
Marine Natural Products as Models to Circumvent Multidrug Resistance





Abstract
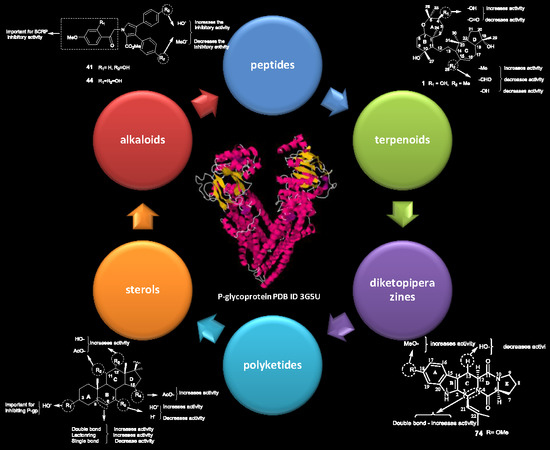
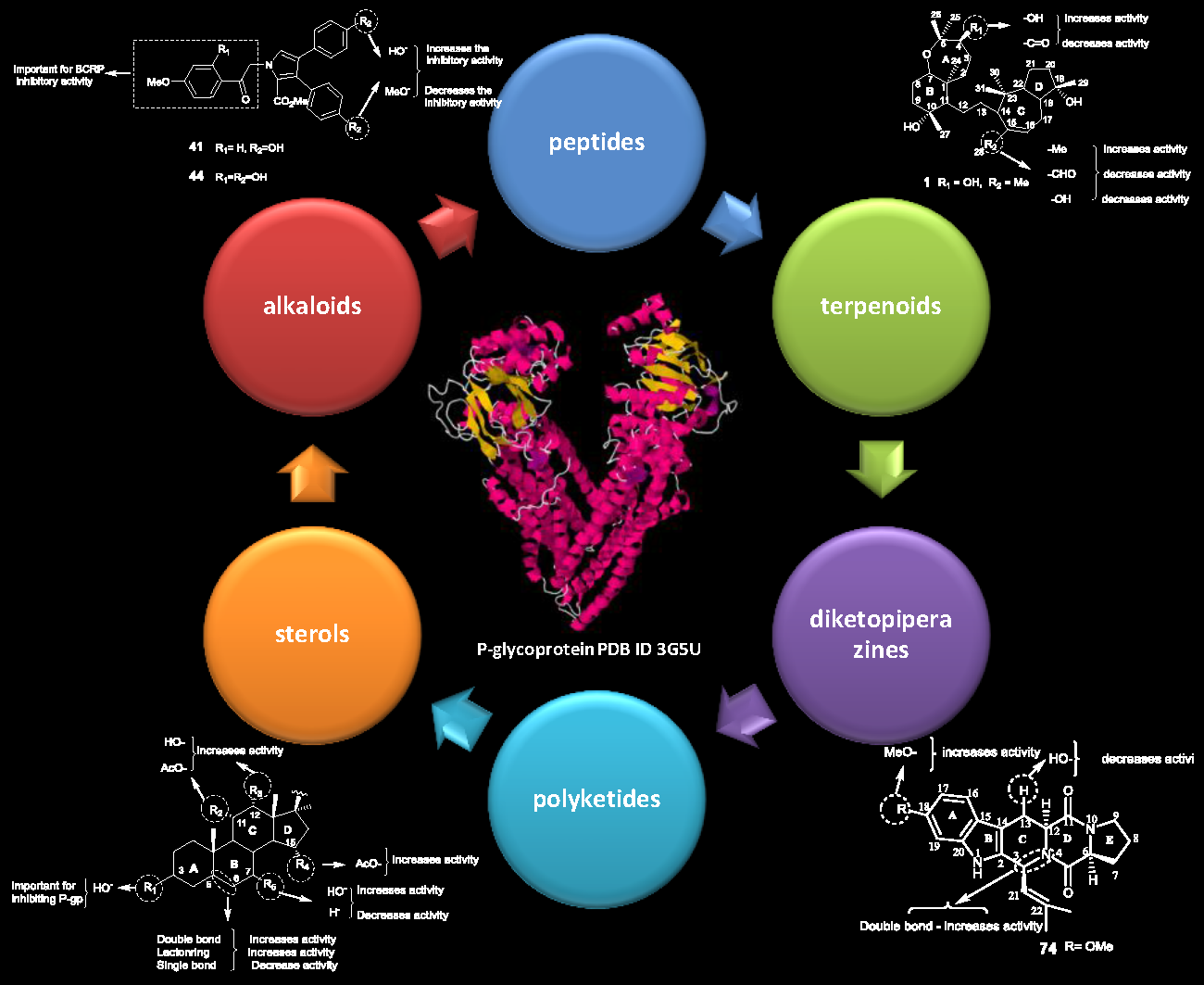
:
{kind=link}
{kind=link}
{kind=link}
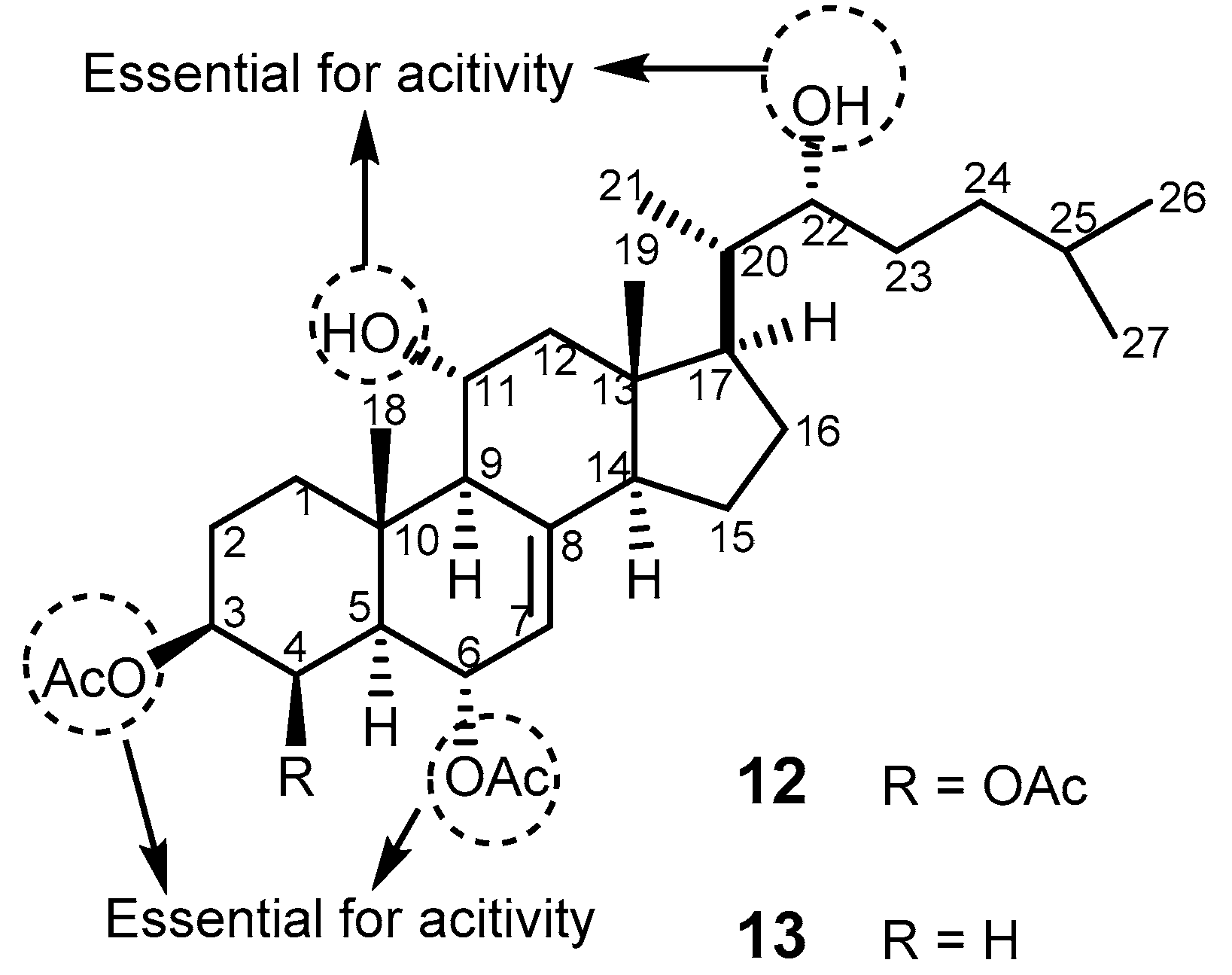{kind=link}
{kind=link}
{kind=link}
{kind=link}
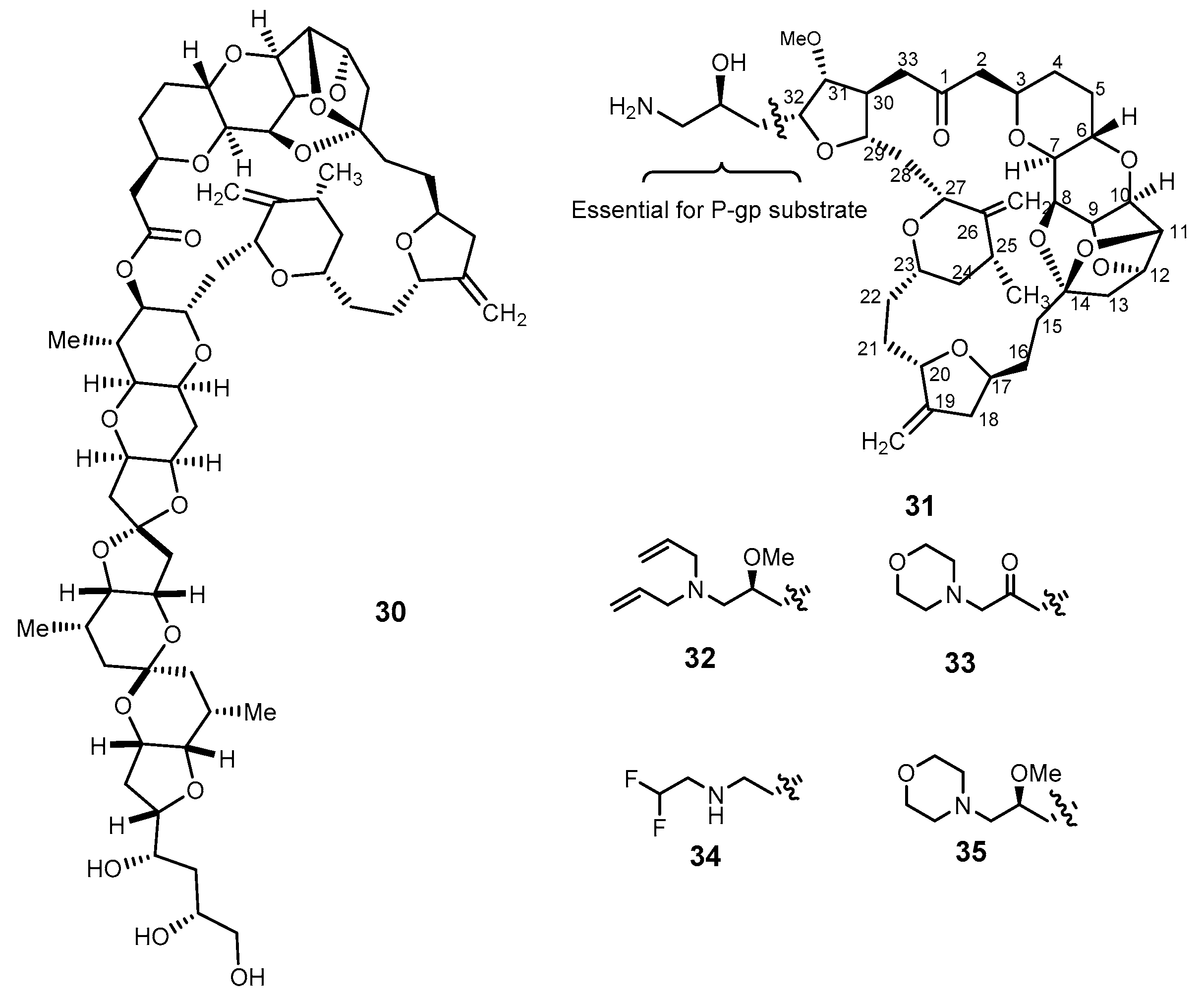{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
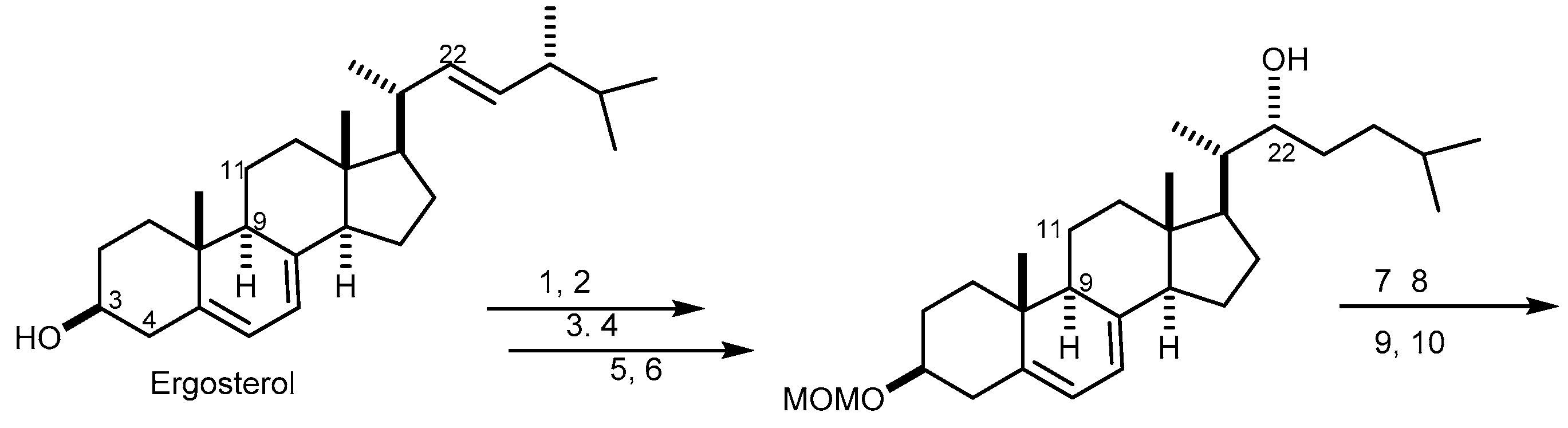{kind=link}
{kind=link}
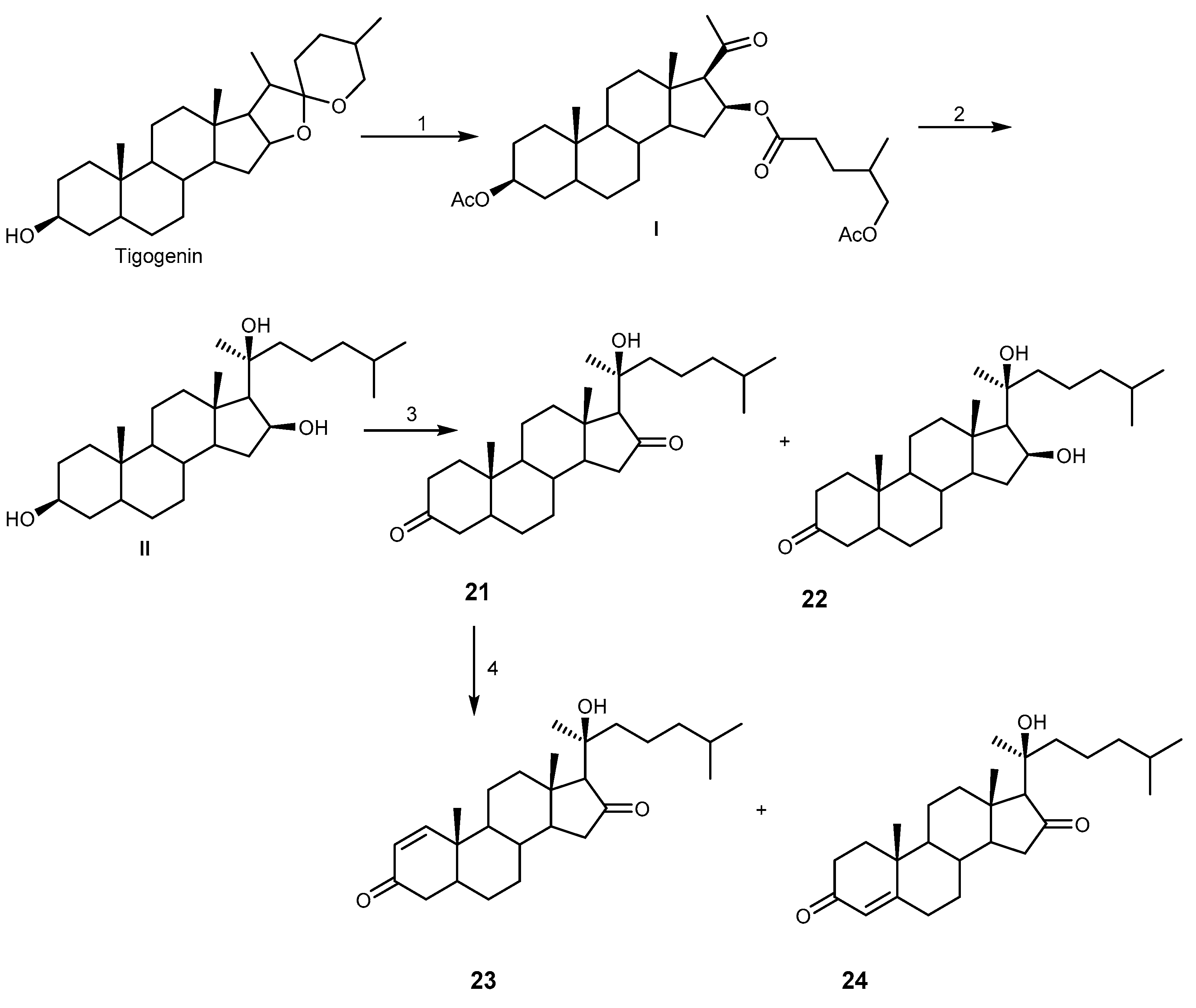{kind=link}
