
Novel 5-Substituted 2-(Aylmethylthio)-4-chloro-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides: Synthesis, Molecular Structure, Anticancer Activity, Apoptosis-Inducing Activity and Metabolic Stability


 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
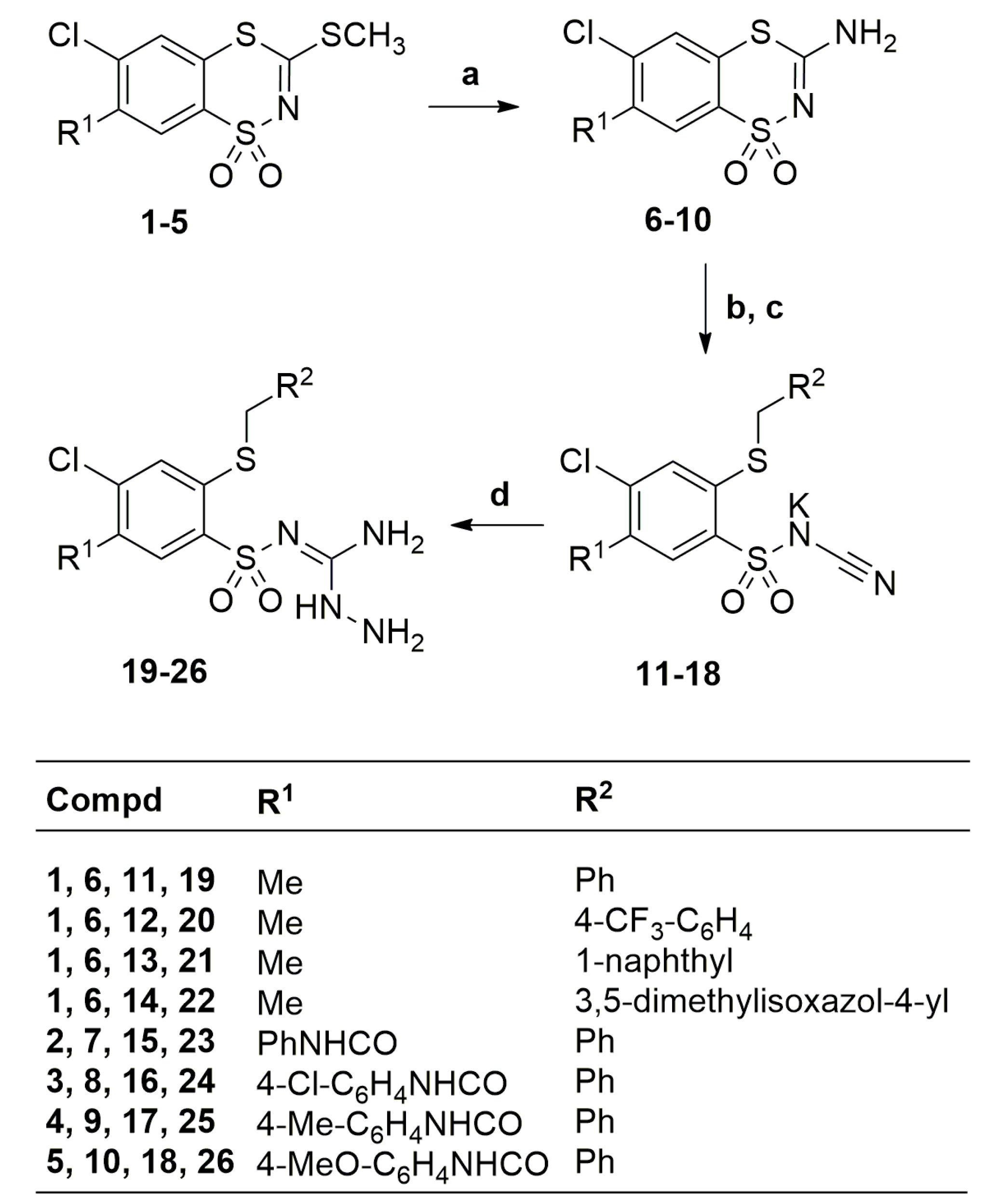
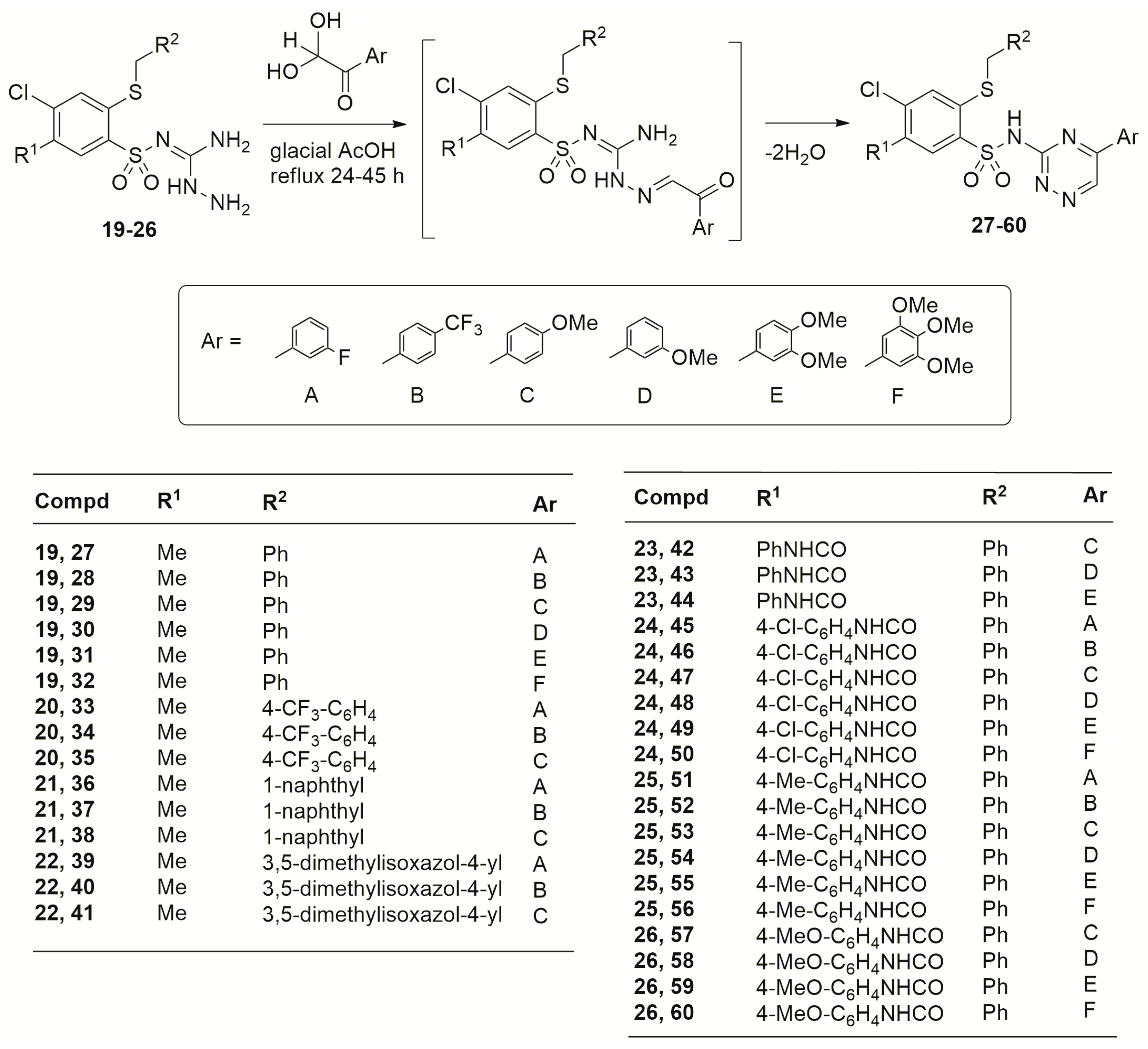
2.1. Chemistry
2.2. Biological Evaluations
2.2.1. Cytotoxic Activity
2.2.2. Investigation of Apoptotic Activity
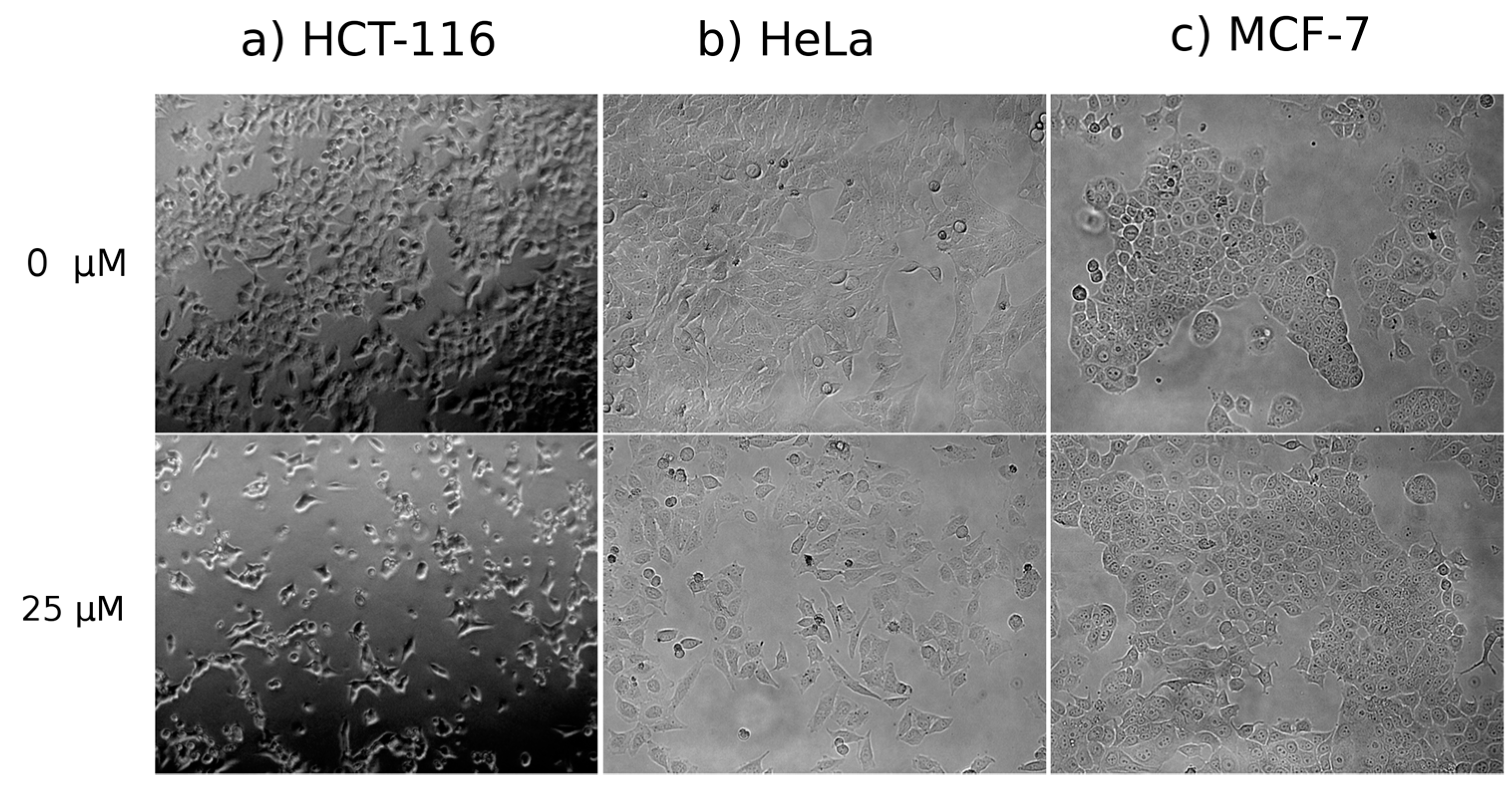
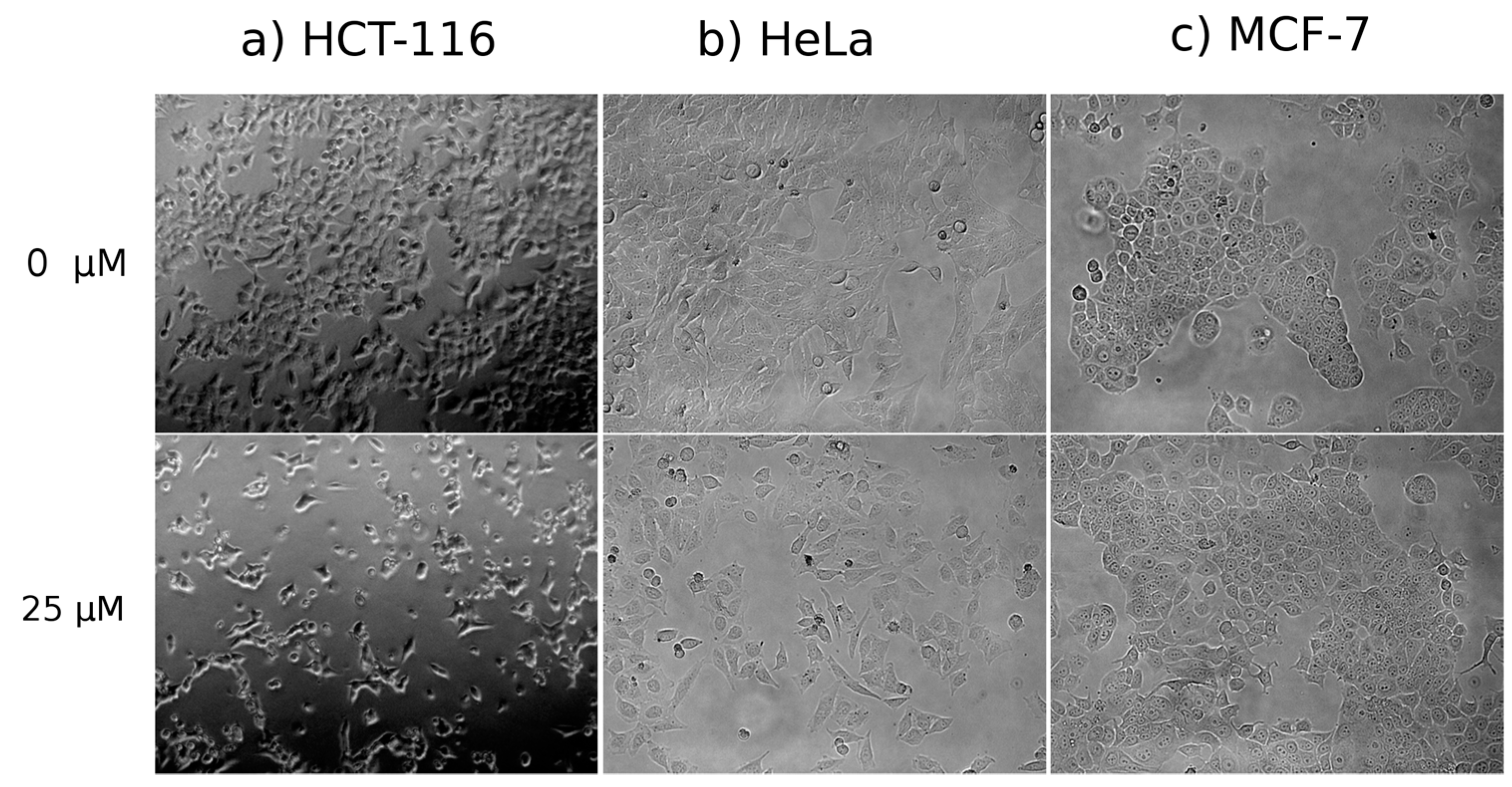
Cell Morphology
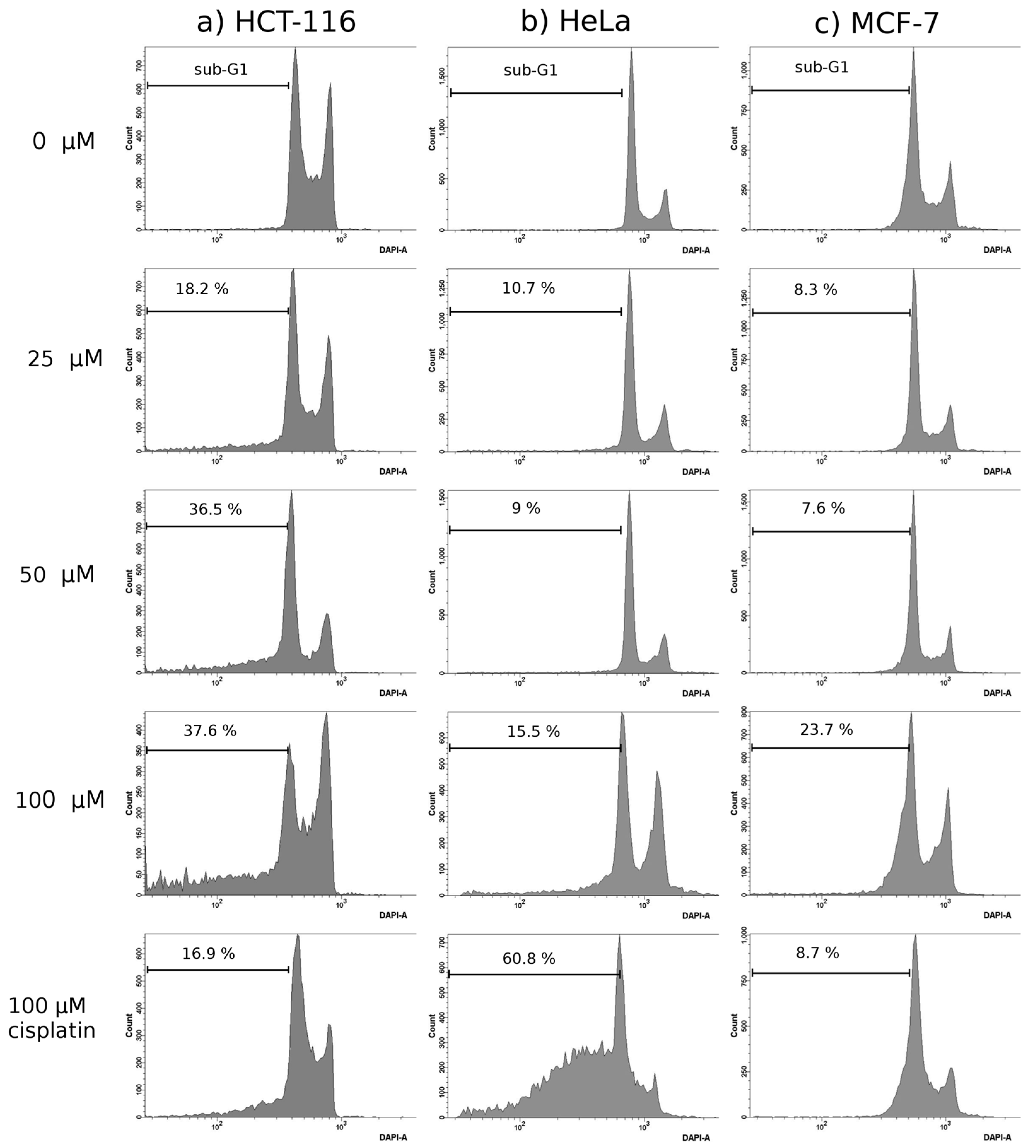
Cell Cycle Analysis
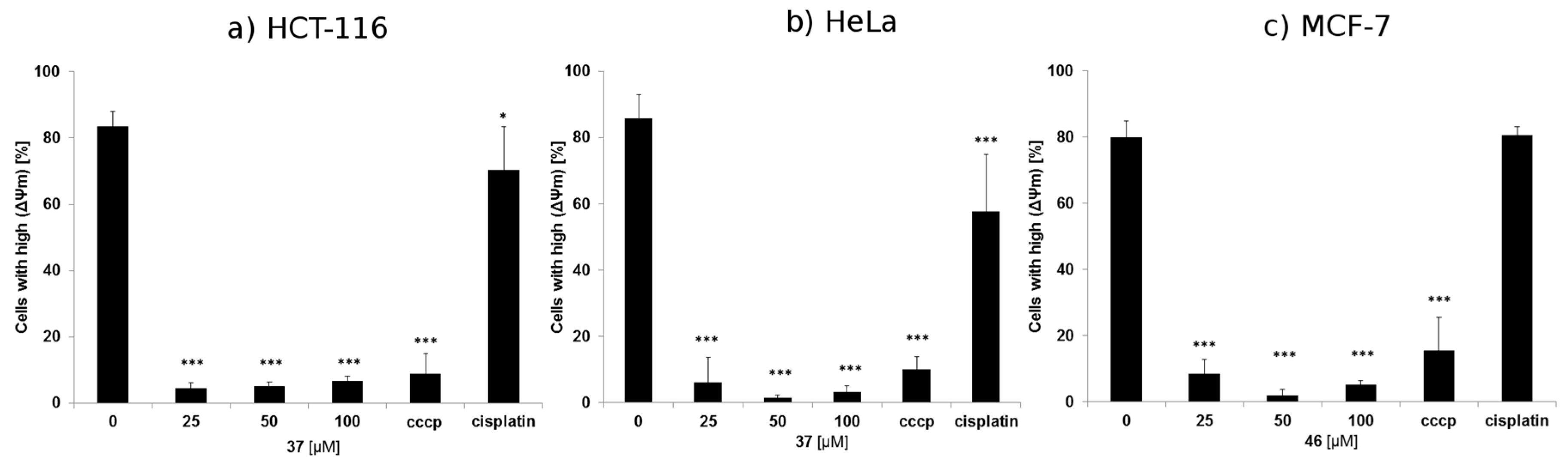
Mitochondrial Membrane Potential (Δψm) Analysis
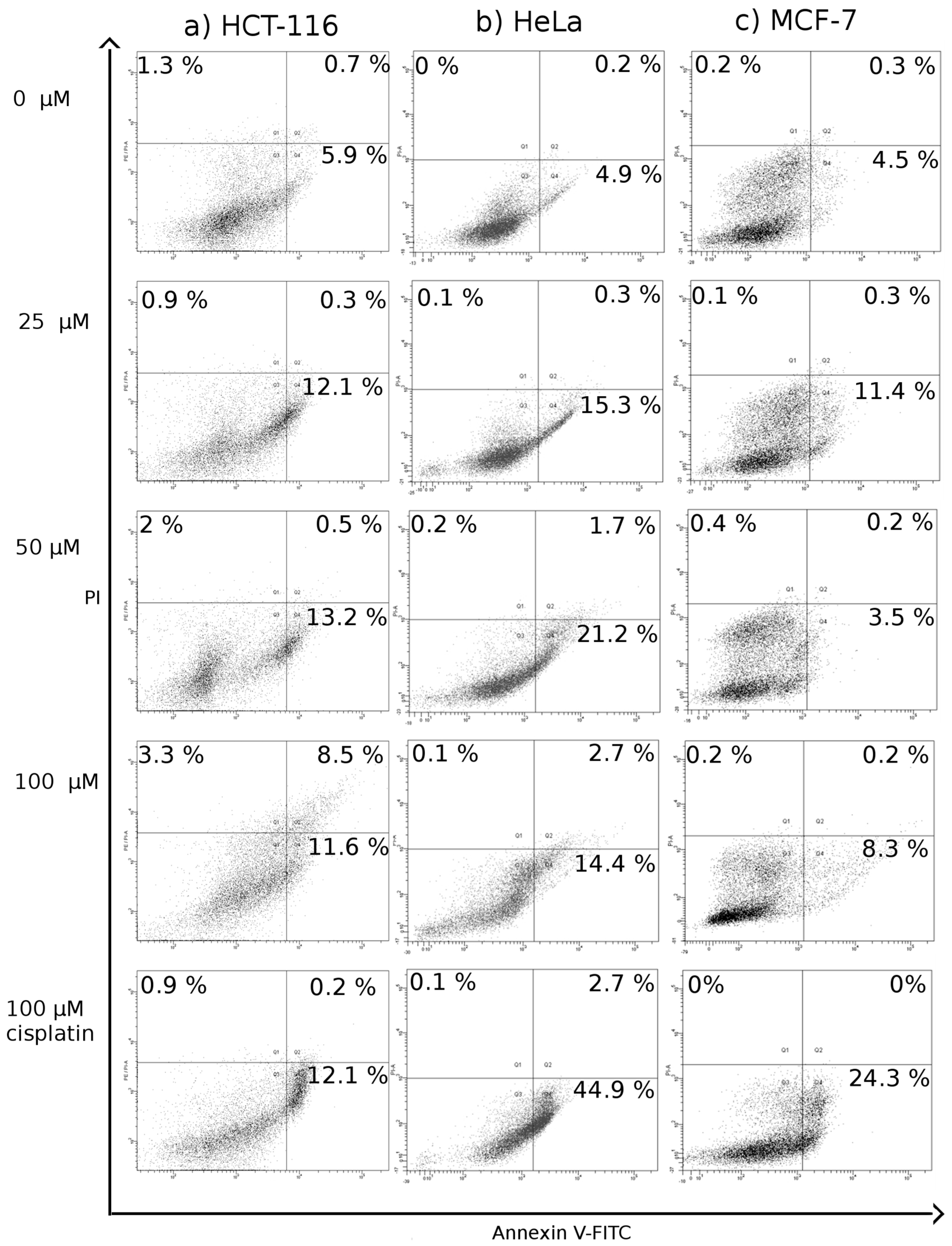
Translocation of Phosphatidylserine to Outer Leaflet of Cell Membrane
Caspase Activation
2.2.3. Metabolic Stability
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Procedures for the Preparation of Aminoguanidines 21–22 and 26
3.2.2. Procedures for the Preparation of 4-Chloro-2-(R2-methylthio)-5-R1-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides 27–60
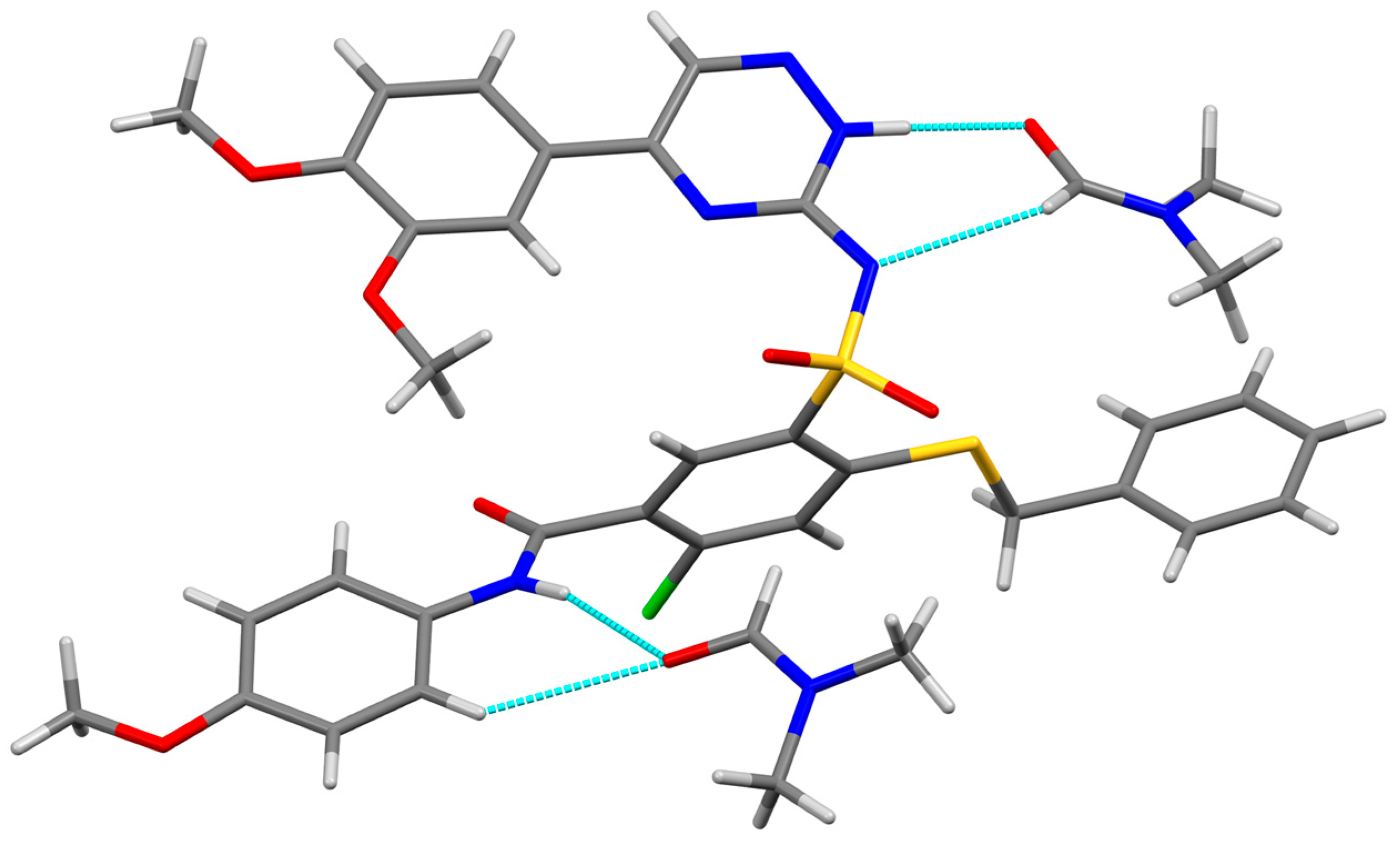
3.3. X-ray Structure Determination
3.4. Cell Culture and Cell Viability Assay
3.4.1. Cell Morphology
3.4.2. Cell Cycle Analysis
3.4.3. Mitochondrial Membrane Potential (Δψm) Analysis
3.4.4. Translocation of Phosphatidylserine to Outer Leaflet of Cell Membrane
3.4.5. Caspase Activity Determination
3.4.6. Statistical Analysis
3.5. Metabolic Stability
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singla, P.; Luxami, V.; Paul, K. Triazine as a promising scaffold for its versatile biological behavior. Eur. J. Med. Chem. 2015, 102, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, A.; Guerrini, G.; Ciciani, G.; Bruni, F.; Selleri, S.; Costa, B.; Martini, C.; Lucacchini, A.; Aiello, P.M.; Ipponi, A. Benzodiazepine receptor ligands. 4. Synthesis and pharmacological evaluation of 3-heteroaryl-8-chloropyrazolo[5,1-c][1,2,4]benzotriazine 5-oxides. J. Med. Chem. 1999, 42, 2218–2226. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, H.; Amini, M.; Khodagholi, F.; Ansari, N.; Tusi, S.K.; Sharifzadeh, M.; Shafiee, A. Synthesis and in vitro evaluation of novel 1,2,4-triazine derivatives as neuroprotective agents. Bioorg. Med. Chem. 2010, 18, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.B.; Babington, R.G.; Deacon, M.A.; Eden, P.L.; Kerestan, S.P.; Leslie, G.H.; Ryan, E.A.; Mason, R.B.; Minor, H.E. A potent, new, sedative-hypnotic agent: 5,7-dihydro-5,5,7,7-tetramethyl-3-(3-nitrophenyl)furo[3,4-e]-as-triazine 4-oxide. J. Med. Chem. 1981, 24, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, G.; Ciciani, G.; Cambi, G.; Bruni, F.; Selleri, S.; Melani, F.; Montali, M.; Martini, C.; Ghelardini, C.; Norcini, M.; et al. Novel 3-aroylpyrazolo[5,1-c][1,2,4]benzotriazine 5-oxides 8-substituted, ligands at GABAA/benzodiazepine receptor complex: Synthesis, pharmacological and molecular modeling studies. Bioorg. Med. Chem. 2008, 16, 4471–4489. [Google Scholar] [CrossRef] [PubMed]
- Szárics, E.; Riedl, Z.; Nyikos, L.; Hajós, G.; Kardos, J. Interaction of novel condensed triazine derivatives with central and peripheral type benzodiazepine receptors: Synthesis, in vitro pharmacology and modelling. Eur. J. Med. Chem. 2006, 41, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Trepanier, D.L.; Shriver, K.L.; Eble, J.N. Aryl-substituted triazines with antidepressant activity. J. Med. Chem. 1969, 12, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lin, J.; Wrobleski, S.T.; Lin, S.; Hynes, J.; Wu, H.; Dyckman, A.J.; Li, T.; Wityak, J.; Gillooly, K.M.; et al. Discovery of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38α MAP kinase inhibitor for the treatment of inflammatory diseases. J. Med. Chem. 2010, 53, 6629–6639. [Google Scholar] [CrossRef] [PubMed]
- Sztanke, K.; Pasternak, K.; Rajtar, B.; Sztanke, M.; Majek, M.; Polz-Dacewicz, M. Identification of antibacterial and antiviral activities of novel fused 1,2,4-triazine esters. Bioorg. Med. Chem. 2007, 15, 5480–5486. [Google Scholar] [CrossRef] [PubMed]
