
Discovery of Novel Allopurinol Derivatives with Anticancer Activity and Attenuated Xanthine Oxidase Inhibition

 , ,
, ,
Abstract
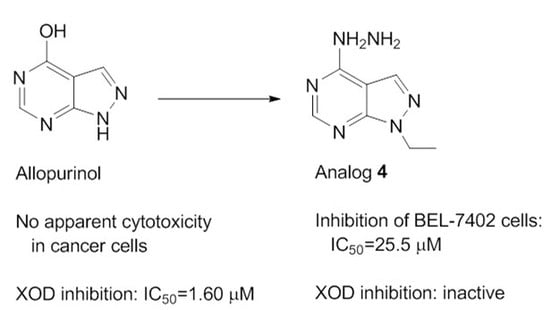
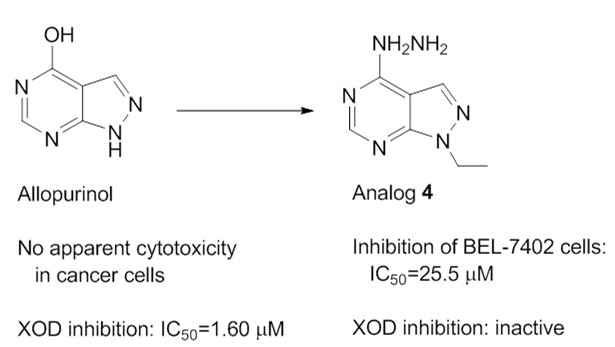
:
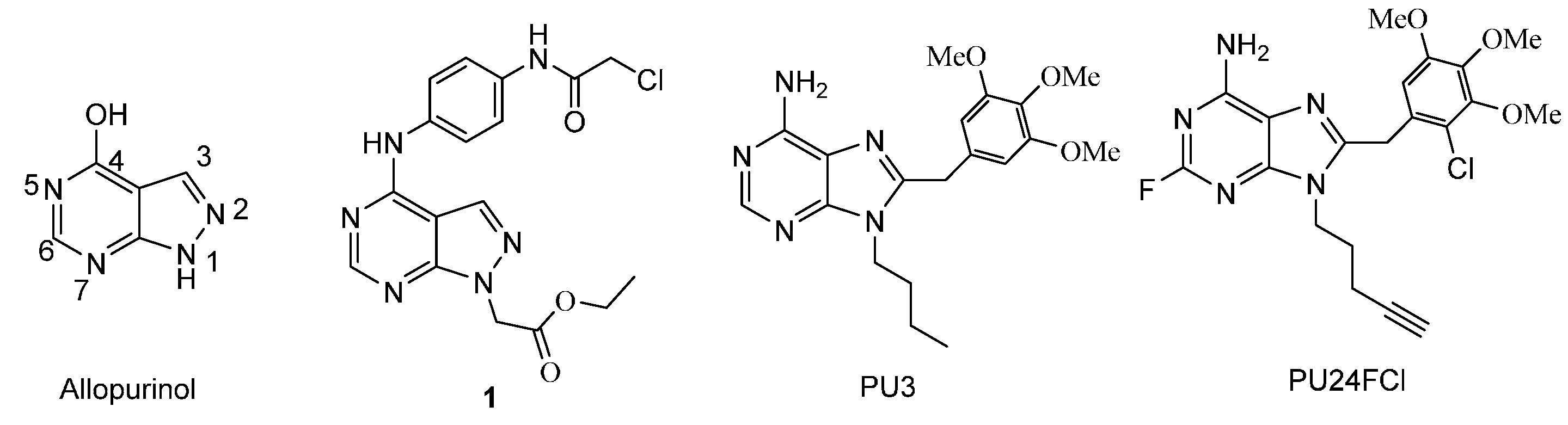
1. Introduction
2. Results and Discussion
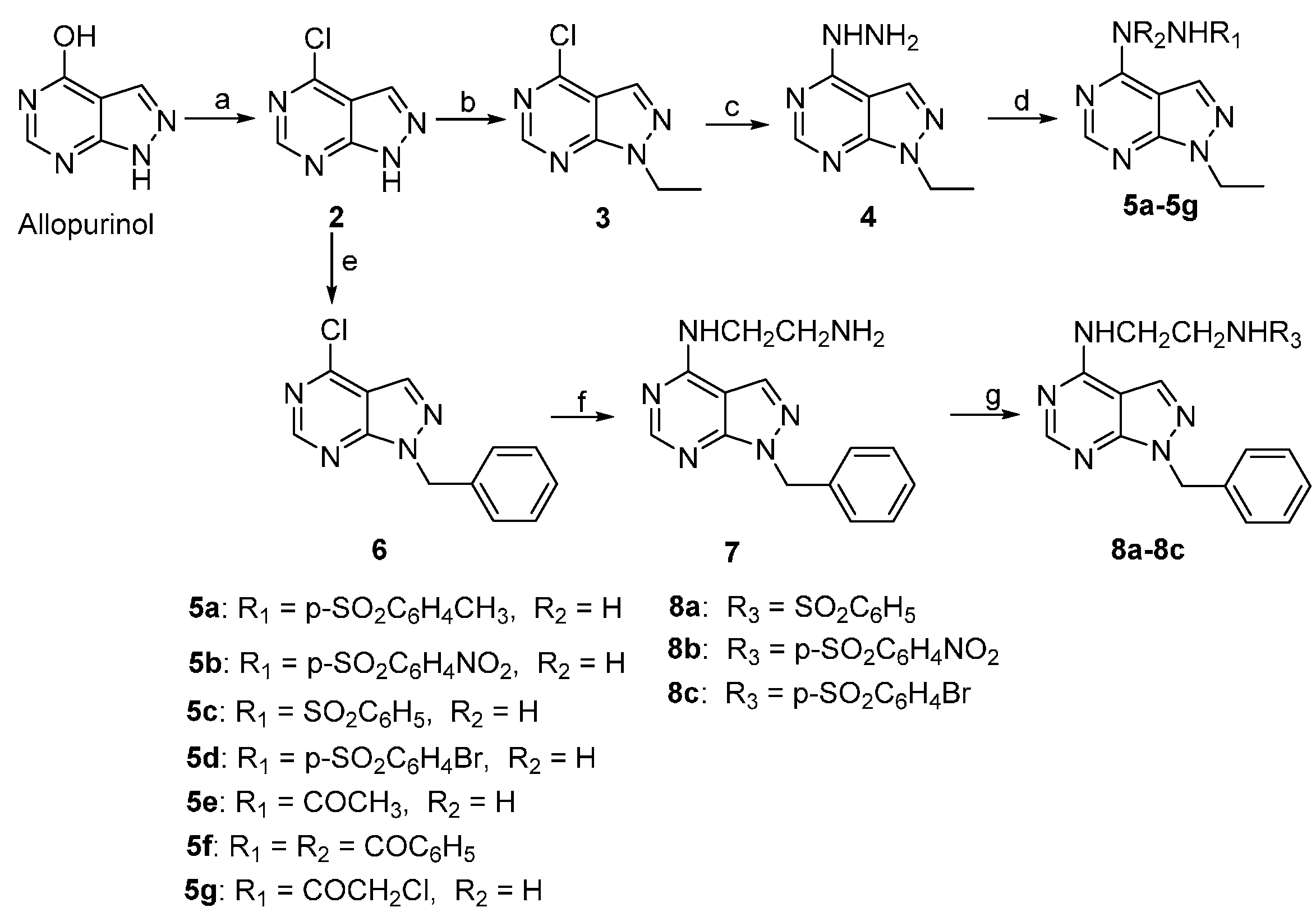
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cytotoxicity against Cancer Cell Lines
2.2.2. Cytotoxicity towards MCF-10A Mammary Epithelial Cells
2.2.3. XOD Inhibitory Activity
3. Materials and Methods
3.1. General Procedures for Chemical Synthesis
3.1.1. 4-Chloro-1H-pyrazolo[3,4-d]pyrimidine (2)
3.1.2. 4-Chloro-1-ethyl-1H-pyrazolo[3,4-d]pyrimidine (3)
3.1.3. 1-Ethyl-4-hydrazinyl-1H-pyrazolo[3,4-d]pyrimidine (4)
3.1.4. Synthesis of Compounds 5a–5g
3.1.5. 1-Benzyl-4-chloro-1H-pyrazolo[3,4-d]pyrimidine (6)
3.1.6. N1-(1-Benzyl-1H/-pyrazolo[3,4-d]pyrimidin-4-yl)ethane-1,2-diamine (7)
3.1.7. Synthesis of Compounds 8a–8c
3.2. Biological Assays
3.2.1. Cell Culture
3.2.2. Cell Survival/Growth Assay
3.2.3. Determination of Xanthine Oxidase (XOD) Inhibitory Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| XOD | Xanthine oxidase |
| 17-AAG | 17-N-allylamino-17-demethoxy geldanamycin |
| Hsp90 | Heat Shock Protein 90 |
| TEA | Triethylamine |
| DMF | Dimethylformamide |
| DMEM | Dulbecco’s modified eagle medium |
| FBS | Fetal bovine Serum |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NIH | National Institute of Health |
| RCMI | Research Centers in Minority Institutions Program |
| NIMHD | National Institute on Minority Health and Health Disparities |
References
- Pacher, P.; Nivorozhkin, A.; Szabó, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Yoshida, T.; Goda, A.E.; Horinaka, M.; Yano, K.; Shiraishi, T.; Wakada, M.; Mizutani, Y.; Miki, T.; Sakai, T. Anti-gout agent allopurinol exerts cytotoxicity to human hormone-refractory prostate cancer