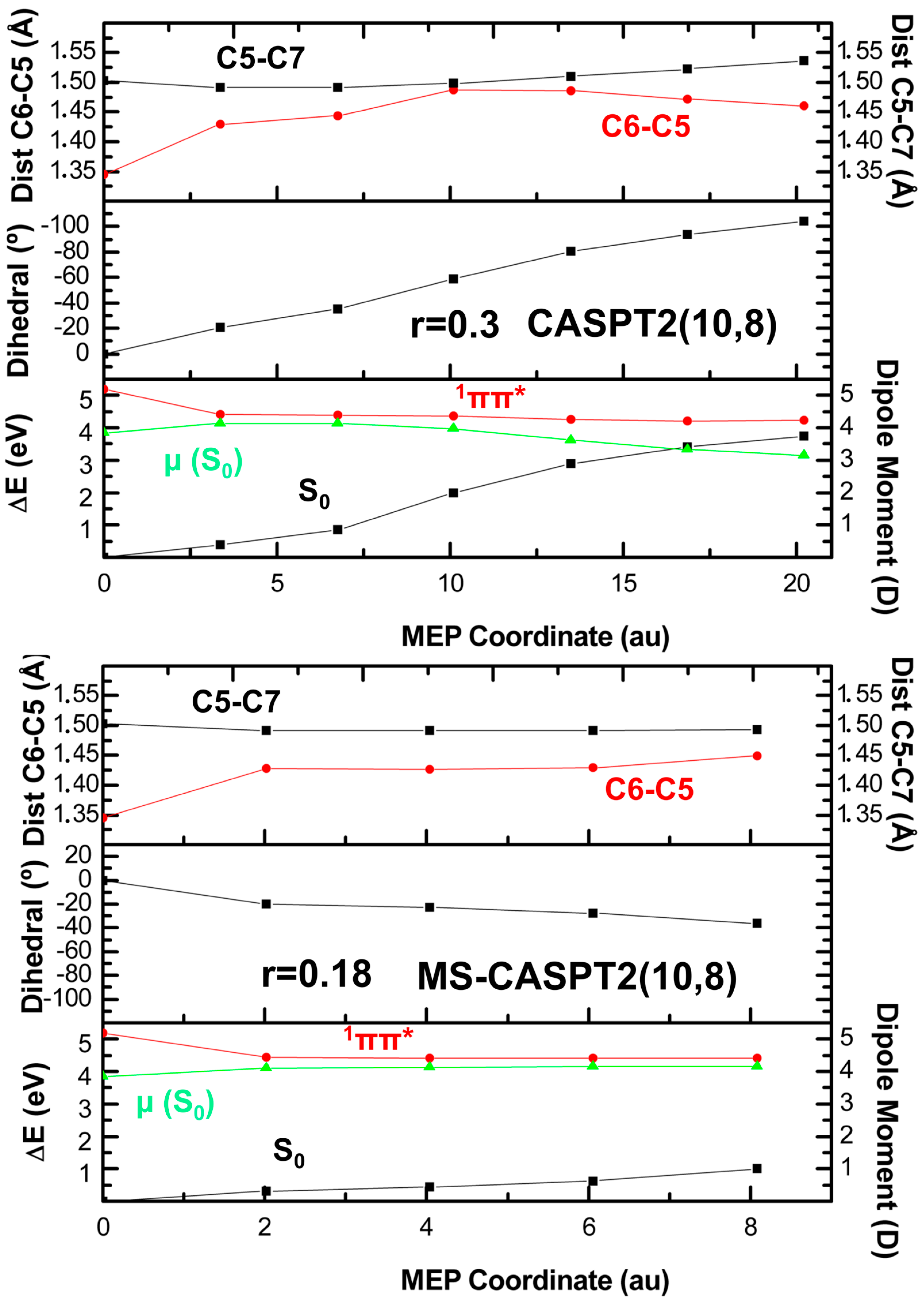
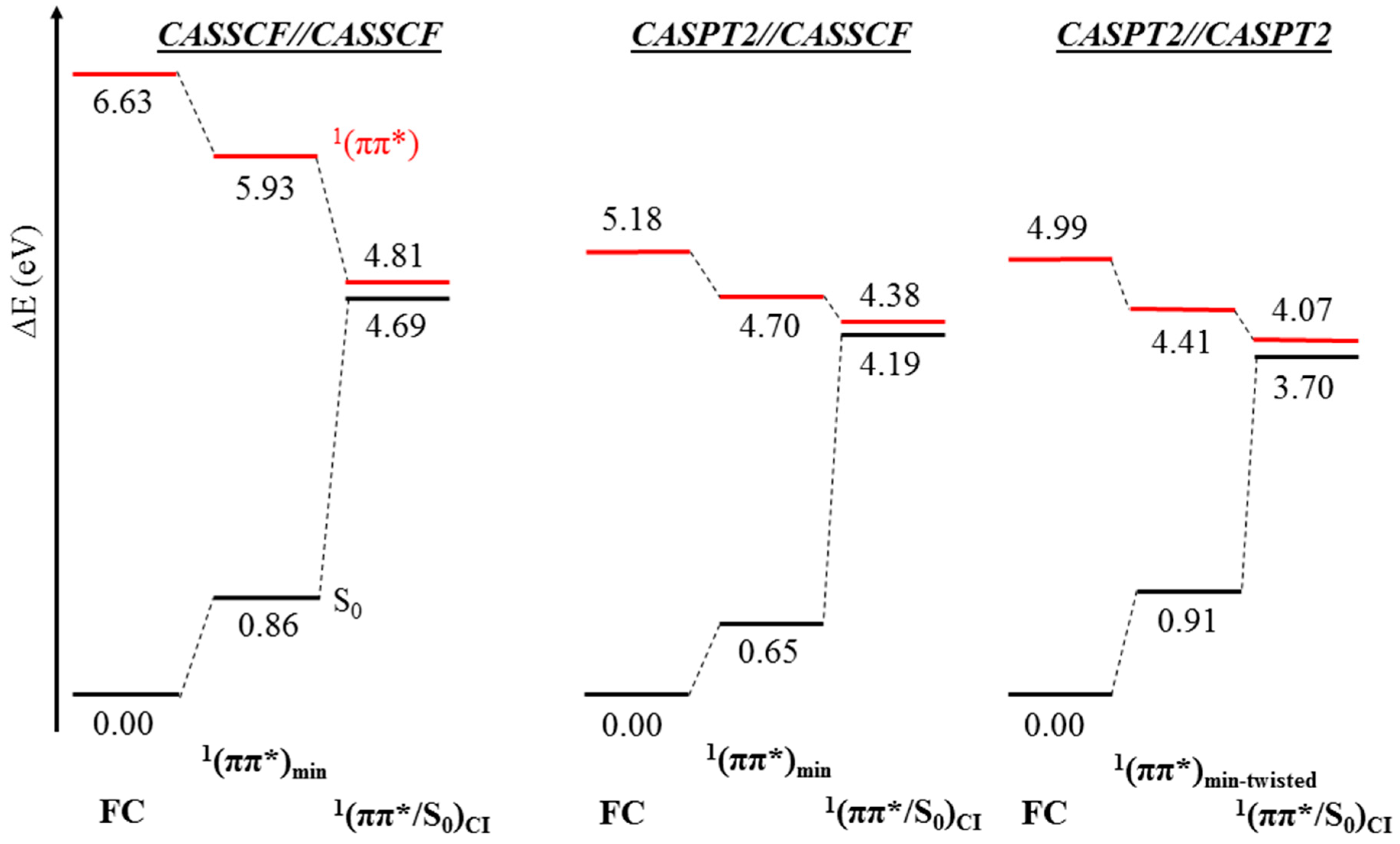
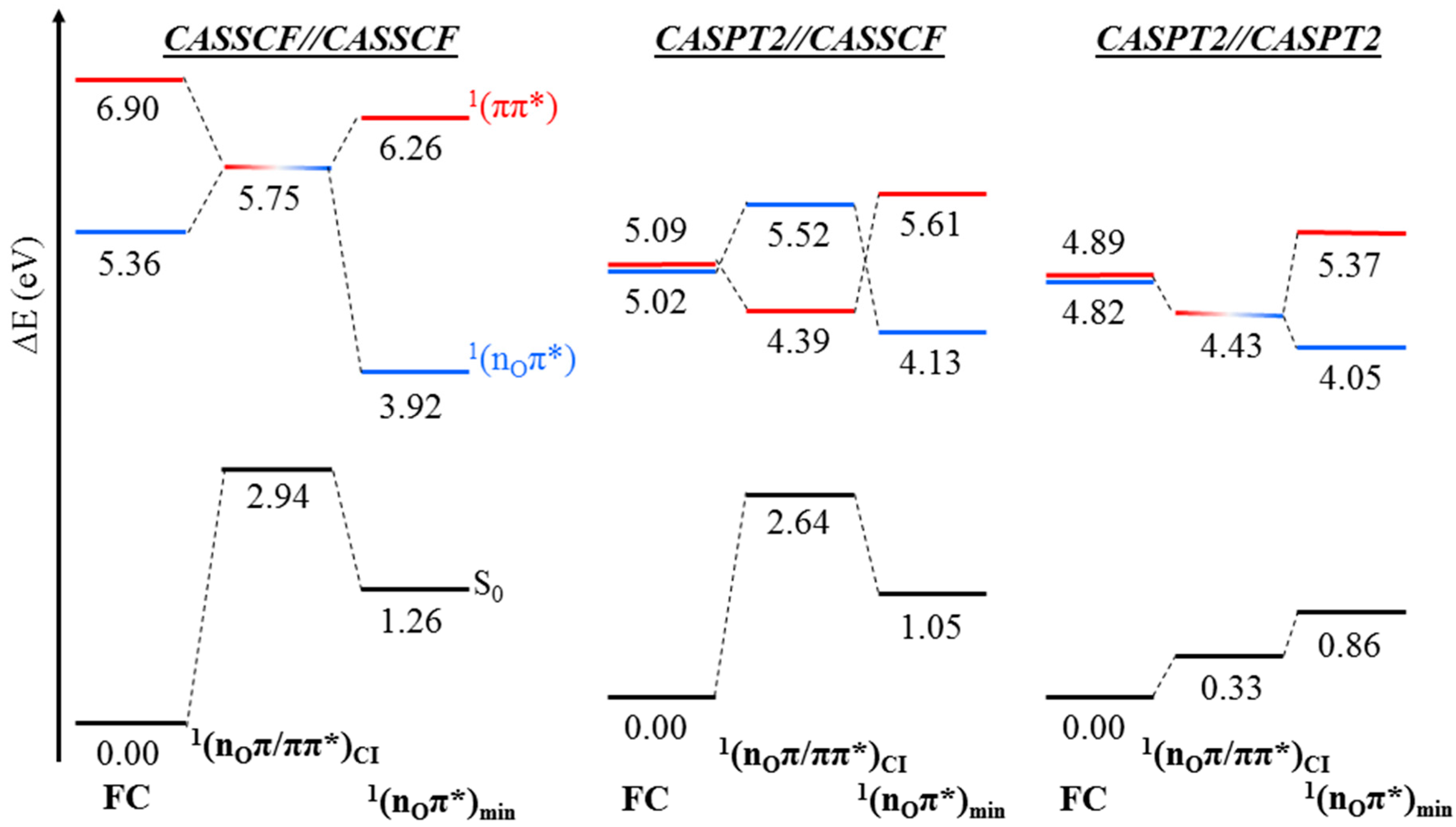
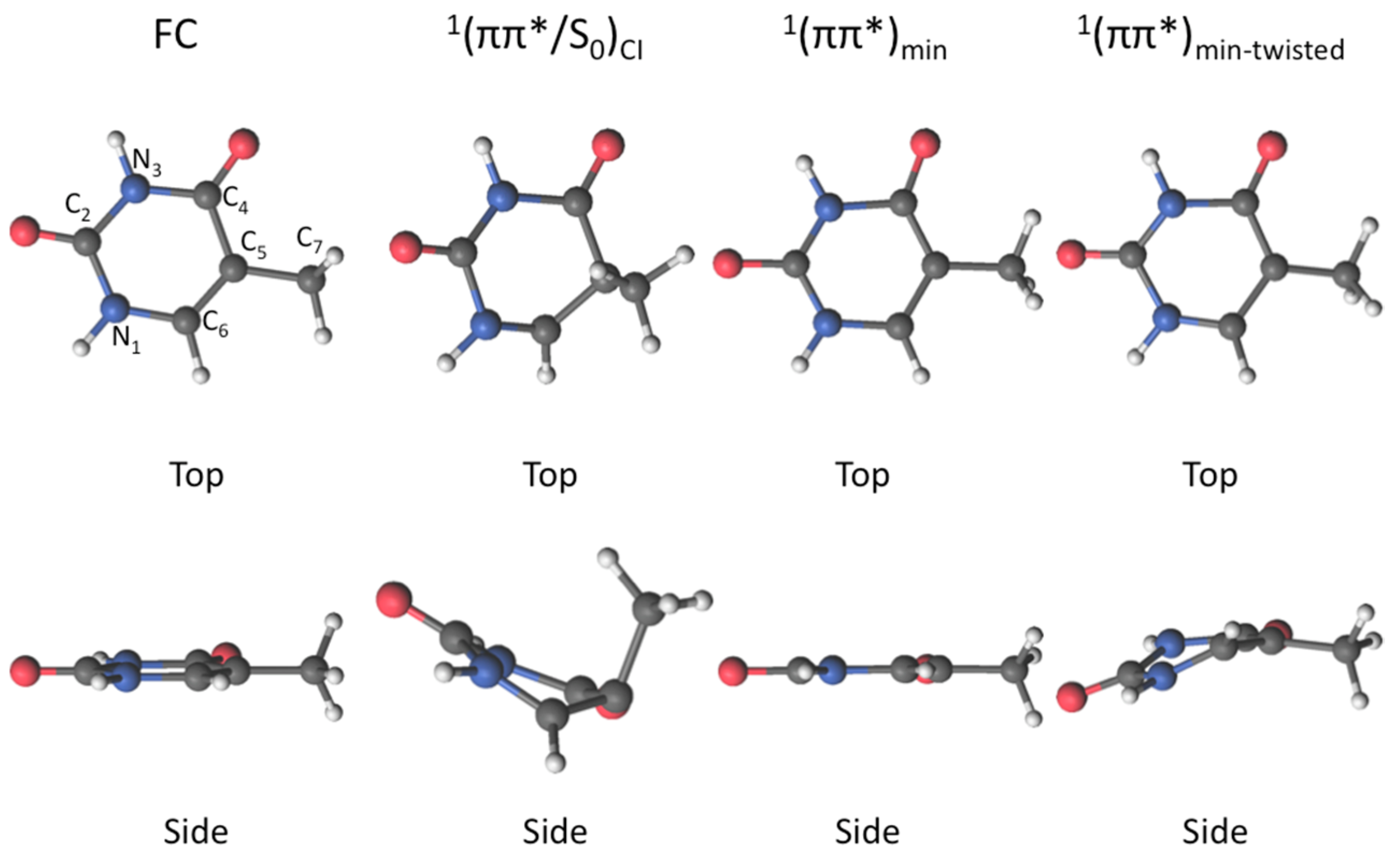
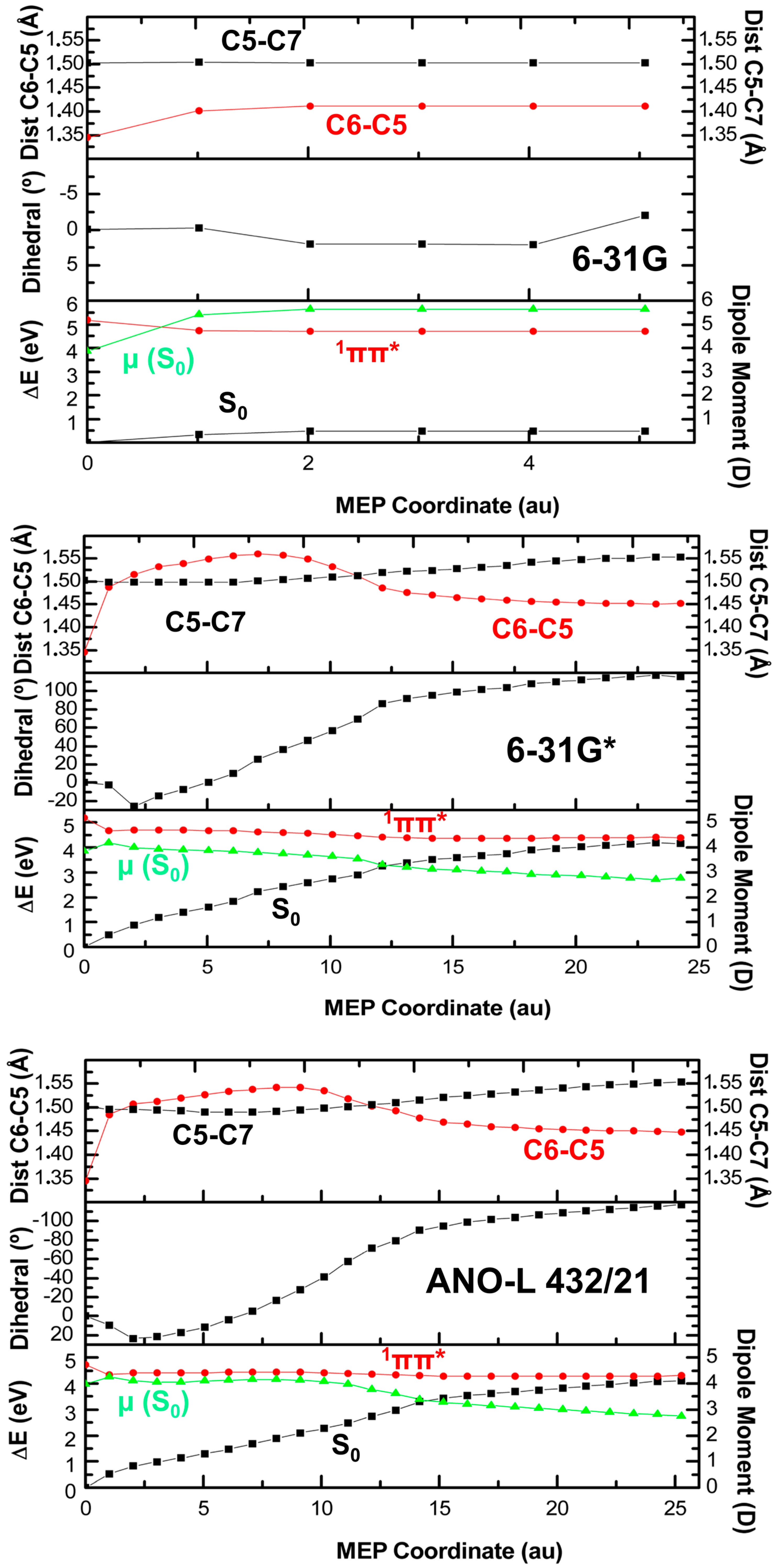
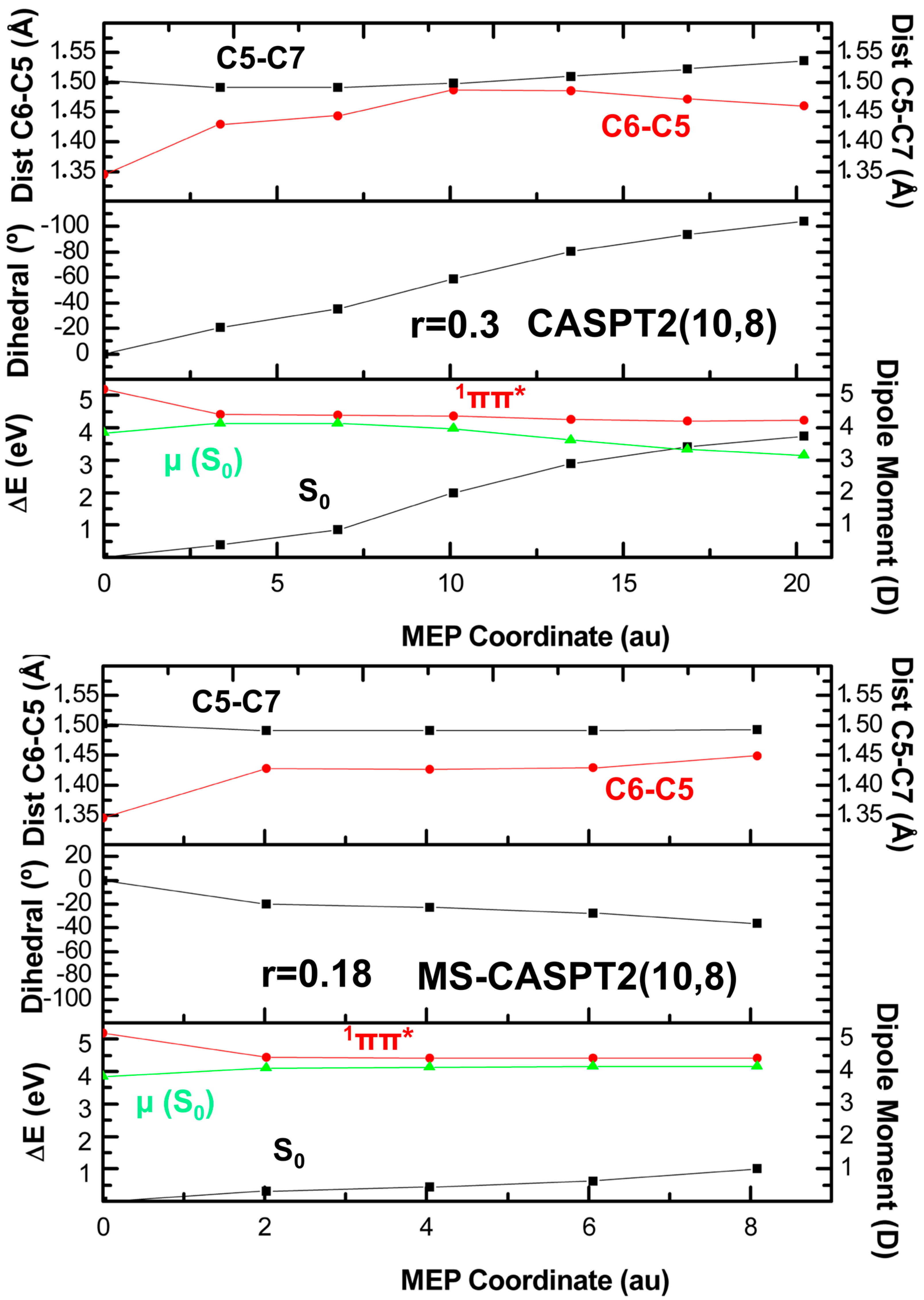
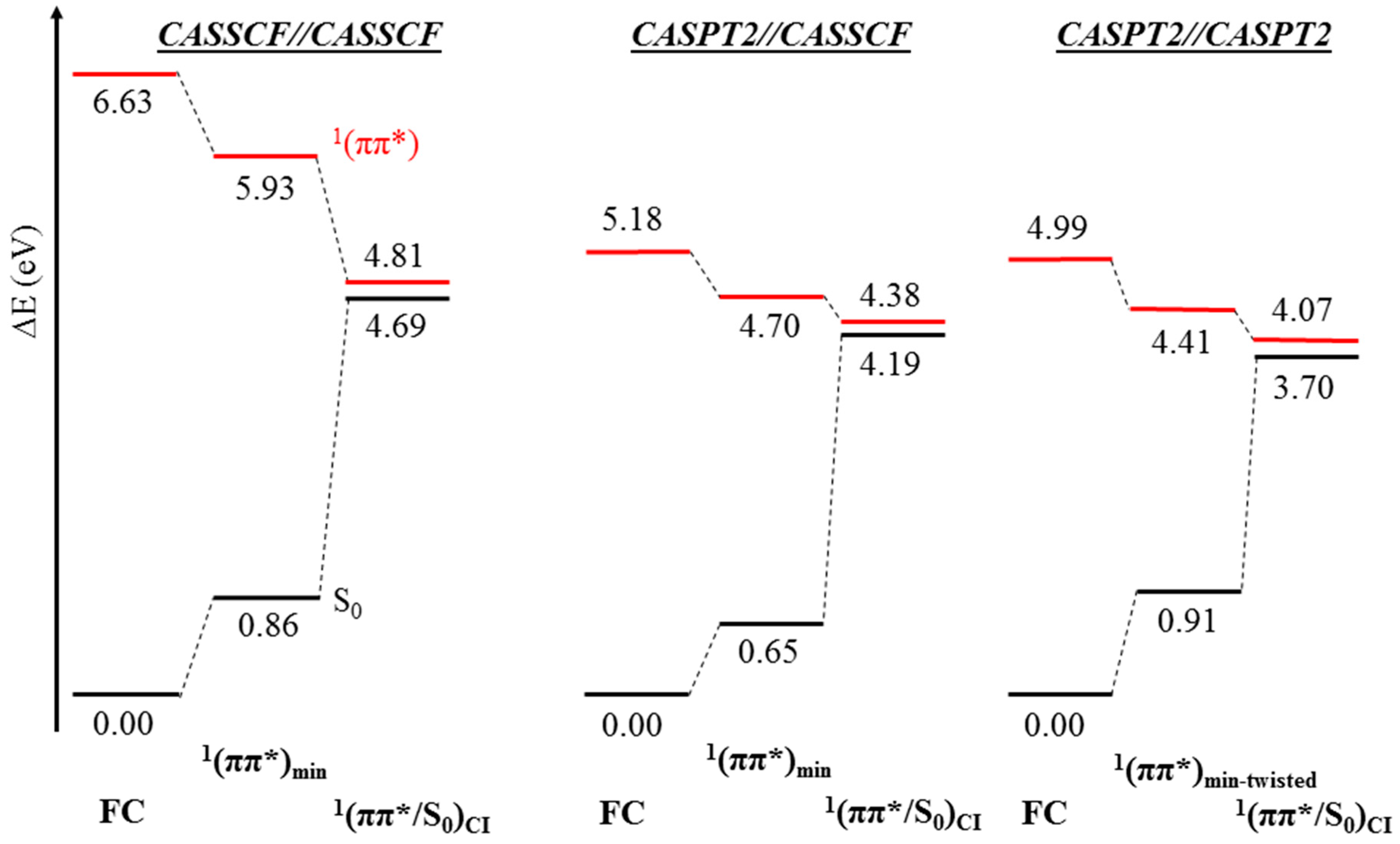
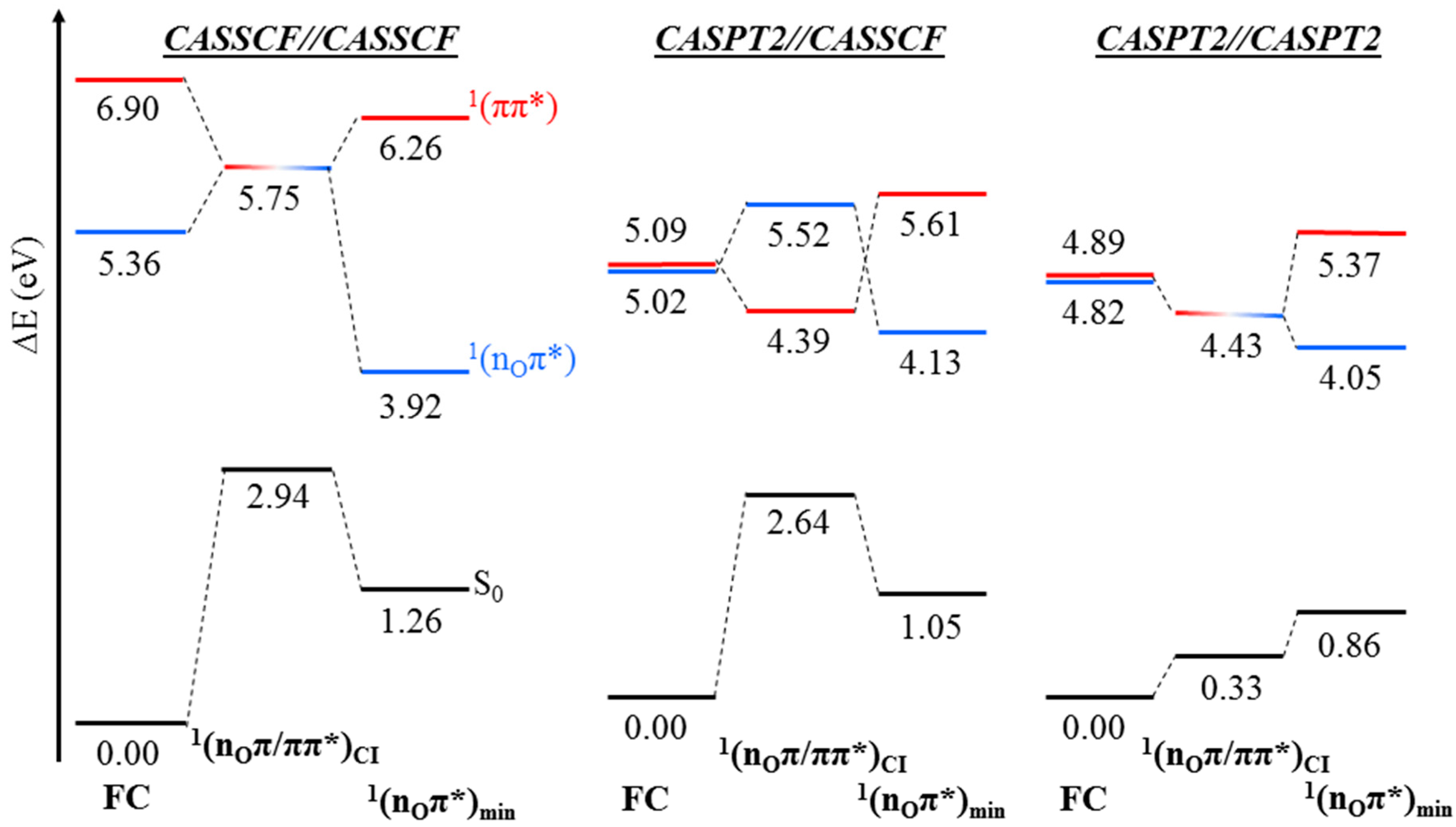
2. Results
2.1. CASPT2//CASSCF
2.2. CASPT2//CASPT2
3. Discussion
4. Computational Details
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kleinermanns, K.; Nachtigallova, D.; de Vries, M.S. Excited state dynamics of DNA bases. Int. Rev. Phys. Chem. 2013, 32, 308–342. [Google Scholar] [CrossRef]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernandez, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 2004, 104, 1977–2019. [Google Scholar] [CrossRef] [PubMed]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernandez, C.E.; Kohler, B. DNA excited-state dynamics: From single bases to the double helix. Annu. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Schreier, W.J.; Gilch, P.; Zinth, W. Early events of DNA photodamage. Annu. Rev. Phys. Chem. 2015, 66, 497–519. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, T.; Improta, R.; Markovitsi, D. DNA/RNA: Building blocks of life under UV irradiation. J. Phys. Chem. Lett. 2010, 1, 2025–2030. [Google Scholar] [CrossRef]
- Barbatti, M.; Borin, A.; Ullrich, S. Photoinduced processes in nucleic acids. Top. Curr. Chem. 2014, 355, 1–32. [Google Scholar]
- Giussani, A.; Segarra-Martí, J.; Roca-Sanjuán, D.; Merchán, M. Excitation of nucleobases from a computational perspective I: Reaction paths. Top. Curr. Chem. 2015, 355, 57–97. [Google Scholar] [PubMed]
- Improta, R.; Barone, V. Excited states behavior of nucleobases in solution: Insights from computational studies. Top. Curr. Chem. 2015, 355, 329–357. [Google Scholar] [PubMed]
- Serrano-Andrés, L.; Merchán, M. Are the five natural DNA/RNA base monomers a good choice from natural selection? A photochemical perspective. J. Photochem. Photobiol. C-Photochem. Rev. 2009, 10, 21–32. [Google Scholar] [CrossRef]
- Merchán, M.; González-Luque, R.; Climent, T.; Serrano-Andrés, L.; Rodriguez, E.; Reguero, M.; Pelaez, D. Unified model for the ultrafast decay of pyrimidine nucleobases. J. Phys. Chem. B 2006, 110, 26471–26476. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L.; Domcke, W. The chemical physics of the photostability of life. Europhys. News 2006, 37, 20–23. [Google Scholar] [CrossRef]
- Serrano-Andrés, L.; Merchán, M.; Borin, A.C. Adenine and 2-aminopurine: Paradigms of modern theoretical photochemistry. Proc. Natl. Acad. Sci. USA 2006, 103, 8691–8696. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Grand, A.; Douki, T. Solar UV radiation-induced DNA bipyrimidine photoproducts: Formation and mechanistic insights. Top. Curr. Chem. 2015, 356, 249–275. [Google Scholar] [PubMed]
- Cadet, J.; Mouret, S.; Ravanat, J.-L.; Douki, T. Photoinduced damage to cellular DNA: Direct and photosensitized reactions. Photochem. Photobiol. 2012, 88, 1048–1065. [Google Scholar] [CrossRef] [PubMed]
- McFarland, B.K.; Farrell, J.P.; Miyabe, S.; Tarantelli, F.; Aguilar, A.; Berrah, N.; Bostedt, C.; Bozek, J.D.; Bucksbaum, P.H.; Castagna, J.C.; et al. Ultrafast X-ray auger probing of photoexcited molecular dynamics. Nat. Commun. 2014, 5, 4235. [Google Scholar] [CrossRef] [PubMed]