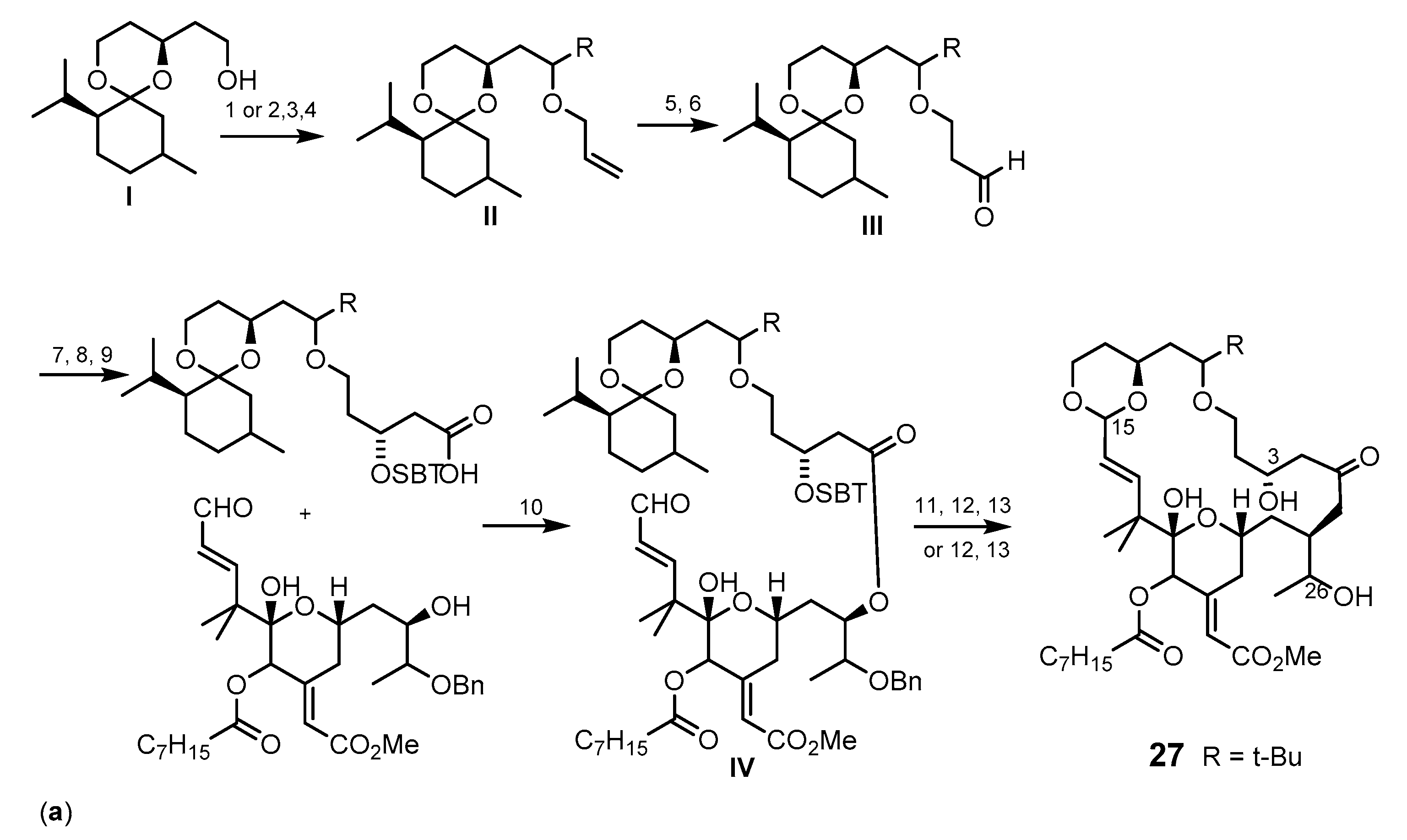{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
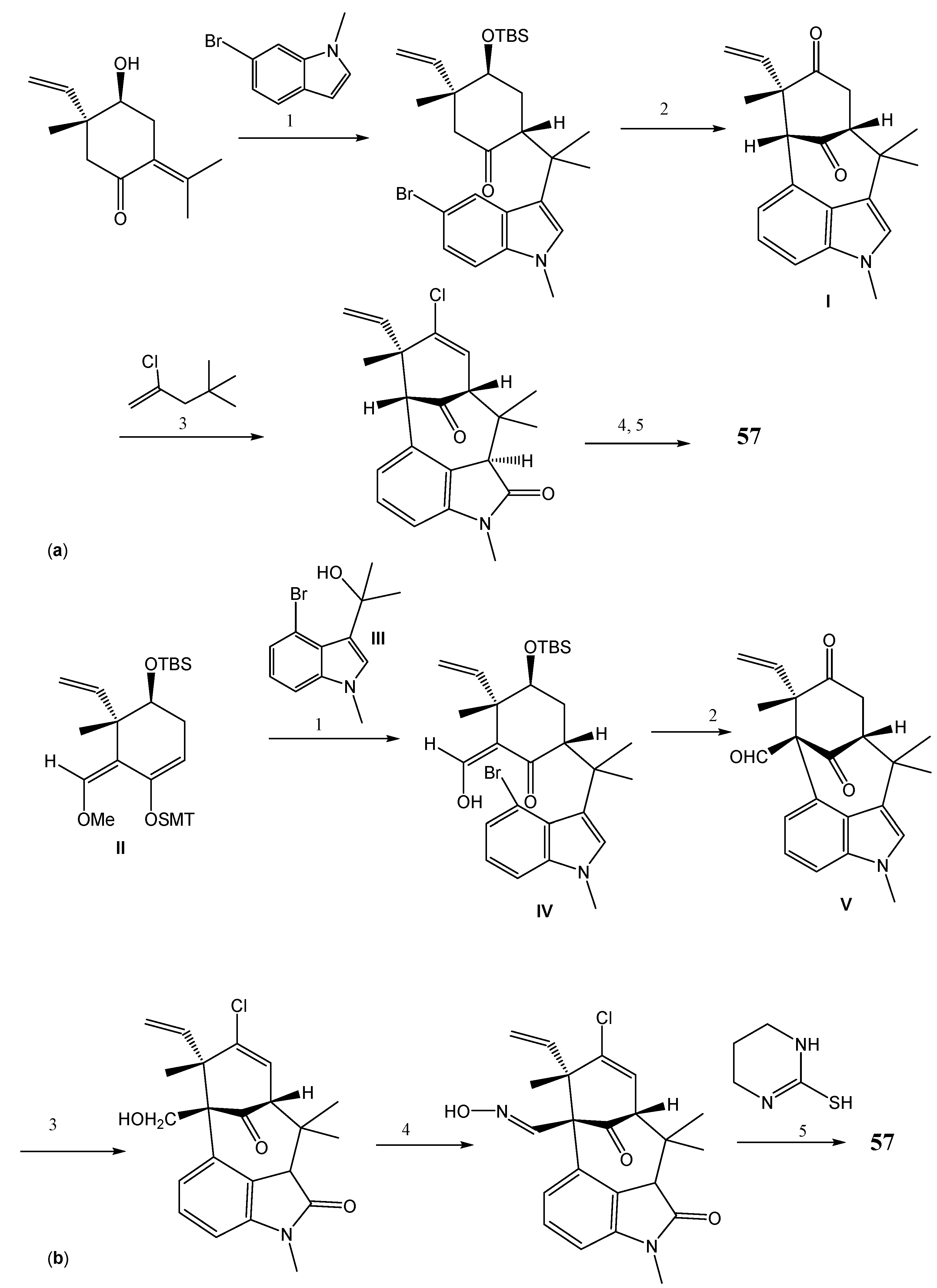{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Marine Natural Products and Derivatives as Inhibitors of ABC Transporters
2.1. Terpenoids
2.1.1. Sipholane Triterpenoids and Derivatives
2.1.2. Parguerenes and Derivatives
2.2. Sterols
2.2.1. Agosterol and Derivatives
2.2.2. Polyoxygenated Steroids and Derivatives
2.3. Polyketides
2.3.1. Bryostatin and Derivatives