cells in combination with tumor necrosis factor–related apoptosis-inducing ligand. Mol. Cancer Res. 2008, 6, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Martina Ferrari, S.; Motta, C.L.; Sartini, S.; Baldini, E.; Materazzi, G.; Politti, U.; Ruffilli, I.; Ulisse, S.; Miccoli, P.; Antonelli, A. Pyrazolopyrimidine derivatives as antineoplastic agents: With a special focus on thyroid cancer. Mini Rev. Med. Chem. 2016, 16, 86–93. [Google Scholar] [CrossRef]
- Yang, L.-L.; Li, G.-B.; Yan, H.-X.; Sun, Q.-Z.; Ma, S.; Ji, P.; Wang, Z.-R.; Feng, S.; Zou, J.; Yang, S.-Y. Discovery of N6-phenyl-1H-pyrazolo [3,4-d] pyrimidine-3,6-diamine derivatives as novel CK1 inhibitors using common-feature pharmacophore model based virtual screening and hit-to-lead optimization. Eur. J. Med. Chem. 2012, 56, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Bannen, L.C.; Chan, D.S.-M.; Dalrymple, L.E.; Jammalamadaka, V.; Khoury, R.G.; Leahy, G.W.; Mac, M.B.; Mann, G.; Mann, L.W.; Nuss, J.M.; et al. C-met modulators and method of use. US Patent 7,977,345 B2, 12 July 2011. [Google Scholar]
- Holzer, P.; Imbach, P.; Furet, P.; Schmiedeberg, N. Preparation of 3-(substituted amino)-pyrazolo[3,4-d]pyrimidines as Ephb4 receptors and VEGFR2 kinase inhibitors. WO 2007062805 A1, 7 June 2007. [Google Scholar]
- Yang, L.-L.; Li, G.-B.; Ma, S.; Zou, C.; Zhou, S.; Sun, Q.-Z.; Cheng, C.; Chen, X.; Wang, L.-J.; Feng, S.; et al. Structure-activity relationship studies of pyrazolo[3,4-d]pyrimidine derivatives leading to the discovery of a novel multikinase inhibitor that potently inhibits FLT3 and VEGFR2 and evaluation of its activity against acute myeloid leukemia in vitro and in vivo. J. Med. Chem. 2013, 56, 1641–1655. [Google Scholar] [PubMed]