- Hudock, H.R.; Levine, B.G.; Thompson, A.L.; Satzger, H.; Townsend, D.; Gador, N.; Ullrich, S.; Stolow, A.; Martinez, T.J. Ab initio molecular dynamics and time-resolved photoelectron spectroscopy of electronically excited uracil and thymine. J. Phys. Chem. A 2007, 111, 8500–8508. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.K.; Leszczynski, J. Electronic spectra, excited state structures and interactions of nucleic acid bases and base assemblies: A review. J. Biomol. Struct. Dyn. 2007, 25, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M. Photorelaxation induced by water–chromophore electron transfer. J. Am. Chem. Soc. 2014, 136, 10246–10249. [Google Scholar] [CrossRef] [PubMed]
- Merchán, M.; Serrano-Andrés, L. Ultrafast internal conversion of excited cytosine via the lowest pp* electronic singlet state. J. Am. Chem. Soc. 2003, 125, 8108–8109. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L.; Domcke, W. Molecular mechanisms of the photostability of life. Phys. Chem. Chem. Phys. 2010, 12, 4897–4898. [Google Scholar] [PubMed]
- Perun, S.; Sobolewski, A.L.; Domcke, W. Conical intersections in thymine. J. Phys. Chem. A 2006, 110, 13238–13244. [Google Scholar] [CrossRef] [PubMed]
- Perun, S.; Sobolewski, A.L.; Domcke, W. Ab initio studies on the radiationless decay mechanisms of the lowest excited singlet states of 9H-adenine. J. Am. Chem. Soc. 2005, 127, 6257–6265. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, T.; Banyasz, A.; Lazzarotto, E.; Markovitsi, D.; Scalmani, G.; Frisch, M.J.; Barone, V.; Improta, R. Singlet excited-state behavior of uracil and thymine in aqueous solution: A combined experimental and computational study of 11 uracil derivatives. J. Am. Chem. Soc. 2006, 128, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Mercier, Y.; Santoro, F.; Reguero, M.; Improta, R. The decay from the dark np* excited state in uracil: An integrated CASPT2/CASSCF and PCM/TD-DFT study in the gas phase and in water. J. Phys. Chem. B 2008, 112, 10769–10772. [Google Scholar] [CrossRef] [PubMed]
- Zechmann, G.; Barbatti, M. Photophysics and deactivation pathways of thymine. J. Phys. Chem. A 2008, 112, 8273–8279. [Google Scholar] [CrossRef] [PubMed]
- Blancafort, L.; Robb, M.A. Key role of a threefold state crossing in the ultrafast decay of electronically excited cytosine. J. Phys. Chem. A 2004, 108, 10609–10614. [Google Scholar] [CrossRef]
- Ismail, N.; Blancafort, L.; Olivucci, M.; Kohler, B.; Robb, M.A. Ultrafast decay of electronically excited singlet cytosine via pp* to nop* state switch. J. Am. Chem. Soc. 2002, 124, 6818–6819. [Google Scholar] [CrossRef] [PubMed]
- Blancafort, L. Energetics of cytosine singlet excited-state decay paths—A difficult case for CASSCF and CASPT2. Photochem. Photobiol. 2007, 83, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Merchán, M.; Serrano-Andrés, L.; Robb, M.A.; Blancafort, L. Triplet-state formation along the ultrafast decay of excited singlet cytosine. J. Am. Chem. Soc. 2005, 127, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Yamazaki, S.; Taketsugu, T. Quantum chemical investigations on the nonradiative deactivation pathways of cytosine derivatives. J. Phys. Chem. A 2014, 118, 9429–9437. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Arai, G.; Yamazaki, S.; Taketsugu, T. Solvent effects on the ultrafast nonradiative deactivation mechanisms of thymine in aqueous solution: Excited-state qm/mm molecular dynamics simulations. J. Chem. Phys. 2013, 139, 214304. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Harabuchi, Y.; Yamazaki, S.; Taketsugu, T. Photophysics of cytosine tautomers: New insights into the nonradiative decay mechanisms from MS-CASPT2 potential energy calculations and excited-state molecular dynamics simulations. Phys. Chem. Chem. Phys. 2013, 15, 12322–12339. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Taketsugu, T. Nonradiative deactivation mechanisms of uracil, thymine, and 5-fluorouracil: A comparative ab initio study. J. Phys. Chem. A 2012, 116, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, J.J.; Barbatti, M.; Hoo, J.T.S.; Adkins, J.A.; Windus, T.L.; Nachtigallova, D.; Lischka, H. Photodynamics simulations of thymine: Relaxation into the first excited singlet state. J. Phys. Chem. A 2009, 113, 12686–12693. [Google Scholar] [CrossRef] [PubMed]
- Asturiol, D.; Lasorne, B.; Worth, G.A.; Robb, M.A.; Blancafort, L. Exploring the sloped-to-peaked S2/S1 seam of intersection of thymine with electronic structure and direct quantum dynamics calculations. Phys. Chem. Chem. Phys. 2010, 12, 4949–4958. [Google Scholar] [CrossRef] [PubMed]
- Asturiol, D.; Lasorne, B.; Robb, M.A.; Blancafort, L. Photophysics of the pp* and np* states of thymine: MS-CASPT2 minimum-energy paths and casscf on-the-fly dynamics. J. Phys. Chem. A 2009, 113, 10211–10218. [Google Scholar] [CrossRef] [PubMed]
- Picconi, D.; Barone, V.; Lami, A.; Santoro, F.; Improta, R. The interplay between ππ*/nπ* excited states in gas-phase thymine: A quantum dynamical study. ChemPhysChem 2011, 12, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Marquetand, P.; González, L. A general method to describe intersystem crossing dynamics in trajectory surface hopping. Int. J. Quantum Chem. 2015, 115, 1215–1231. [Google Scholar] [CrossRef]
- Mai, S.; Richter, M.; Marquetand, P.; González, L. Excitation of nucleobases from a computational perspective II: Dynamics. Top. Curr. Chem. 2015, 355, 99–153. [Google Scholar] [PubMed]
- Buchner, F.; Nakayama, A.; Yamazaki, S.; Ritze, H.-H.; Lübcke, A. Excited-state relaxation of hydrated thymine and thymidine measured by liquid-jet photoelectron spectroscopy: Experiment and simulation. J. Am. Chem. Soc. 2015, 137, 2931–2938. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O. The complete active space self-consistent field method and its applications in electronic structure calculations. Adv. Chem. Phys. 1987, 69, 399–445. [Google Scholar]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O. 2nd-order perturbation-theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Blancafort, L. Photochemistry and photophysics at extended seams of conical intersection. ChemPhysChem 2014, 15, 3166–3181. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. Multiconfigurational Quantum Chemistry; Wiley: Hoboken, NJ, USA, 2016; p. 240. [Google Scholar]
- Serrano-Andrés, L.; Merchán, M. Quantum chemistry of the excited state: 2005 overview. Theochem-J. Mol. Struct. 2005, 729, 99–108. [Google Scholar] [CrossRef]
- Merchán, M.; Serrano-Andrés, L. Ab initio methods for excited states. In Computational Photochemistry; Olivucci, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 16, pp. 35–91. [Google Scholar]