- Kusch, P.; Deininger, S.; Specht, S.; Maniako, R.; Haubrich, S.; Pommerening, T.; Lin, P.K.; Hoerauf, A.; Kaiser, A. In vitro and in vivo antimalarial activity assays of seeds from Balanites aegyptiaca: Compounds of the extract show growth inhibition and activity against Plasmodial Aminopeptidase. J. Parasitol. Res. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Morrey, J.D.; Smee, D.F.; Sidwell, R.W.; Tseng, C. Identification of active antiviral compounds against a New York isolate of West Nile virus. Antivir. Res. 2002, 55, 107–116. [Google Scholar] [CrossRef]
- Centorino, M.B.; Catalano, G.; Catalano, M.C. Lamotrigine induced whole body tics: A case report and literature review. Curr. Drug Saf. 2016, 11, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Grennan, D.M.; McLeod, M.; Watkins, C.; Dick, W.C. Clinical assessment of azapropazone in rheumatoid arthritis. Curr. Med. Res. Opin. 1976, 4, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Patil, U.N.; Jambulingappa, K.L. A combination strategy of Ceftriaxone, Sulbactam and disodium edetate for the treatment of multi-drug resistant (MDR) septicaemia: A retrospective, observational study in Indian Tertiary Care Hospital. J. Clin. Diagn. Res. 2015, 9, FC29–FC32. [Google Scholar] [CrossRef] [PubMed]
- Ucherek, M.M.; Wroblewska, J.; Modzelewska-Banachiewicz, B.; Gospodarek, E. Biological activity of methyl 2-[5-oxo-3,4-di-(2-pirydyl)-1,4,5,6-tetrahydro-1,2,4-triazine-6-ylidene]acetate. Acta Polon. Pharm. Drug Res. 2008, 65, 789–791. [Google Scholar]
- Fahmy, U.A. Nanoethosomal transdermal delivery of vardenafil for treatment of erectile dysfunction: Optimization, characterization, and in vivo evaluation. Drug Des. Dev. Ther. 2015, 9, 6129–6137. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.P.; Pruijn, F.B.; Gamage, S.A.; Liyanage, H.D.; Kovacs, M.S.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A. DNA-targeted 1,2,4-benzotriazine 1,4-dioxide: Potent analogues of the hypoxia selective cytotoxin tirapazamine. J. Med. Chem. 2004, 47, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.R.; Aur, R.J.A.; Hernandez, K.; Vietti, T.; Pinkel, D. 6-Azauridine in combination chemotherapy of childhood acute myelocytic leukemia. Cancer 1972, 29, 1057–1060. [Google Scholar] [CrossRef]