- Sartini, S.; Coviello, V.; Bruno, A.; La Pietra, V.; Marinelli, L.; Simorini, F.; Taliani, S.; Salerno, S.; Marini, A.M.; Fioravanti, A. Structure-based optimization of tyrosine kinase inhibitor CLM3. Design, synthesis, functional evaluation, and molecular modeling studies. J. Med. Chem. 2014, 57, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ji, P.; Yang, H.-W.; Yang, L.-L.; Zhou, S.; Zhong, L.; Ma, S.; Fu, X.-Y.; Zhou, C.; Li, G.-B. Sc-535, a novel oral multikinase inhibitor, showed potent antitumor activity in human melanoma models. Cell. Physiol. Biochem. 2013, 32, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Bocci, G.; Motta, C.L.; Ferrari, S.M.; Fallahi, P.; Corrado, A.; Fioravanti, A.; Sartini, S.; Orlandi, P.; Piaggi, S. CLM29, a multi-target pyrazolopyrimidine derivative, has anti-neoplastic activity in medullary thyroid cancer in vitro and in vivo. Mol. Cell. Endocrinol. 2014, 393, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yang, L.-L.; Niu, T.; Cheng, C.; Zhong, L.; Zheng, M.-W.; Xiong, Y.; Li, L.-L.; Xiang, R.; Chen, L.-J.; et al. SKLB-677, an FLT3 and Wnt/β-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, G.; Diasio, R.B.; Cheng, Y.C. Pyrimidine and purine antimetabolites. In Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C.J., Gansler, T.S., Holland, J.F., Frei, E.I., Eds.; BC Decker: Hamiltom, ON, Canada, 2003. [Google Scholar]
- Chiosis, G.; Timaul, M.N.; Lucas, B.; Munster, P.N.; Zheng, F.F.; Sepp-Lorenzino, L.; Rosen, N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol. 2001, 8, 289–299. [Google Scholar] [CrossRef]
- Chiosis, G.; Lucas, B.; Shtil, A.; Huezo, H.; Rosen, N. Development of a purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and induce the degradation of Her2 tyrosine kinase. Bioorg. Med. Chem. 2002, 10, 3555–3564. [Google Scholar] [CrossRef]