- Roca-Sanjuán, D.; Aquilante, F.; Lindh, R. Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 585–603. [Google Scholar] [CrossRef]
- Giussani, A.; Segarra-Martí, J.; Nenov, A.; Rivalta, I.; Tolomelli, A.; Mukamel, S.; Garavelli, M. Spectroscopic fingerprints of DNA/RNA pyrimidine nucleobases in third-order nonlinear electronic spectra. Theor. Chem. Acc. 2016, 135, 1–18. [Google Scholar] [CrossRef]
- Olaso-González, G.; Merchán, M.; Serrano-Andrés, L. The role of adenine excimers in the photophysics of oligonucleotides. J. Am. Chem. Soc. 2009, 131, 4368–4377. [Google Scholar] [CrossRef] [PubMed]
- González-Ramírez, I.; Segarra-Martí, J.; Serrano-Andrés, L.; Merchán, M.; Rubio, M.; Roca-Sanjuán, D. On the N1-H and N3-H bond dissociation in uracil by low energy electrons: A CASSCF/CASPT2 study. J. Chem. Theor. Comput. 2012, 8, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Francés-Monerris, A.; Segarra-Martí, J.; Merchán, M.; Roca-Sanjuán, D. Complete-active-space second-order perturbation theory (CASPT2//CASSCF) study of the dissociative electron attachment in canonical DNA nucleobases caused by low-energy electrons (0–3 eV). J. Chem. Phys. 2015, 143, 215101. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.J.; Miller, A.M.; Kohler, B. Singlet excited-state lifetimes of cytosine derivatives measured by femtosecond transient absorption. Photochem. Photobiol. 2003, 77, 158–164. [Google Scholar] [CrossRef]
- Pecourt, J.M.L.; Peon, J.; Kohler, B. DNA excited-state dynamics: Ultrafast internal conversion and vibrational cooling in a series of nucleosides. J. Am. Chem. Soc. 2001, 123, 10370–10378. [Google Scholar] [CrossRef] [PubMed]
- Hare, P.M.; Crespo-Hernández, C.E.; Kohler, B. Internal conversion to the electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution. Proc. Natl. Acad. Sci. USA 2007, 104, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Hare, P.M.; Middleton, C.T.; Mertel, K.I.; Herbert, J.M.; Kohler, B. Time-resolved infrared spectroscopy of the lowest triplet state of thymine and thymidine. Chem. Phys. 2008, 347, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Kohler, B. Excited states in DNA strands investigated by ultrafast laser spectroscopy. Top. Curr. Chem. 2015, 356, 39–87. [Google Scholar] [PubMed]
- Doorley, G.W.; Wojdyla, M.; Watson, G.W.; Towrie, M.; Parker, A.W.; Kelly, J.M.; Quinn, S.J. Tracking DNA excited states by picosecond-time-resolved infrared spectroscopy: Signature band for a charge-transfer excited state in stacked adenine—Thymine systems. J. Phys. Chem. Lett. 2013, 4, 2739. [Google Scholar] [CrossRef]
- Quinn, S.; Doorley, G.W.; Watson, G.W.; Cowan, A.J.; George, M.W.; Parker, A.W.; Ronayne, K.L.; Towrie, M.; Kelly, J.M. Ultrafast IR spectroscopy of the short-lived transients formed by UV excitation of cytosine derivatives. Chem. Commun. 2007, 2130–2132. [Google Scholar] [CrossRef] [PubMed]
- Chatterley, A.S.; West, C.W.; Stavros, V.G.; Verlet, J.R.R. Time-resolved photoelectron imaging of the isolated deprotonated nucleotides. Chem. Sci. 2014, 5, 3963–3975. [Google Scholar] [CrossRef]