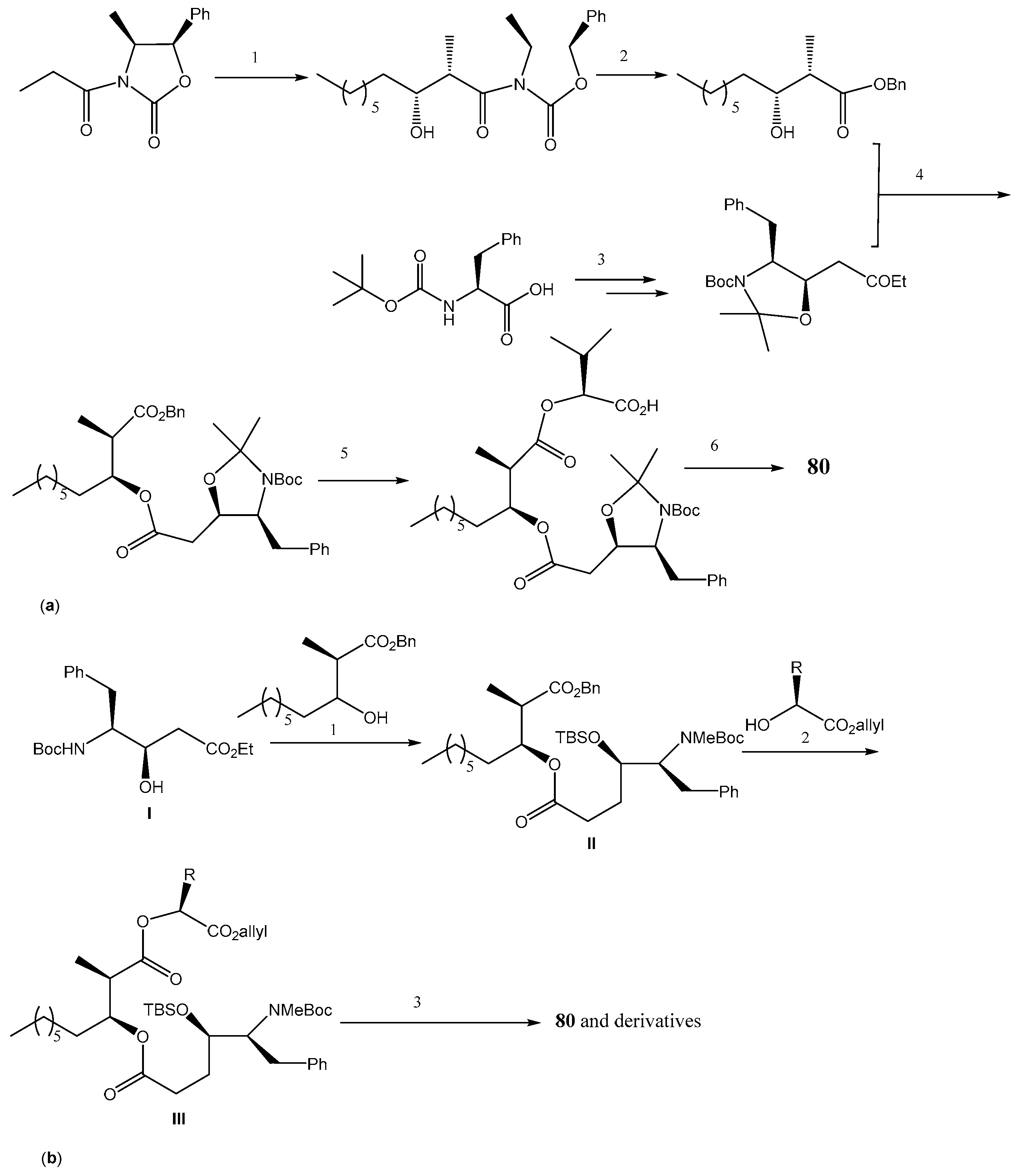
2.3.2. Discodermolide
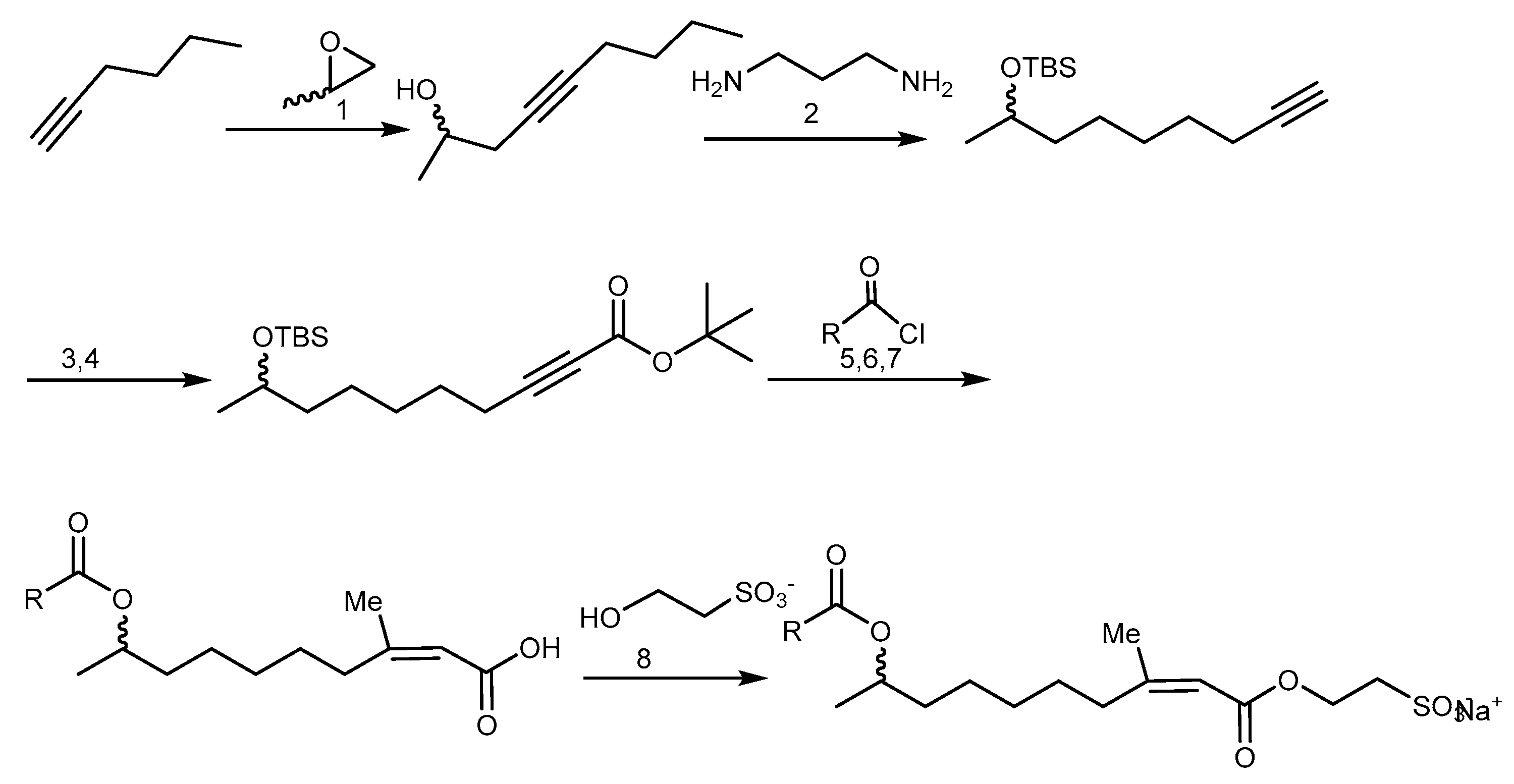
2.3.3. Halicondrin B and Derivatives
2.4. Alkaloids
2.4.1. Lamellarins and Derivatives
2.4.2. Ningalins and Derivatives
2.4.3. Welwitindolinones and Derivatives
2.4.4. Harmine and Derivatives
2.4.5. Indolcarbazoles and Derivatives
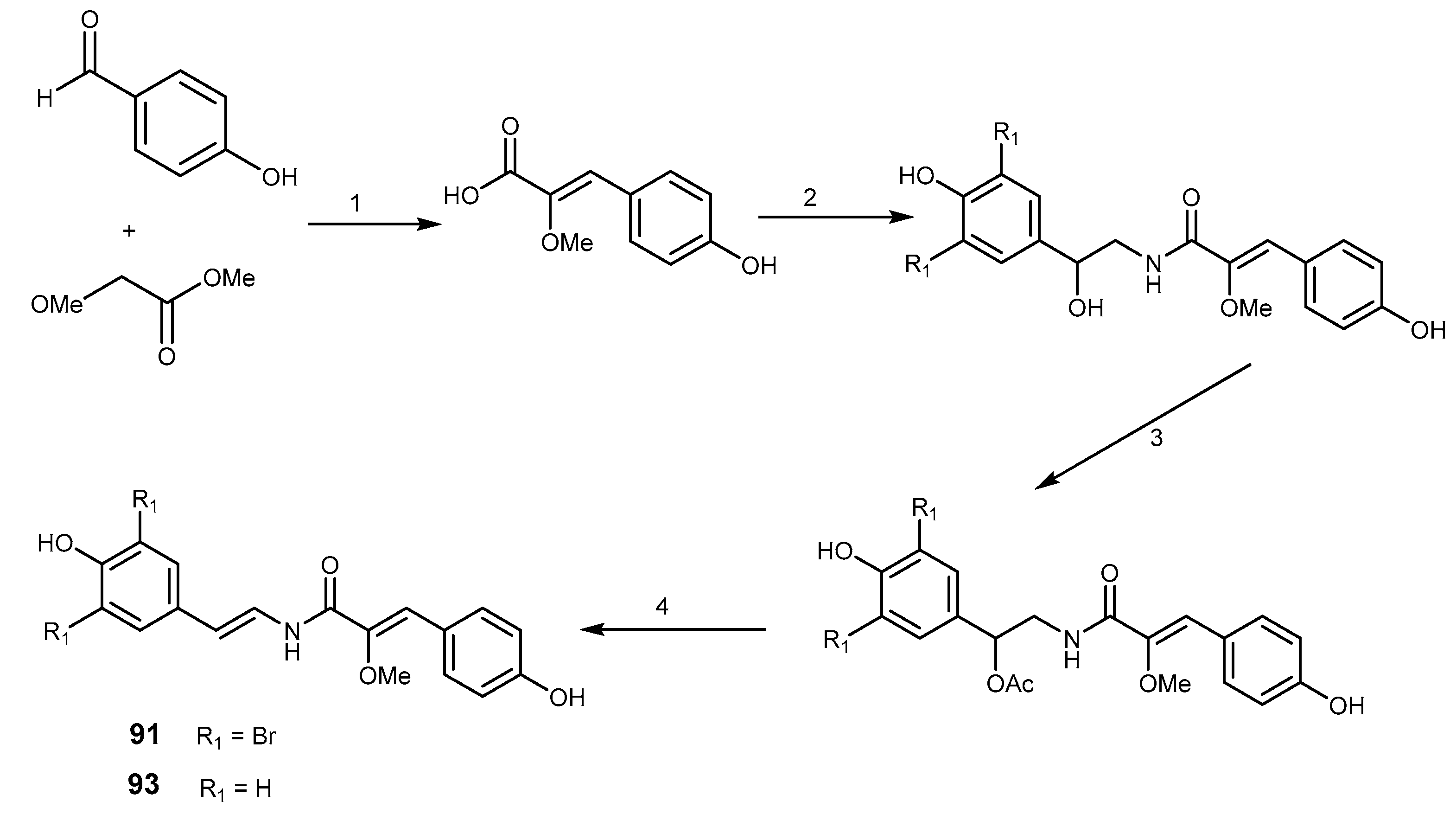
2.4.6. Ecteinascidin 743 (Trabectedin) and Derivatives
2.5. Diketopiperazines
2.5.1. Nocardioazines