- Saad, H.A.; Moustafa, A.H. Synthesis and anticancer activity of some new S-glycosyl and S-alkyl 1,2,4-triazinone derivatives. Molecules 2011, 16, 5682–5700. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, M.R.; Mohamed, A.A. The Reaction of cyanoacetylhydrazine with ω-bromo(4-methyl)acetophenone: Synthesis of heterocyclic derivatives with antitumor activity. Molecules 2010, 15, 3602–3617. [Google Scholar] [CrossRef] [PubMed]
- Żołnowska, B.; Sławiński, J.; Belka, M.; Bączek, T.; Kawiak, A.; Chojnacki, J.; Pogorzelska, A.; Szafrański, K. Synthesis, molecular structure, metabolic stability and QSAR studies of a novel series of anticancer N-acylbenzenesulfonamides. Molecules 2015, 20, 19101–19129. [Google Scholar] [CrossRef] [PubMed]
- Żołnowska, B.; Sławiński, J.; Pogorzelska, A.; Chojnacki, J.; Vullo, D.; Supuran, C.T. Carbonic anhydrase inhibitors. Synthesis, and molecular structure of novel series N-substituted N’-(2-arylmethylthio-4-chloro-5-methylbenzenesulfonyl)guanidines and their inhibition of human cytosolic isozymes I and II and the transmembrane tumor-associated isozymes IX and XII. Eur. J. Med. Chem. 2014, 71, 135–147. [Google Scholar]
- Sławiński, J.; Brzozowski, Z.; Żołnowska, B.; Szafrański, K.; Pogorzelska, A.; Vullo, D.; Supuran, C.T. Synthesis of a new series of N4-substituted 4-(2-aminoethyl)benzenesulfonamides and their inhibitory effect on human carbonic anhydrase cytosolic isozymes I and II and transmembrane tumor-associated isozymes IX and XII. Eur. J. Med. Chem. 2014, 84, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Sławiński, J.; Żołnowska, B.; Orlewska, C.; Chojnacki, J. Synthesis and molecular structure of novel 2-(alkylthio)-4-chloro-N-(4,5-dihydro-5-oxo-1H-1,2,4-triazol-3-yl)-5-methylbenzenesulfonamides with potential anticancer activity. Mon. Chem. 2012, 143, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Brożewicz, K.; Sławiński, J. Synthesis and in vitro activity of novel 2-(benzylthio)-4-chloro-5-(1,3,4-oxadiazol-2-yl)benzenesulfonamide