- Chatterley, A.S.; West, C.W.; Roberts, G.M.; Stavros, V.G.; Verlet, J.R.R. Mapping the ultrafast dynamics of adenine onto its nucleotide and oligonucleotides by time-resolved photoelectron imaging. J. Phys. Chem. Lett. 2014, 5, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Onidas, D.; Markovitsi, D.; Marguet, S.; Sharonov, A.; Gustavsson, T. Fluorescence properties of DNA nucleosides and nucleotides: A refined steady-state and femtosecond investigation. J. Phys. Chem. B 2002, 106, 11367–11374. [Google Scholar] [CrossRef]
- Segarra-Martí, J.; Garavelli, M.; Aquilante, F. Multiconfigurational second-order perturbation theory with frozen natural orbitals extended to the treatment of photochemical problems. J. Chem. Theor. Comput. 2015, 11, 3772–3784. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Giussani, A.; Segarra-Martí, J.; Nenov, A.; Rivalta, I.; Voityuk, A.A.; Mukamel, S.; Roca-Sanjuán, D.; Garavelli, M.; Blancafort, L. Multiple decay mechanisms and 2D-UV spectroscopic fingerprints of singlet excited solvated adenine-uracil monophosphate. Chem. Eur. J. 2016, 22, 7497–7507. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nun, M.; Martínez, T.J. Photodynamics of ethylene: Ab initio studies of conical intersections. Chem. Phys. 2000, 259, 237–248. [Google Scholar] [CrossRef]
- Molina, V.; Merchán, M.; Roos, B.O.; Malmqvist, P.-A. On the low-lying singlet excited states of styrene: A theoretical contribution. Phys. Chem. Chem. Phys. 2000, 2, 2211–2217. [Google Scholar] [CrossRef]
- Richter, M.; Mai, S.; Marquetand, P.; Gonzalez, L. Ultrafast intersystem crossing dynamics in uracil unravelled by ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2014, 16, 24423–24436. [Google Scholar] [CrossRef] [PubMed]
- González-Luque, R.; Climent, T.; González-Ramírez, I.; Merchán, M.; Serrano-Andrés, L. Singlet-triplet states interaction regions in DNA/RNA nucleobase hypersurfaces. J. Chem. Theor. Comput. 2010, 6, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pérez, J.J.; González-Luque, R.; Merchán, M.; Serrano-Andrés, L. On the intrinsic population of the lowest triplet state of thymine. J. Phys. Chem. B 2007, 111, 11880–11883. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.A. Spin—Orbit coupling and the radiationless processes in nitrogen heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- Szymczak, J.J.; Barbatti, M.; Lischka, H. Influence of the active space on CASSCF nonadiabatic dynamics simulations. Int. J. Quantum Chem. 2011, 111, 3307–3315. [Google Scholar] [CrossRef]
- Plasser, F.; Crespo-Otero, R.; Pederzoli, M.; Pittner, J.; Lischka, H.; Barbatti, M. Surface hopping dynamics with correlated single-reference methods: 9H-adenine as a case study. J. Chem. Theor. Comput. 2014, 10, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Pepino, A.J.; Segarra-Martí, J.; Banyasz, A.; Garavelli, M.; Improta, R. Computing the absorption and emission spectra of 5-methylcytidine in different solvents: A test-case for different solvation models. J. Chem. Theor. Comput. 2016, 12, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Szalay, P.G.; Watson, T.; Perera, A.; Lotrich, V.F.; Bartlett, R.J. Benchmark studies on the building blocks of DNA. 1. Superiority of coupled cluster methods in describing the excited states of nucleobases in the Franck-Condon region. J. Phys. Chem. A 2012, 116, 6702–6710. [Google Scholar] [CrossRef] [PubMed]