2.5.2. Fumitremorgins and Derivatives
2.5.3. Halimide and Derivatives
2.6. Peptides
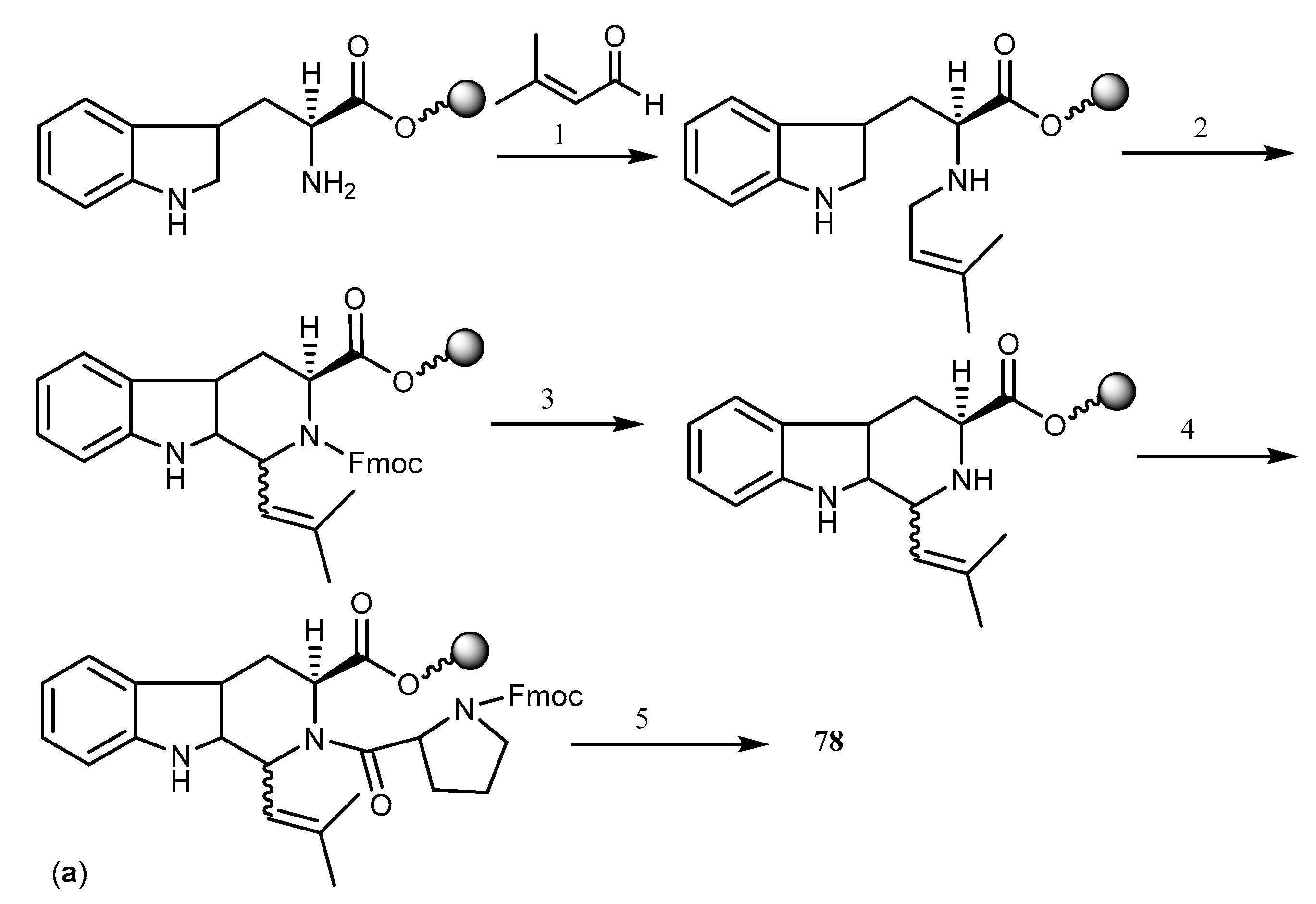
2.6.1. Hapalosin and Derivatives
2.6.2. Botryllamides and Derivatives
2.6.3. Kendarimide
2.6.4. Patellamide and Derivatives
2.7. Miscellaneous
2.7.1. Irciniasulfonic Acid Derivatives
2.7.2. Secalonic Acid D
2.7.3. Terrein
2.7.4. Shornephine A
3. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cancer. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 16 March 2016).