derivatives. Mon. Chem. 2012, 143, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Brożewicz, K.; Sławiński, J. 1-(2-Mercaptobenzenesulfonyl)-3-hydroxyguanidines—Novel potent antiproliferatives, synthesis and in vitro biological activity. Eur. J. Med. Chem. 2012, 55, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Sławiński, J.; Brożewicz, K.; Fruziński, A.; Główka, M.L. Synthesis and antitumor activity of novel N′-(2-benzylthiobenzenesulfonyl)-1H-pyrazole-1-amidine derivatives. Heterocycles 2011, 83, 1093–1109. [Google Scholar] [CrossRef]
- Sławiński, J.; Gdaniec, M. Synthesis, molecular structure and in vitro antitumor activity of new 4-chloro-2-mercaptobenzenesulfonamide derivatives. Eur. J. Med. Chem. 2005, 40, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, Z.; Sławiński, J. 1,1-Dioxo-1,4,2-benzodithiazine derivatives. I. Synthesis of various 7-carboxy-3-mercapto-1,1-dioxo-1,4,2-benzodithiazine. Acta Polon. Pharm. 1984, 41, 5–13. [Google Scholar]
- Brzozowski, Z.; Sławiński, J. 1,1-Dioxo-1,4,2-benzodithiazine derivatives. II Synthesis of some 3-mercapto-1,1-dioxo-1,4,2-benzodithiazine derivatives. Acta Polon. Pharm. 1984, 41, 133–139. [Google Scholar]
- Sławiński, J. Syntheses and Some Reactions of 3-Amino-6-choro-7-methyl-1,1-dioxo-1,4,2-benzodithiazine. Pol. J. Chem. 2001, 75, 1309–1316. [Google Scholar]
- Brzozowski, Z.; Sławiński, J. Syntheses and some reactions of 3-amino-6-chloro-7-methyl-1,1-dioxo-1,4,2-benzodithiazine. Pol. J. Chem. 2007, 81, 1419–1426. [Google Scholar]
- Sławiński, J.; Żołnowska, B.; Pirska, D.; Kędzia, A.; Kwapisz, E. Synthesis of 2-(4-chloro-2-mercaptobenzenesulfonyl)-3,5-dihydroxy-1,1-dioxo-2H-1,2,4,6-thiatriazine derivatives with potential anti-HIV and anticancer activities. J. Enzyme Inhib. Med. Chem. 2013, 28, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Sławiński, J.; Pogorzelska, A.; Żołnowska, B.; Kędzia, A.; Ziółkowska-Klinkosz, M.; Kwapisz, E. Synthesis and anti-yeast evaluation of novel 2-alkylthio-4-chloro-5-methyl-N-[imino-(1-oxo-(1H)-phthalazin-2-yl)methyl]benzenesulfonamide derivatives. Molecules 2014, 19, 13704–13723. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Modi, A.; Narayan, G.; Singh, S.K. Benzothiazole derivatives bearing amide moiety: Potential cytotoxic and apoptosis-inducing agents against cervical cancer. Anticancer Drugs 2016, 27, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Perchellet, E.M.; Ward, M.M.; Lou, K.; Hua, D.H.; Perchellet, J.P. Rapid collapse of mitochondrial transmembrane potential in HL-60 cells and isolated mitochondria treated with anti-tumor 1,4-anthracenediones. Anticancer Drugs 2005, 16, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Kassel, D.B. Applications of high-throughput ADME in drug discovery. Curr. Opin. Chem. Biol. 2004, 8, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Korfmacher, W.A. Advances in the integration of drug metabolism into the lead optimization paradigm. Mini Rev. Med. Chem. 2009, 9, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Baranczewski, P.; Stanczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar] [PubMed]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- STOE & Cie GmbH. X-Area 1.75, Software Package for Collecting Single-Crystal Data on STOE Area-Detector Diffractometers, for Image Processing, Scaling Reflection Intensities and for Outlier Rejection; STOE & Cie: Darmstadt, Germany, 2015. [Google Scholar]
- Blessing, R.H. Routines employed in the SORTAV program for identifying and downweighting mismeasured outliers in merging multiply measured data sets are described. J. Appl. Cryst. 1997, 30, 421–426. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Stasiłojć, G.; Pinto, S.; Wyszkowska, R.; Wejda, M.; Słomińska, E.M.; Filipska, M.; Koszałka, P.; Swierczyński, J.; O’Connor, J.E.; Bigda, J.J. U937 variant cells as a model of apoptosis without cell disintegration. Cell. Mol. Biol. Lett. 2013, 18, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds 21–22, 26–60 are available from the authors.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C38H42ClN7O8S2 |
|---|---|
| Formula weight | 824.35 |
| Temperature | 120(2) K |
| Wavelength | 1.54186 Å |
| Crystal system | Triclinic |