- Altavilla, S.F.; Segarra-Martí, J.; Nenov, A.; Conti, I.; Rivalta, I.; Garavelli, M. Deciphering the photochemical mechanisms describing the UV-induced processes occurring in solvated guanine monophosphate. Front. Chem. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Conti, I.; Altoe, P.; Stenta, M.; Garavelli, M.; Orlandi, G. Adenine deactivation in DNA resolved at the CASPT2//CASSCF/AMBER level. Phys. Chem. Chem. Phys. 2010, 12, 5016–5023. [Google Scholar] [CrossRef] [PubMed]
- Altoé, P.; Stenta, M.; Bottoni, A.; Garavelli, M. A tunable QM/MM approach to chemical reactivity, structure and physico-chemical properties prediction. Theor. Chem. Acc. 2007, 118, 219–240. [Google Scholar] [CrossRef]
- De Vico, L.; Olivucci, M.; Lindh, R. New general tools for constrained geometry optimizations. J. Chem. Theor. Comput. 2005, 1, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Canuel, C.; Mons, M.; Piuzzi, F.; Tardivel, B.; Dimicoli, I.; Elhanine, M. Excited states dynamics of DNA and RNA bases: Characterization of a stepwise deactivation pathway in the gas phase. J. Chem. Phys. 2005, 122, 074316. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.G.; Ko, C.; Quenneville, J.; MartÍnez, T.J. Conical intersections and double excitations in time-dependent density functional theory. Mol. Phys. 2006, 104, 1039–1051. [Google Scholar] [CrossRef]
- Serrano-Andrés, L.; Merchán, M.; Lindh, R. Computation of conical intersections by using perturbation techniques. J. Chem. Phys. 2005, 122, 104107. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, A.A. Extended multi-configuration quasi-degenerate perturbation theory: The new approach to multi-state multi-reference perturbation theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef] [PubMed]
- Gozem, S.; Melaccio, F.; Lindh, R.; Krylov, A.I.; Granovsky, A.A.; Angeli, C.; Olivucci, M. Mapping the excited state potential energy surface of a retinal chromophore model with multireference and equation-of-motion coupled-cluster methods. J. Chem. Theor. Comput. 2013, 9, 4495–4506. [Google Scholar] [CrossRef] [PubMed]
- Gozem, S.; Huntress, M.; Schapiro, I.; Lindh, R.; Granovsky, A.A.; Angeli, C.; Olivucci, M. Dynamic electron correlation effects on the ground state potential energy surface of a retinal chromophore model. J. Chem. Theor. Comput. 2012, 8, 4069–4080. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, T.; Woywod, C.; Werner, H.-J. Pyrazine excited states revisited using the extended multi-state complete active space second-order perturbation method. Phys. Chem. Chem. Phys. 2013, 15, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, T.; Werner, H.-J. Multireference explicitly correlated F12 theories. Mol. Phys. 2013, 111, 607–630. [Google Scholar] [CrossRef]
- Shiozaki, T.; Gyrffy, W.; Celani, P.; Werner, H.-J. Communication: Extended multi-state complete active space second-order perturbation theory: Energy and nuclear gradients. J. Chem. Phys. 2011, 135, 081106. [Google Scholar] [CrossRef] [PubMed]