- Che, X.F.; Nakajima, Y.; Sumizawa, T.; Ikeda, R.; Ren, X.Q.; Zheng, C.L.; Mukai, M.; Furukawa, T.; Haraguchi, M.; Gao, H.; et al. Reversal of p-glycoprotein mediated multidrug resistance by a newly synthesized 1,4-benzothiazipine derivative, jtv-519. Cancer Lett. 2002, 187, 111–119. [Google Scholar] [CrossRef]
- Gustav, L. P-glycoprotein as a drug target in the treatment of multidrug resistant cancer. Curr. Drug Targets 2000, 1, 85–99. [Google Scholar]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Kim, I.-W.; Sauna, Z.E. The power of the pump: Mechanisms of action of P-glycoprotein (ABCB1). Eur. J. Pharm. Sci. 2006, 27, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar] [PubMed]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide d, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Li, X.; Yuan, H.; Wu, J.; Li, J.; Qu, X.; Xu, W.; Tang, W. Strategies to overcome or circumvent P-glycoprotein mediated multidrug resistance. Curr. Med. Chem. 2008, 15, 470–476. [Google Scholar] [CrossRef]
- Li, X.; Li, J.P.; Yuan, H.Y.; Gao, X.; Qu, X.J.; Xu, W.F.; Tang, W. Recent advances in P-glycoprotein-mediated multidrug resistance reversal mechanisms. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Baguley, B.C. Multiple drug resistance mechanisms in cancer. Mol. Biotechnol. 2010, 46, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.-P.; Gottesman, M.M. Overcoming multidrug resistance in cancer: 35 years after the discovery of abcb1. Drug Resist. Updates 2012, 15, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Armstrong, M.A.; Oehler, M.K.; Ricciardelli, C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit. Rev. Oncol. Hematol. 2015, 2, 220–256. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P.C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Seca, H.; Sousa, D.; Sousa, E.; Lima, R.T.; Vasconcelos, M.H. The network of P-glycoprotein and micrornas interactions. Int. J. Cancer 2014, 135, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Gibson, B.; Smith, F.O. Acute myeloid leukemia. Pediatr. Clin. N. Am. 2008, 55, 21–51. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, D.; Legrand, O. ABC transporters and drug resistance in leukemia: Was P-gp nothing but the first head of the hydra? Leukemia 2007, 21, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E. How I treat pediatric acute myeloid leukemia. Blood 2012, 119, 5980–5988. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Botterill, V.; Wodinsky, I. Uptake and retention of daunomycin by mouse leukemic cells as factors in drug response. Cancer Res. 1968, 28, 938–941. [Google Scholar] [PubMed]
- Srivastava, S.; Choudhary, B.S.; Sharma, M.; Malik, R. Pharmacophore modeling and 3D-QSAR studies of galloyl benzamides as potent P-gp inhibitors. Med. Chem. Res. 2016, 25, 1140–1147. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Perez-Victorias, F.J.; Conseil, G.; Munoz-Martinez, F.; Perez-Victoria, J.M.; Dayan, G.; Marsaud, V.; Castanys, S.; Gamarro, F.; Renoir, J.M.; di Pietro, A. Ru49953: A non-hormonal steroid derivative that potently inhibits P-glycoprotein and reverts cellular multidrug resistance. Cell. Mol. Life Sci. 2003, 60, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Modok, S.; Mellor, H.R.; Callaghan, R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr. Opin. Pharmacol. 2006, 6, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer—Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Coley, H.M. Overcoming multidrug resistance in cancer: Clinical studies of P-glycoprotein inhibitors. In Multi-Drug Resistance in Cancer; Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 341–358. [Google Scholar]
- Discontinuation of two phase III trials of tariquidar in non-small-cell lung cancer. Inpharma Wkly 2013, 1387, 10. [CrossRef]
- Wandel, C.; Kim, R.B.; Kajiji, S.; Guengerich, F.P.; Wilkinson, G.R.; Wood, A.J.J. P-glycoprotein and cytochrome p-450 3a inhibition: Dissociation of inhibitory potencies. Cancer Res. 1999, 59, 3944–3948. [Google Scholar] [PubMed]
- Cripe, L.D.; Uno, H.; Paietta, E.M.; Litzow, M.R.; Ketterling, R.P.; Bennett, J.M.; Rowe, J.M.; Lazarus, H.M.; Luger, S.; Tallman, M.S. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: A randomized, placebo-controlled trial of the eastern cooperative oncology group 3999. Blood 2010, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, J.; Ji, C. Quercetin: A potential drug to reverse multidrug resistance. Life Sci. 2010, 87, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Cherigo, L.; Lopez, D.; Martinez-Luis, S. Marine natural products as breast cancer resistance protein inhibitors. Mar. Drugs 2015, 13, 2010–2029. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can some marine-derived fungal metabolites become actual anticancer agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Bekeredjian, R.; Schmidt, J.; Büchler, M.W.; Märten, A. Effects of the high-affinity peptide reversin 121 on multidrug resistance proteins in experimental pancreatic cancer. Tumor Biol. 2009, 29, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Bogman, K.; Erne-Brand, F.; Alsenz, J.; Drewe, J. The role of surfactants in the reversal of active transport mediated by multidrug resistance proteins. J. Pharm. Sci. 2003, 92, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Capon, R.J. Marine bioprospecting—Trawling for treasure and pleasure. Eur. J. Org. Chem. 2001, 633–645. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhu, T.; Gu, Q.; Li, D. Advanced tools in marine natural drug discovery. Curr. Opin. Biotechnol. 2016, 42, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Martinez-Luis, S. Marine natural products with P-glycoprotein inhibitor properties. Mar. Drugs 2014, 12, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; El Sayed, K.; Chen, Z.S.; Guo, H. Current status on marine products with reversal effect on cancer multidrug resistance. Mar. Drugs 2012, 10, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updates 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Stonik, V.A.; Fedorov, S.N. Marine low molecular weight natural products as potential cancer preventive compounds. Mar. Drugs 2014, 12, 636–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.K.; Wang, Y.J.; Vispute, S.G.; Jain, S.; Chen, Y.; Li, J.; Youssef, D.T.A.; El Sayed, K.A.; Chen, Z.S. Esters of the marine-derived triterpene sipholenol a reverse P-gp-mediated drug resistance. Mar. Drugs 2015, 13, 2267–2286. [Google Scholar] [CrossRef] [PubMed]
- Shanthi, J.; Senthil, A.; Gopikrishnan, V.; Balagurunathan, R. Characterization of a potential β-lactamase inhibitory metabolite from a marine streptomyces sp. Pm49 active against multidrug-resistant pathogens. Appl. Biochem. Biotechnol. 2015, 175, 3696–3708. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Kumar, P.; Anreddy, N.; Xiao, X.; Yang, D.H.; Chen, Z.S. P-gp inhibitory activity from marine sponges, tunicates and algae. In Handbook of Anticancer Drugs from Marine Origin; Springer: Berlin, Germany, 2015; pp. 593–619. [Google Scholar]
- Dinić, J.; Podolski-Renić, A.; Stanković, T.; Banković, J.; Pešić, M. New approaches with natural product drugs for overcoming multidrug resistance in cancer. Curr. Pharm. Design 2015, 21, 5589–5604. [Google Scholar] [CrossRef]
- Patel, A.; Wang, D.-S.; Sim, H.-M.; Ambudkar, S.V.; Chen, Z.-S. ABC transporter modulatory drugs from marine sources: A new approach to overcome drug resistance in cancer. In Resistance to Targeted abc Transporters in Cancer; Efferth, T., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 183–208. [Google Scholar]
- Shi, Z.; Jain, S.; Kim, I.W.; Peng, X.X.; Abraham, I.; Youssef, D.T.A.; Fu, L.W.; El Sayed, K.; Ambudkar, S.V.; Chen, Z.S. Sipholenol a, a marine-derived sipholane triterpene, potently reverses P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Cancer Sci. 2007, 98, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Jain, S.; Wu, C.P.; Khanfar, M.A.; Kuang, Y.; Dai, C.L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.R.; Foudah, A.I.; Ebrahim, H.Y.; Meyer, S.A.; El Sayed, K.A. The marine-derived sipholenol A-4-O-3′,4′-dichlorobenzoate inhibits breast cancer growth and motility in vitro andin vivo through the suppression of brk and fak signaling. Mar. Drugs 2014, 12, 2282–2304. [Google Scholar] [CrossRef] [PubMed]
- Foudah, A.I.; Sallam, A.A.; Akl, M.R.; El Sayed, K.A. Optimization, pharmacophore modeling and 3D-QSAR studies of sipholanes as breast cancer migration and proliferation inhibitors. Eur. J. Med. Chem. 2014, 73, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Laphookhieo, S.; Shi, Z.; Fu, L.W.; Akiyama, S.I.; Chen, Z.S.; Youssef, D.T.A.; van Soest, R.W.M.; El Sayed, K.A. Reversal of p-glycoprotein-mediated multidrug resistance by sipholane triterpenoids. J. Nat. Prod. 2007, 70, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Abraham, I.; Carvalho, P.; Kuang, Y.H.; Shaala, L.A.; Youssef, D.T.A.; Avery, M.A.; Chen, Z.S.; El Sayed, K.A. Sipholane triterpenoids: Chemistry, reversal of ABCB1/P-glycoprotein-mediated multidrug resistance, and pharmacophore modeling. J. Nat. Prod. 2009, 72, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, S.J.; Capon, R.J. Parguerenes revisited: New brominated diterpenes from the southern australian marine red alga laurencia filiformis. Aust. J. Chem. 1996, 49, 19–26. [Google Scholar]