- Chiosis, G.; Lucas, B.; Huezo, H.; Solit, D.; Basso, A.; Rosen, N. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr. Cancer Drug Targets 2003, 3, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.; Barril, X.; Dymock, B.; Sheridan, L.; Surgenor, A.; Beswick, M.; Drysdale, M.; Collier, A.; Massey, A.; Davies, N. Structure-activity relationships in purine-based inhibitor binding to Hsp90 isoforms. Chem. Biol. 2004, 11, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Dymock, B.; Barril, X.; Beswick, M.; Collier, A.; Davies, N.; Drysdale, M.; Fink, A.; Fromont, C.; Hubbard, R.E.; Massey, A. Adenine derived inhibitors of the molecular chaperone HSP90-SAR explained through multiple X-ray structures. Bioorg. Med. Chem. Lett. 2004, 14, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.; Solit, D.; Basso, A.; Huezo, H.; Lucas, B.; He, H.; Rosen, N.; Spampinato, C.; Modrich, P.; Chiosis, G. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem. Biol. 2004, 11, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Song, C.; Li, C.; Li, Y.; Dong, L.; Yin, S. Synthesis and biological evaluation of pyrazolo[4,3-d]pyrimidine analogues. Eur. J. Med. Chem. 2013, 67, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.-A.; Mitsiades, C.S.; Anderson, K.C.; Richardson, P.G. Tanespimycin as antitumor therapy. Clin. Lymphoma Myeloma Leuk. 2011, 11, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, G.; Yang, C.; Wang, B. Novel rhein analogues as potential anticancer agents. Chem. Med. Chem. 2011, 6, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Shimizu, T.; Ohyama, S.-I.; Murooka, H.; Iwai, A.; Nakamura, K.; Hasegawa, K.; Kobayashi, Y.; Takahashi, N.; Takahashi, K. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: Synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N′-{4-(4-quinolyloxy) phenyl} ureas. J. Med. Chem. 2005, 48, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Cos, P.; Ying, L.; Calomme, M.; Hu, J.P.; Cimanga, K.; van Poel, B.; Pieters, L.; Vlietinck, A.J.; Berghe, D.V. Structure-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J. Nat. Prod. 1998, 61, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.