- Vlaisavljevich, B.; Shiozaki, T. Nuclear energy gradients for internally contracted complete active space second-order perturbation theory: Multistate extensions. J. Chem. Theor. Comput. 2016, 12, 3781–3787. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, J.; Martinez, T.J. Dynamical correlation effects on photoisomerization: Ab initio multiple spawning dynamics with MS-CASPT2 for a model trans-protonated schiff base. J. Phys. Chem. B 2016, 120, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Marquetand, P.; González, L. Intersystem crossing pathways in the noncanonical nucleobase 2-thiouracil: A time-dependent picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Levine, B.G.; Martínez, T.J. Ab initio multiple spawning dynamics using multi-state second-order perturbation theory. J. Phys. Chem. A 2009, 113, 13656–13662. [Google Scholar] [CrossRef] [PubMed]
- Frutos, L.M.; Andruniów, T.; Santoro, F.; Ferré, N.; Olivucci, M. Tracking the excited-state time evolution of the visual pigment with multiconfigurational quantum chemistry. Proc. Natl. Acad. Sci. USA 2007, 104, 7764–7769. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Richter, M.; Marquetand, P.; González, L. The DNA nucleobase thymine in motion—Intersystem crossing simulated with surface hopping. Chem. Phys. 2016. In press. [Google Scholar]
- Nenov, A.; Giussani, A.; Segarra-martí, J.; Vishal, K.; Rivalta, I.; Cerullo, G.; Mukamel, S. Modeling the high-energy electronic state manifold of adenine: Calibration for nonlinear electronic spectroscopy. J. Chem. Phys. 2015, 142, 212443. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Aquino, A.J.A.; Szymczak, J.J.; Nachtigallova, D.; Hobza, P.; Lischka, H. Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. USA 2010, 107, 21453–21458. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; de Vico, L.; Ferré, N.; Ghigo, G.; Malmqvist, P.A.; Neogrady, P.; Pedersen, T.B.; Pitonak, M.; Reiher, M.; Roos, B.O.; et al. Software news and update MOLCAS 7: The next generation. J. Computat. Chem. 2010, 31, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Autschbach, J.; Carlson, R.; Chibotaru, L.; Delcey, M.G.; De Vico, L.; Fernández Galvan, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. MOLCAS 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [PubMed]
- Pierloot, K.; Dumez, B.; Widmark, P.O.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. IV. Medium size basis sets for the atoms h-kr. Theor. Chim. Acta 1995, 90, 87–114. [Google Scholar] [CrossRef]
- Widmark, P.O.; Persson, B.J.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. II. Second row atoms. Theor. Chim. Acta 1991, 79, 419–432. [Google Scholar] [CrossRef]
- Forsberg, N.; Malmqvist, P.A. Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett. 1997, 274, 196–204. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.A.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Delcey, M.G.; Pedersen, T.B.; Aquilante, F.; Lindh, R. Analytical gradients of the state-average complete active space self-consistent field method with density fitting. J. Chem. Phys. 2015, 143, 044110. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision b.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Bearpark, M.J.; Robb, M.A.; Bernhard Schlegel, H. A direct method for the location of the lowest energy point on a potential surface crossing. Chem. Phys. Lett. 1994, 223, 269–274. [Google Scholar] [CrossRef]
- Nenov, A.; Segarra-Martí, J.; Giussani, A.; Conti, I.; Rivalta, I.; Dumont, E.; Jaiswal, V.K.; Altavilla, S.F.; Mukamel, S.; Garavelli, M. Fd 177: Probing deactivation pathways of DNA nucleobases by two-dimensional electronic spectroscopy: First principles simulations. Faraday Discuss. 2015, 177, 345–362. [Google Scholar] [CrossRef] [PubMed]