- Awad, N.E. Bioactive brominated diterpenes from the marine red alga Jania Rubens (L.) lamx. Phytother. Res. 2004, 18, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Kurosawa, E.; Komiyama, K.; Suzuki, T. The structures of cytotoxic diterpenes containing bromine from the marine red alga Laurencia obtusa (hudson) lamouroux. Bull. Chem. Soc. Jpn. 1990, 63, 3066–3072. [Google Scholar] [CrossRef]
- Huang, X.-C.; Sun, Y.-L.; Salim, A.A.; Chen, Z.-S.; Capon, R.J. Parguerenes: Marine red alga bromoditerpenes as inhibitors of P-glycoprotein (ABCB1) in multidrug resistant human cancer cells. Biochem. Pharmacol. 2013, 85, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Chen, Z.-S.; Higasiyama, K.; Setiawan, I.; Akiyama, S.-I.; Kobayashi, M. Reversing effect of agosterol a, a spongean sterol acetate, on multidrug resistance in human carcinoma cells. Jpn. J. Cancer Res. 2001, 92, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Yoshioka, Y.; Miyamoto, Y.; Higuchi, K.; Setiawan, A.; Murakami, N.; Chen, Z.S.; Sumizawa, T.; Akiyama, S.; Kobayashi, M. Agosterol a, a novel polyhydroxylated sterol acetate reversing multidrug resistance from a marine sponge of spongia sp. Tetrahedron Lett. 1998, 39, 6303–6306. [Google Scholar] [CrossRef]
- Chen, Z.-S.; Aoki, S.; Komatsu, M.; Ueda, K.; Sumizawa, T.; Furukawa, T.; Okumura, H.; Ren, X.-Q.; Belinsky, M.G.; Lee, K.; et al. Reversal of drug resistance mediated by multidrug resistance protein (MRP) 1 by dual effects of agosterol a on MRP1 function. Int. J. Cancer 2001, 93, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Rodríguez, A.D.; Berlinck, R.G.S.; Hamann, M.T. Marine pharmacology in 2003-4: Marine compounds with anthelmintic antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiplatelet, antiprotozoal, antituberculosis, and antiviral activities; affecting the cardiovascular, immune and nervous systems, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 145, 553–581. [Google Scholar] [PubMed]
- Aoki, S.; Setiawan, A.; Yoshioka, Y.; Higuchi, K.; Fudetani, R.; Chen, Z.-S.; Sumizawa, T.; Akiyama, S.-I.; Kobayashi, M. Reversal of multidrug resistance in human carcinoma cell line by agosterols, marine spongean sterols. Tetrahedron 1999, 55, 13965–13972. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Gustafson, K.R. Marine pharmacology in 2001–2: Antitumour and cytotoxic compounds. Eur. J. Cancer 2004, 40, 2676–2704. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Sugimoto, M.; Morita, M.; Akiyama, S.-I.; Kobayashi, M. Synthesis and evaluation of 4-deacetoxyagosterol a as an mdr-modulator. Bioorg. Med. Chem. Lett. 2000, 10, 2521–2524. [Google Scholar] [CrossRef]
- Murakami, N.; Sugimoto, M.; Morita, M.; Kobayashi, M. Total synthesis of agosterol a: An MDR-modulator from a marine sponge. Chem. Eur. J. 2001, 7, 2663–2670. [Google Scholar] [CrossRef]
- Tanaka, J.; Trianto, A.; Musman, M.; Issa, H.H.; Ohtani, I.I.; Ichiba, T.; Higa, T.; Yoshida, W.Y.; Scheuer, P.J. New polyoxygenated steroids exhibiting reversal of multidrug resistance from the gorgonian isis hippuris. Tetrahedron 2002, 58, 6259–6266. [Google Scholar] [CrossRef]
- Boonananwong, S.; Kongkathip, B.; Kongkathip, N. First synthesis of 3,16,20-polyoxygenated cholestanes, new cytotoxic steroids from the gorgonian leptogorgia sarmentosa. Steroids 2008, 73, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, H.; Wang, P.; Gong, W.; Xue, M.; Zhang, H.; Liu, T.; Liu, B.; Yi, Y.; Zhang, W. Bioactive polyoxygenated steroids from the south china sea soft coral, sarcophyton sp. Mar. Drugs 2013, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shao, C.L.; Qi, X.; Li, X.B.; Li, J.; Sun, L.L.; Wang, C.Y. Polyoxygenated sterols from the south China sea soft coral sinularia sp. Mar. Drugs 2012, 10, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Tang, H.; Liu, B.-S.; Li, T.-J.; Sun, P.; Zhu, W.; Luo, Y.-P.; Zhang, W. Tumor cell growth inhibitory activity and structure-activity relationship of polyoxygenated steroids from the gorgonian menella kanisa. Steroids 2013, 78, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sutton, S.C.; Guo, C.; LaCour, T.G.; Fuchs, P.L. Synthesis of the north 1 unit of the cephalostatin family from hecogenin acetate1. J. Am. Chem. Soc. 1999, 121, 2056–2070. [Google Scholar] [CrossRef]
- Musumeci, D.; Sica, D.; Zollo, F. Synthesis of polyoxygenated steroids with transition metal-based oxidants: Methyltrioxorhenium-hydrogen peroxide system, ruthenium tetraoxide, osmium tetraoxide and potassium permanganate. Curr. Org. Synth. 2005, 2. [Google Scholar] [CrossRef]
- Weiss, J.M.; Hoffmann, H.M.R. Synthesis of the c1–c9 segment of bryostatin. Tetrahedron Asymmetry 1997, 8, 3913–3920. [Google Scholar] [CrossRef]
- Ohmori, K.; Ogawa, Y.; Obitsu, T.; Ishikawa, Y.; Nishiyama, S.; Yamamura, S. Total synthesis of bryostatin 3. Angew. Chem. Int. Ed. 2000, 39, 2290–2294. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Hogan, F. Chapter 12—Biosynthetic products for anticancer drug design and treatment: The bryostatins. In Anticancer Drug Development; Kerr, B.C.B.J., Ed.; Academic Press: San Diego, CA, USA, 2002; pp. 203–235. [Google Scholar]
- Spitaler, M.; Utz, I.; Hilbe, W.; Hofmann, J.; Grunicke, H. PKC-independent modulation of multidrug resistance in cells with mutant (V185) but not wild-type (G185) P-glycoprotein by bryostatin 1. Biochem. Pharmacol. 1998, 56, 861–869. [Google Scholar] [CrossRef]
- Utz, I.; Hofmann, J.; Grunicke, H. Bryostatin 1 regulates multi drug resistance by a PKC-independent mechanism. Eur. J. Cancer 1995, 31 (Suppl. 3), S13. [Google Scholar] [CrossRef]
- Scala, S.; Dickstein, B.; Regis, J.; Szallasi, Z.; Blumberg, P.M.; Bates, S.E. Bryostatin 1 affects P-glycoprotein phosphorylation but not function in multidrug-resistant human breast cancer cells. Clin. Cancer Res. 1995, 1, 1581–1587. [Google Scholar] [PubMed]
- Kedei, N.; Telek, A.; Czap, A.; Lubart, E.S.; Czifra, G.; Yang, D.; Chen, J.; Morrison, T.; Goldsmith, P.K.; Lim, L.; et al. The synthetic bryostatin analog merle 23 dissects distinct mechanisms of bryostatin activity in the lncap human prostate cancer cell line. Biochem. Pharmacol. 2011, 81, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Kedei, N.; Telek, A.; Michalowski, A.M.; Kraft, M.B.; Li, W.; Poudel, Y.B.; Rudra, A.; Petersen, M.E.; Keck, G.E.; Blumberg, P.M. Comparison of transcriptional response to phorbol ester, bryostatin 1, and bryostatin analogs in lncap and u937 cancer cell lines provides insight into their differential mechanism of action. Biochem. Pharmacol. 2013, 85, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; De Brabander, J.; Harran, P.G.; Hinkle, K.W.; Lippa, B.; Pettit, G.R. Synthesis and biological evaluation of fully synthetic bryostatin analogues. Tetrahedron Lett. 1998, 39, 8625–8628. [Google Scholar] [CrossRef]
- Trost, B.M.; Yang, H.; Dong, G. Total syntheses of bryostatins: Synthesis of two ring-expanded bryostatin analogues and the development of a new-generation strategy to access the c7–c27 fragment. Chem. Eur. J. 2011, 17, 9789–9805. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Yang, H.; Thiel, O.R.; Frontier, A.J.; Brindle, C.S. Synthesis of a ring-expanded bryostatin analogue. J. Am. Chem. Soc. 2007, 129, 2206–2207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roy, R.; Rey, A.W.; Charron, M.; Molino, R. Enantiospecific synthesis of the c-17–c-20 and c-21–c-27 synthons of the antineoplastic macrolide bryostatins. J. Chem. Soc. Chem. Commun. 1989, 1308–1310. [Google Scholar] [CrossRef]
- Honore, S.; Kamath, K.; Braguer, D.; Horwitz, S.B.; Wilson, L.; Briand, C.; Jordan, M.A. Synergistic suppression of microtubule dynamics by discodermolide and paclitaxel in non-small cell lung carcinoma cells. Cancer Res. 2004, 64, 4957–4964. [Google Scholar] [CrossRef] [PubMed]
- Loggley, R.E.; Ccddigan, D.; Harmody, D.; Gunasekera, M.; Gunasekera, S.P. Discodermolide-a new, marine-derived immunosuppressive compound: I. in vitro studies. Transplantation 1991, 52, 650–655. [Google Scholar] [CrossRef]
- Longley, R.E.; Gunasekera, S.P.; Faherty, D.; McLane, J.; Dumont, F. Immunosuppression by discodermolide. Ann. N. Y. Acad. Sci. 1993, 696, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, R.J.; Giannakakou, P.; Gunasekera, S.P.; Longley, R.E.; Day, B.W.; Hamel, E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol. Pharmacol. 1997, 52, 613–622. [Google Scholar] [PubMed]
- Betzer, J.F.; Ardisson, J. Discodermolide: Total synthesis of natural product and analogues. In Strategies and Tactics in Organic Synthesis; Elsevier: San Diego, CA, USA, 2015; Volume 11, pp. 51–84. [Google Scholar]