{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50(μM) a | |
|---|---|---|
| BEL-7402 | SMMC-7221 | |
| 17-AAG | 12.4 ± 0.1 | 9.85 ± 0.08 |
| 1 | 18.7 [19] | 9.03 [19] |
| 3 | >100 b | >100 b |
| 4 | 25.5 ± 0.4 | 35.2 ± 0.4 |
| 5a | >100 b | >100 b |
| 5b | >100 b | >100 b |
| 5c | >100 b | >100 b |
| 5d | >100 b | >100 b |
| 5e | >100 b | >100 b |
| 5f | >100 b | >100 b |
| 5g | >100 b | >100 b |
| 6 | >100 b | >100 b |
| 7 | >100 b | >100 b |
| 8a | 66.61 ± 0.62 | >100 b |
| 8b | >100 b | 81.46 ± 0.78 |
| 8c | >100 b | >100 b |
| Compounds | Survival Rate (%) | Compounds | Survival Rate (%) | ||
|---|---|---|---|---|---|
| 10 μM | 1 μM | 10 μM | 1 μM | ||
| 17-AAG | 4.8 ± 0.5 | 6.7 ± 0.5 | 5e | 61.3 ± 1.0 | 94.8 ± 0.5 |
| allopurinol | 51.7 ± 6.7 | 84.3 ± 7.7 | 5f | 88.1 ± 1.0 | 102.5 ± 11.0 |
| 1 | 1.0 ± 0.6 | 40.2 ± 3.9 | 5g | 105.4 ± 2.9 | 105.4 ± 4.8 |
| 3 | 67.0 ± 4.8 | 86.2 ± 8.6 | 6 | 75.7 ± 1.5 | 77.6 ± 9.1 |
| 4 | 27.8 ± 2.4 | 66.1 ± 0.5 | 7 | 57.5 ± 1.0 | 81.4 ± 7.2 |
| 5a | 72.8 ± 3.9 | 89.1 ± 1.5 | 8a | 58.4 ± 2.4 | 114..0 ± 9.1 |
| 5b | 68.0 ± 2.4 | 92.9 ± 1.5 | 8b | 71.8 ± 8.5 | 75.7 ± 2.4 |
| 5c | 74.7 ± 0.1 | 75.7 ± 11.0 | 8c | 78.5 ± 3.9 | 105.4 ± 8.6 |
| 5d | 70.9 ± 1.0 | 80.5 ± 2.0 | DMSO | 98.7 ± 6.3 | 100.6 ± 1.4 |
| Compounds | IC50 (μM) |
|---|---|
| 1 | 5.59 ± 0.27 |
| 4 | 17.36 ± 0.69 |
| 5e | 20.25 ± 0.81 |
| 5g | 23.29 ± 0.71 |
| 17-AAG | 0.09 ± 0.01 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Cao, T.-T.; Guo, S.; Zhong, Q.; Li, C.-H.; Li, Y.; Dong, L.; Zheng, S.; Wang, G.; Yin, S.-F. Discovery of Novel Allopurinol Derivatives with Anticancer Activity and Attenuated Xanthine Oxidase Inhibition. Molecules 2016, 21, 771. https://doi.org/10.3390/molecules21060771
Li Y, Cao T-T, Guo S, Zhong Q, Li C-H, Li Y, Dong L, Zheng S, Wang G, Yin S-F. Discovery of Novel Allopurinol Derivatives with Anticancer Activity and Attenuated Xanthine Oxidase Inhibition. Molecules. 2016; 21(6):771. https://doi.org/10.3390/molecules21060771
Chicago/Turabian StyleLi, Yong, Ting-Ting Cao, Shanchun Guo, Qiu Zhong, Cai-Hu Li, Ying Li, Lin Dong, Shilong Zheng, Guangdi Wang, and Shu-Fan Yin. 2016. "Discovery of Novel Allopurinol Derivatives with Anticancer Activity and Attenuated Xanthine Oxidase Inhibition" Molecules 21, no. 6: 771. https://doi.org/10.3390/molecules21060771
APA StyleLi, Y., Cao, T.-T., Guo, S., Zhong, Q., Li, C.-H., Li, Y., Dong, L., Zheng, S., Wang, G., & Yin, S.-F. (2016). Discovery of Novel Allopurinol Derivatives with Anticancer Activity and Attenuated Xanthine Oxidase Inhibition. Molecules, 21(6), 771. https://doi.org/10.3390/molecules21060771