- West, B.A.; Moran, A.M. Two-dimensional electronic spectroscopy in the ultraviolet wavelength range. J. Phys. Chem. Lett. 2012, 3, 2575–2581. [Google Scholar] [CrossRef] [PubMed]
- West, B.A.; Womick, J.M.; Moran, A.M. Probing ultrafast dynamics in adenine with mid-UV four-wave mixing spectroscopies. J. Phys. Chem. A 2011, 115, 8630–8637. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basis Set | Type of Calc. | Active Space (e− in Orbs) | |
|---|---|---|---|
| 10in8 | 10in11 | ||
| 6-31G | Opt | 1(ππ*)min | 1(ππ*)min |
| Opt Tight 1 | 1(ππ*)min | 1(ππ*)min | |
| MEP | 1(ππ*)min | 1(ππ*)min | |
| 6-31G* | Opt | 1(ππ*)min | 1(ππ*/S0)CI |
| Opt Tight 1 | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| MEP | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| ANO-S 321/21 | Opt | 1(ππ*)min | 1(ππ*)min |
| Opt Tight 1 | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| MEP | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| ANO-L 321/21 | Opt | 1(ππ*)min | 1(ππ*/S0)CI |
| Opt Tight 1 | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| MEP | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| ANO-L 432/21 | Opt | 1(ππ*)min | 1(ππ*/S0)CI |
| Opt Tight 1 | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| MEP | 1(ππ*/S0)CI | 1(ππ*/S0)CI | |
| Type of Calculation | Method | Structure |
|---|---|---|
| Opt | CASPT2 | 1(ππ*)min |
| MS-CASPT2 | 1(ππ*)min | |
| Opt Tight 1 | CASPT2 | 1(ππ*/S0)CI |
| MS-CASPT2 | 1(ππ*)min-twisted | |
| MEP | CASPT2 | |
| r = 0.09 | 1(ππ*)min-twisted | |
| r = 0.18 | 1(ππ*/S0)CI | |
| r = 0.30 | 1(ππ*/S0)CI | |
| MS-CASPT2 | ||
| r = 0.18 | 1(ππ*)min-twisted | |
| r = 0.30 | 1(ππ*/S0)CI |
| Type of Calc. | Method | Dihedral/HC6C5C7 | Dist C6-C5 | Dist C5-C7 |
|---|---|---|---|---|
| Opt | CASPT2 | 0.0487 | 1.4331 | 1.4915 |
| MS-CASPT2 | 0.079 | 1.4302 | 1.4913 | |
| Opt Tight 1 | CASPT2 | 98.2401 | 1.4666 | 1.5283 |
| MS-CASPT2 | −30.063 | 1.4288 | 1.4902 | |
| MEP | CASPT2 | |||
| r = 0.09 | −33.8446 | 1.4505 | 1.4924 | |
| r = 0.18 | −102.8496 | 1.4625 | 1.5341 | |
| r = 0.30 | −103.7962 | 1.4607 | 1.5363 | |
| MS-CASPT2 | ||||
| r = 0.18 | −35.9473 | 1.4497 | 1.4924 | |
| r = 0.30 | −111.5802 | 1.4505 | 1.5405 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segarra-Martí, J.; Francés-Monerris, A.; Roca-Sanjuán, D.; Merchán, M. Assessment of the Potential Energy Hypersurfaces in Thymine within Multiconfigurational Theory: CASSCF vs. CASPT2. Molecules 2016, 21, 1666. https://doi.org/10.3390/molecules21121666
Segarra-Martí J, Francés-Monerris A, Roca-Sanjuán D, Merchán M. Assessment of the Potential Energy Hypersurfaces in Thymine within Multiconfigurational Theory: CASSCF vs. CASPT2. Molecules. 2016; 21(12):1666. https://doi.org/10.3390/molecules21121666
Chicago/Turabian StyleSegarra-Martí, Javier, Antonio Francés-Monerris, Daniel Roca-Sanjuán, and Manuela Merchán. 2016. "Assessment of the Potential Energy Hypersurfaces in Thymine within Multiconfigurational Theory: CASSCF vs. CASPT2" Molecules 21, no. 12: 1666. https://doi.org/10.3390/molecules21121666
APA StyleSegarra-Martí, J., Francés-Monerris, A., Roca-Sanjuán, D., & Merchán, M. (2016). Assessment of the Potential Energy Hypersurfaces in Thymine within Multiconfigurational Theory: CASSCF vs. CASPT2. Molecules, 21(12), 1666. https://doi.org/10.3390/molecules21121666