- Yu, Z.; Ely, R.J.; Morken, J.P. Synthesis of (+)-discodermolide by catalytic stereoselective borylation reactions. Angew. Chem. Int. Ed. 2014, 53, 9632–9636. [Google Scholar] [CrossRef] [PubMed]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001, 61, 1013–1021. [Google Scholar] [PubMed]
- Cortes, J.; Montero, A.J.; Glück, S. Eribulin mesylate, a novel microtubule inhibitor in the treatment of breast cancer. Cancer Treat. Rev. 2012, 38, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Huyck, T.K.; Gradishar, W.; Manuguid, F.; Kirkpatrick, P. Eribulin mesylate. Nat. Rev. Drug Discov. 2011, 10, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Carlson, E.M.; Cheng, H.; Du, H.; Hu, Y.; Jiang, Y.; Lewis, B.M.; Seletsky, B.M.; Tendyke, K.; Zhang, H.; et al. Novel second generation analogs of eribulin. Part I: Compounds containing a lipophilic c32 side chain overcome P-glycoprotein susceptibility. Bioorg. Med. Chem. Lett. 2011, 21, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
- Garrone, O.; Montemurro, F.; Saggia, C.; la Verde, N.; Vandone, A.M.; Airoldi, M.; de Conciliis, E.; Donadio, M.; Lucio, F.; Polimeni, M.A.; et al. Eribulin in pretreated metastatic breast cancer patients: Results of the trotter trial—A multicenter retrospective study of eribulin in real life. SpringerPlus 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, M.J.; Zheng, W.; Seletsky, B.M. From micrograms to grams: Scale-up synthesis of eribulin mesylate. Nat. Prod. Rep. 2013, 30, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Imbri, D.; Tauber, J.; Opatz, T. Synthetic approaches to the lamellarins—A comprehensive review. Mar. Drugs 2014, 12, 6142–6177. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, F.; Tanabe, S.; Oda, T.; Iwao, M. Synthesis and structure-activity relationship study of lamellarin derivatives. J. Nat. Prod. 2002, 65, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Anticancer properties of lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef] [PubMed]
- Ploypradith, P.; Jinaglueng, W.; Pavaro, C.; Ruchirawat, S. Further developments in the synthesis of lamellarin alkaloids via direct metal-halogen exchange. Tetrahedron Lett. 2003, 44, 1363–1366. [Google Scholar] [CrossRef]
- Facompre, M.; Tardy, C.; Bal-Mahieu, C.; Colson, P.; Perez, C.; Manzanares, I.; Cuevas, C.; Bailly, C. Lamellarin d: A novel potent inhibitor of topoisomerase i. Cancer Res. 2003, 63, 7392–7399. [Google Scholar] [PubMed]
- Ridley, C.P.; Reddy, M.V.; Rocha, G.; Bushman, F.D.; Faulkner, D.J. Total synthesis and evaluation of lamellarin alpha 20-sulfate analogues. Bioorg. Med. Chem. 2002, 10, 3285–3290. [Google Scholar] [CrossRef]
- Ohta, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. Design and synthesis of lamellarin d analogues targeting topoisomerase i. J. Org. Chem. 2009, 74, 8143–8153. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Guitián, E.; Castedo, L. Syntheses of lamellarins i and k by [3 + 2] cycloaddition of a nitrone to an alkyne. Synlett 2001, 1164–1166. [Google Scholar] [CrossRef]
- Quesada, A.R.; García Grávalos, M.D.; Fernández Puentes, J.L. Polyaromatic alkaloids from marine invertebrates as cytotoxic compounds and inhibitors of multidrug resistance caused by P-glycoprotein. Br. J. Cancer 1996, 74, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Xiao, X.; Zhang, Y.K.; Talele, T.T.; Salim, A.A.; Chen, Z.S.; Capon, R.J. Lamellarin o, a pyrrole alkaloid from an australian marine sponge, ianthella sp., reverses bcrp mediated drug resistance in cancer cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Fuerstner, A.; Weintritt, H.; Hupperts, A. A new, titanium-mediated approach to pyrroles: First synthesis of lukianol a and lamellarin o dimethyl ether. J. Org. Chem. 1995, 60, 6637–6641. [Google Scholar] [CrossRef]
- Banwell, M.G.; Flynn, B.L.; Hamel, E.; Hockless, D.C.R. Convergent syntheses of the pyrrolic marine natural products lamellarin-O, lamellarin-Q, lukianol-A and some more highly oxygenated congeners. Chem. Commun. 1997, 207–208. [Google Scholar] [CrossRef]
- Marfil, M.; Albericio, F.; Álvarez, M. Solid-phase synthesis of lamellarins Q and O. Tetrahedron 2004, 60, 8659–8668. [Google Scholar] [CrossRef]
- Fukuda, T.; Sudo, E.-I.; Shimokawa, K.; Iwao, M. Palladium-catalyzed cross-coupling of N-benzenesulfonyl-3,4-dibromopyrrole and its application to the total syntheses of lamellarins O, P, Q, and R. Tetrahedron 2008, 64, 328–338. [Google Scholar] [CrossRef]
- Boger, D.L.; Boyce, C.W.; Labroli, M.A.; Sehon, C.A.; Jin, Q. Total syntheses of ningalin A, lamellarin O, lukianol A, and permethyl storniamide a utilizing heterocyclic azadiene diels-alder reactions. J. Am. Chem. Soc. 1999, 121, 54–62. [Google Scholar] [CrossRef]
- Pla, D.; Marchal, A.; Olsen, C.A.; Albericio, F.; Álvarez, M. Modular total synthesis of lamellarin d. J. Org. Chem. 2005, 70, 8231–8234. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, N.; Ohta, T.; Yamaguchi, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. Total synthesis of lamellarins D, L, and N. Tetrahedron 2006, 62, 594–604. [Google Scholar] [CrossRef]
- Ueda, K.; Amaike, K.; Maceiczyk, R.M.; Itami, K.; Yamaguchi, J. B-selective C–H arylation of pyrroles leading to concise syntheses of lamellarins c and i. J. Am. Chem. Soc. 2014, 136, 13226–13232. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Fenical, W. Ningalins A–D: Novel aromatic alkaloids from a western australian ascidian of the genus didemnum. J. Org. Chem. 1997, 62, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total synthesis of lamellarins D, H, and R and ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Soenen, D.R.; Hwang, I.; Hedrick, M.P.; Boger, D.L. Multidrug resistance reversal activity of key ningalin analogues. Bioorg. Med. Chem. Lett. 2003, 13, 1777–1781. [Google Scholar] [CrossRef]
- Tao, H.; Hwang, I.; Boger, D.L. Multidrug resistance reversal activity of permethyl ningalin b amide derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5979–5981. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Guan, Y.; Soenen, D.R.; Danishefsky, S.J.; Boger, D.L. Potent reversal of multidrug resistance by ningalins and its use in drug combinations against human colon carcinoma xenograft in nude mice. Cancer Chemother. Pharmacol. 2005, 56, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.Y.; Wong, I.L.K.; Yan, C.S.W.; Zhang, X.Y.; Jiang, T.; Chow, L.M.C.; Wan, S.B. Design and syntheses of permethyl ningalin B analogues: Potent multidrug resistance (MDR) reversal agents of cancer cells. J. Med. Chem. 2010, 53, 5108–5120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wong, I.L.K.; Li, F.X.; Yang, C.; Liu, Z.; Jiang, T.; Jiang, T.F.; Chow, L.M.C.; Wan, S.B. Optimization of permethyl ningalin B analogs as P-glycoprotein inhibitors. Bioorg. Med. Chem. 2015, 23, 5566–5573. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wong, I.L.K.; Jin, W.B.; Jiang, T.; Chow, L.M.C.; Wan, S.B. Modification of marine natural product ningalin B and SAR study lead to potent P-glycoprotein inhibitors. Mar. Drugs 2014, 12, 5209–5221. [Google Scholar] [CrossRef] [PubMed]
- Bin, J.W.; Wong, I.L.K.; Hu, X.; Yu, Z.X.; Xing, L.F.; Jiang, T.; Chow, L.M.C.; Biao, W.S. Structure-activity relationship study of permethyl ningalin B analogues as P-glycoprotein chemosensitizers. J. Med. Chem. 2013, 56, 9057–9070. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, A.; Zimpleman, J.M.; Hwang, I.; Boger, D.L. Total synthesis of ningalin D. J. Am. Chem. Soc. 2005, 127, 10767–10770. [Google Scholar] [CrossRef] [PubMed]
- Saracoglu, N. Recent advances and applications in 1,2,4,5-tetrazine chemistry. Tetrahedron 2007, 63, 4199–4236. [Google Scholar] [CrossRef]
- Fu, T.H.; McElroy, W.T.; Shamszad, M.; Martin, S.F. Formal syntheses of naturally occurring welwitindolinones. Org. Lett. 2012, 14, 3834–3837. [Google Scholar] [CrossRef] [PubMed]
- MacKay, J.A.; Bishop, R.L.; Rawal, V.H. Rapid synthesis of the N-methylwelwitindolinone skeleton. Org. Lett. 2005, 7, 3421–3424. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Zilfou, J.T.; Stratmann, K.; Patterson, G.M.L.; Moore, R.E. Welwitindolinone analogues that reverse P-glycoprotein-mediated multiple drug resistance. Mol. Pharmacol. 1995, 47, 241–247. [Google Scholar] [PubMed]
- Grundmann, A.; Kuznetsova, T.; Afiyatullov, S.; Li, S.M. FTMPT2, an N-prenyltransferase from aspergillus fumigatus, catalyses the last step in the biosynthesis of fumitremorgin B. Chembiochem 2008, 9, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.H.; McElroy, W.T.; Shamszad, M.; Heidebrecht, R.W., Jr.; Gulledge, B.; Martin, S.F. Studies toward welwitindolinones: Formal syntheses of N-methylwelwitindolinone C isothiocyanate and related natural products. Tetrahedron 2013, 69, 5588–5603. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L. Total synthesis: Welwitindolinone is well worth it. Nat. Chem. 2012, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Komine, K.; Nomura, Y.; Ishihara, J.; Hatakeyama, S. Total synthesis of (−)-N-methylwelwitindolinone C isothiocyanate based on a Pd-catalyzed tandem enolate coupling strategy. Org. Lett. 2015, 17, 3918–3921. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Gadewar, M.; Tripathi, R.; Prasad, S.K.; Patel, D.K. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid “harmine”. Asian Pac. J. Trop. Biomed. 2012, 2, 660–664. [Google Scholar] [CrossRef]