| Space group | |
| Unit cell dimensions | a = 7.8336(6) Å, α = 84.84(3)° |
| b = 22.4284(8) Å, β = 86.95(4)° | |
| c = 22.2739(9) Å, γ = 85.13(5)° | |
| Volume | 3879.4(5) Å3 |
| Z | 4 |
| Density (calculated) | 1.411 Mg/m3 |
| Absorption coefficient | 2.399 mm−1 |
| F(000) | 1728 |
| Crystal size | 0.03 × 0.03 × 0.25 mm3 |
| Theta range for data collection | 1.984 to 64.981° |
| Index ranges | −8 ≤ h ≤ 8, −23 ≤ k ≤ 26, −25 ≤ l ≤ 23 |
| Reflections collected | 19645 |
| Independent reflections | 11676 [R(int) = 0.1757] |
| Completeness to θ = 67.686° | 83.1% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 11,676/0/1014 |
| Goodness-of-fit on F2 | 1.253 |
| Final R indices [I > 2σ(I)] | R1 = 0.1476, wR2 = 0.3359 |
| R indices (all data) | R1 = 0.2313, wR2 = 0.4260 |
| Extinction coefficient | 0.0114(11) |
| Compound | IC50 (μM) | ||
|---|---|---|---|
| HCT-116 | HeLa | MCF-7 | |
| 27 | 75 ± 1 | 84 ± 1 | 80 ± 2 |
| 28 | 200 ± 4 | 100 ± 6 | 120 ± 5 |
| 29 | 720 ± 36 | 190 ± 7 | 110 ± 2 |
| 30 | 85 ± 2 | 90 ± 4 | 67 ± 1 |
| 31 | 140 ± 7 | 150 ± 6 | 77 ± 2 |
| 32 | 88 ± 3 | 87 ± 4 | 87 ± 4 |
| 33 | 67 ± 1 | 76 ± 3 | 88 ± 1 |
| 34 | 51 ± 1 | 73 ± 1 | 68 ± 3 |
| 35 | 68 ± 3 | 69 ± 1 | 78 ± 2 |
| 36 | 49 ± 2 | 55 ± 2 | 96 ± 4 |
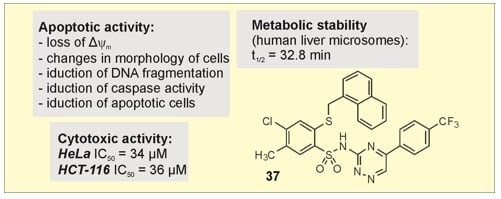
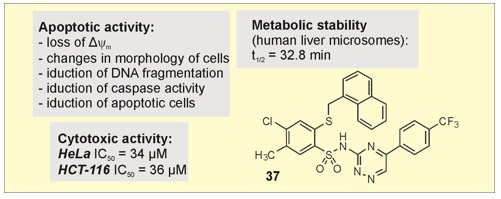
| 37 | 36 ± 1 | 34 ± 2 | 70 ± 3 |
| 38 | 38 ± 2 | 42 ± 1 | 69 ± 1 |
| 39 | 83 ± 2 | 93 ± 1 | 95 ± 3 |
| 40 | 77 ± 1 | 85 ± 2 | 98 ± 3 |
| 41 | 97 ± 2 | 85 ± 1 | 99 ± 1 |
| 42 | 77 ± 2 | 82 ± 1 | 83 ± 2 |
| 43 | 92 ± 3 | 130 ± 10 | 87 ± 2 |
| 44 | 160 ± 6 | 160 ± 8 | 90 ± 5 |
| 45 | 97 ± 2 | 82 ± 1 | 90 ± 1 |
| 46 | 64 ± 1 | 80 ± 2 | 59 ± 2 |
| 47 | 75 ± 5 | 79 ± 1 | 78 ± 3 |
| 48 | 80 ± 3 | 80 ± 2 | 87 ± 3 |
| 49 | 110 ± 2 | 120 ± 6 | 81 ± 5 |
| 50 | 115 ± 3 | 130 ± 8 | 120 ± 4 |
| 51 | 190 ± 13 | * | 95 ± 5 |
| 52 | 70 ± 2 | * | 69 ± 1 |
| 53 | 76 ± 2 | 74 ± 2 | 88 ± 2 |
| 54 | 84 ± 5 | 82 ± 1 | 96 ± 5 |
| 55 | 150 ± 3 | 105 ± 4 | 150 ± 6 |
| 56 | 75 ± 2 | 80 ± 2 | 89 ± 3 |
| 57 | 73 ± 1 | 80 ± 1 | 90 ± 1 |
| 58 | 98 ± 2 | 88 ± 6 | 92 ± 6 |
| 59 | 140 ± 5 | 150 ± 2 | 100 ± 5 |
| 60 | 230 ± 18 | 230 ± 5 | 270 ± 13 |
| Cisplatin | 3.8 ± 0.2 | 2.2 ± 0.2 | 3 ± 0.1 |
| Compound | In Vitro Metabolic Half-Life t1/2 (min) |
|---|---|
| 30 | 24.9 |
| 31 | 17.6 |
| 34 | >60 (154) |
| 35 | 42.4 |
| 36 | 13.9 |
| 37 | 32.8 |
| 38 | <5 |
| 46 | >60 (78.1) |
| 47 | 17.5 |
| 52 | 26.5 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żołnowska, B.; Sławiński, J.; Pogorzelska, A.; Szafrański, K.; Kawiak, A.; Stasiłojć, G.; Belka, M.; Ulenberg, S.; Bączek, T.; Chojnacki, J. Novel 5-Substituted 2-(Aylmethylthio)-4-chloro-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides: Synthesis, Molecular Structure, Anticancer Activity, Apoptosis-Inducing Activity and Metabolic Stability. Molecules 2016, 21, 808. https://doi.org/10.3390/molecules21060808
Żołnowska B, Sławiński J, Pogorzelska A, Szafrański K, Kawiak A, Stasiłojć G, Belka M, Ulenberg S, Bączek T, Chojnacki J. Novel 5-Substituted 2-(Aylmethylthio)-4-chloro-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides: Synthesis, Molecular Structure, Anticancer Activity, Apoptosis-Inducing Activity and Metabolic Stability. Molecules. 2016; 21(6):808. https://doi.org/10.3390/molecules21060808
Chicago/Turabian StyleŻołnowska, Beata, Jarosław Sławiński, Aneta Pogorzelska, Krzysztof Szafrański, Anna Kawiak, Grzegorz Stasiłojć, Mariusz Belka, Szymon Ulenberg, Tomasz Bączek, and Jarosław Chojnacki. 2016. "Novel 5-Substituted 2-(Aylmethylthio)-4-chloro-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides: Synthesis, Molecular Structure, Anticancer Activity, Apoptosis-Inducing Activity and Metabolic Stability" Molecules 21, no. 6: 808. https://doi.org/10.3390/molecules21060808
APA StyleŻołnowska, B., Sławiński, J., Pogorzelska, A., Szafrański, K., Kawiak, A., Stasiłojć, G., Belka, M., Ulenberg, S., Bączek, T., & Chojnacki, J. (2016). Novel 5-Substituted 2-(Aylmethylthio)-4-chloro-N-(5-aryl-1,2,4-triazin-3-yl)benzenesulfonamides: Synthesis, Molecular Structure, Anticancer Activity, Apoptosis-Inducing Activity and Metabolic Stability. Molecules, 21(6), 808. https://doi.org/10.3390/molecules21060808