- Khan, A.M.; Noreen, S.; Imran, Z.P.; Attaur, R.; Choudhary, M.I. A new compound, jolynamine, from marine brown alga jolyna laminarioides. Nat. Prod. Res. 2011, 25, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, A.; Gu, F.; Wang, Z.; Tian, C.; Qian, Z.; Tang, L.; Gu, Y. Novel harmine derivatives for tumor targeted therapy. Oncotarget 2015, 6, 8988–9001. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Q.; Chao, R.; Chen, H.; Hou, X.; Yan, H.; Zhou, S.; Peng, W.; Xu, A. Antitumor and neurotoxic effects of novel harmine derivatives and structure-activity relationship analysis. Int. J. Cancer 2005, 114, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Fan, W.; Guo, L.; Ma, Q.; Zhang, G.; Li, J.; Chen, X.; Ren, Z.; Qiu, L. Synthesis and structure-activity relationships of harmine derivatives as potential antitumor agents. Eur. J. Med. Chem. 2013, 60, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wink, M. The β-carboline alkaloid harmine inhibits BCRP and can reverse resistance to the anticancer drugs mitoxantrone and camptothecin in breast cancer cells. Phytother. Res. 2010, 24, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Bai, Z.; Ma, Q.; Fan, W.; Guo, L.; Zhang, G.; Qiu, L.; Yu, H.; Shao, G.; Cao, R. Synthesis and biological evaluation of novel bivalent β-carbolines as potential antitumor agents. Med. Chem. Commun. 2014, 5, 953–957. [Google Scholar] [CrossRef]
- Frédérick, R.; Bruyére, C.; Vancraeynest, C.; Reniers, J.; Meinguet, C.; Pochet, L.; Backlund, A.; Masereel, B.; Kiss, R.; Wouters, J. Novel trisubstituted harmine derivatives with original in vitro anticancer activity. J. Med. Chem. 2012, 55, 6489–6501. [Google Scholar] [CrossRef] [PubMed]
- Dighe, S.U.; Khan, S.; Soni, I.; Jain, P.; Shukla, S.; Yadav, R.; Sen, P.; Meeran, S.M.; Batra, S. Synthesis of β-carboline-based n-heterocyclic carbenes and their antiproliferative and antimetastatic activities against human breast cancer cells. J. Med. Chem. 2015, 58, 3485–3499. [Google Scholar] [CrossRef] [PubMed]
- Pohl, B.; Luchterhandt, T.; Bracher, F. Total syntheses of the chlorinated β-carboline alkaloids bauerine a, b, and c. Synth. Commun. 2007, 37, 1273–1280. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Shultz, E.E.; Chernov, S.V.; Turmukhambetov, A.Z.; Seydakhmetova, R.B.; Shakirov, M.M.; Tolstikov, G.A.; Adekenov, S.M. Synthesis of substituted indolizino[8,7-b]indoles from harmine and their biological activity. Chem. Heterocycl. Compd. 2011, 46, 1494–1499. [Google Scholar] [CrossRef]
- Mukusheva, G.K.; Nurmaganbetov, Z.S.; Ismagulova, N.M.; Ponamareva, O.A.; Burdel’naya, E.V.; Turmukhambetov, A.Z.; Kazantsev, A.V.; Adekenov, S.M. Synthesis and phagocytosis-stimulating activity of harmine and glaucine n-oxides. Pharm. Chem. J. 2011, 45, 458–460. [Google Scholar] [CrossRef]
- Begum, S.; Ali, S.; Farhat, F.; Hassan, S.; Siddiqui, B. A simple, rapid and mild one pot synthesis of benzene ring acylated and demethylated analogues of harmine under solvent-free conditions. Molecules 2008, 13, 1584–1598. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Mendez, C.; Salas, J.A. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045. [Google Scholar] [CrossRef] [PubMed]
- Prudhomme, M. Biological targets of antitumor indolocarbazoles bearing a sugar moiety. Curr. Med. Chem. Anticancer Agents 2004, 4, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhai, F.; Zhao, L.; Guo, Q.; You, Q. Design and synthesis of N-methylmaleimide indolocarbazole bearing modified 2-acetamino acid moieties as topoisomerase i inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Shukla, S.; Steadman, K.; Obrzut, T.; Finley, E.M.; Ambudkar, S.V.; Bates, S.E. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol. Cancer Ther. 2007, 6, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Slater, M.J.; Cockerill, S.; Baxter, R.; Bonser, R.W.; Gohil, K.; Gowrie, C.; Robinson, J.E.; Littler, E.; Parry, N.; Randall, R.; et al. Indolocarbazoles: Potent, selective inhibitors of human cytomegalovirus replication. Bioorg. Med. Chem. 1999, 7, 1067–1074. [Google Scholar] [CrossRef]
- Slater, M.J.; Baxter, R.; Bonser, R.W.; Cockerill, S.; Gohil, K.; Parry, N.; Robinson, E.; Randall, R.; Yeates, C.; Snowden, W.; et al. Synthesis of n-alkyl substituted indolocarbazoles as potent inhibitors of human cytomegalovirus replication. Bioorg. Med. Chem. Lett. 2001, 11, 1993–1995. [Google Scholar] [CrossRef]
- Cuevas, C.; Pérez, M.; Martín, M.J.; Chicharro, J.L.; Fernândez-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de la Calle, F.; et al. Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, A.; Takebayashi, Y.; Ren, X.Q.; Miyashita, H.; Mori, S.; Akiyama, S.; Pommier, Y. Overcoming multidrug drug resistance in P-glycoprotein/MDR1-overexpressing cell lines by ecteinascidin 743. Mol. Cancer Ther. 2002, 1, 1327–1334. [Google Scholar] [PubMed]
- Beumer, J.H.; Buckle, T.; Ouwehand, M.; Franke, N.E.F.; Lopez-Lazaro, L.; Schellens, J.H.M.; Beijnen, J.H.; van Tellingen, O. Trabectedin (ET-743, yondelis™) is a substrate for P-glycoprotein, but only high expression of P-glycoprotein confers the multidrug resistance phenotype. Investig. New Drugs 2007, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.G.; Machado, M.S.; Rocca, C.J.; Poindessous, V.; Ouaret, D.; Sarasin, A.; Galmarini, C.M.; Henriques, J.A.P.; Escargueil, A.E.; Larsen, A.K. Trabectedin and its c subunit modified analogue pm01183 attenuate nucleotide excision repair and show activity toward platinum-resistant cells. Mol. Cancer Ther. 2011, 10, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, J.; de Paolis, M.; Zhu, J. Synthetic studies toward ecteinascidin 743. J. Org. Chem. 2005, 70, 4397–4408. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Yasui, Y.; Takemoto, Y. Synthetic study toward ecteinascidin 743: Concise construction of the diazabicyclo[3.3.1]nonane skeleton and assembly of the pentacyclic core. J. Org. Chem. 2010, 75, 4876–4879. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Chan, C.; Furuuchi, T.; Wright, B.J.; Zhou, B.; Guo, J.; Danishefsky, S.J. Stereospecific formal total synthesis of ecteinascidin 743. Angew. Chem. Int. Ed. Engl. 2006, 45, 1754–1759. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, X.; Willot, M.; Zhu, J. Asymmetric total syntheses of ecteinascidin 597 and ecteinascidin 583. Angew. Chem. Int. Ed. Engl. 2006, 45, 8028–8032. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, F.; Toma, T.; Inui, T.; Yokoshima, S.; Fukuyama, T. Total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 2013, 135, 13684–13687. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, X.; Bois-Choussy, M.; Zhu, J. Total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 2006, 128, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.; Piggott, A.M.; Huang, X.C.; Capon, R.J. Nocardioazines: A novel bridged diketopiperazine scaffold from a marine-derived bacterium inhibits P-glycoprotein. Org. Lett. 2011, 13, 2770–2773. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Reisman, S.E. Enantioselective total synthesis of (−)-lansai b and (+)-nocardioazines a and b. Angew. Chem. Int. Ed. 2014, 53, 6206–6210. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Suzuki, H.; Okumura, H.; Takahashi, S.; Osada, H. A point mutation in ftmd blocks the fumitremorgin biosynthetic pathway in aspergillus fumigatus strain af293. Biosci. Biotechnol. Biochem. 2013, 77, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Van Loevezijn, A.; van Maarseveen, J.H.; Stegman, K.; Visser, G.M.; Koomen, G.-J. Solid phase synthesis of fumitremorgin, verruculogen and tryprostatin analogs based on a cyclization/cleavage strategy. Tetrahedron Lett. 1998, 39, 4737–4740. [Google Scholar] [CrossRef]
- Rabindran, S.K.; He, H.; Singh, M.; Brown, E.; Collins, K.I.; Annable, T.; Greenberger, L.M. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin c. Cancer Res. 1998, 58, 5850–5858. [Google Scholar] [PubMed]
- Rabindran, S.K.; Ross, D.D.; Doyle, L.A.; Yang, W.; Greenberger, L.M. Fumitremorgin c reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000, 60, 47–50. [Google Scholar] [PubMed]
- Gonzalez-Lobato, L.; Real, R.; Prieto, J.G.; Alvarez, A.I.; Merino, G. Differential inhibition of murine BCRP1/ABCG2 and human BCRP/ABCG2 by the mycotoxin fumitremorgin c. Eur. J. Pharmacol. 2010, 644, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Suzuki, H.; Takagi, H.; Asami, Y.; Kakeya, H.; Uramoto, M.; Usui, T.; Takahashi, S.; Sugimoto, Y.; Osada, H. Identification of cytochrome p450s required for fumitremorgin biosynthesis in aspergillus fumigatus. ChemBioChem 2009, 10, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin c. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar] [PubMed]
- Van Loevezijn, A.; Allen, J.D.; Schinkel, A.H.; Koomen, G.J. Inhibition of bcrp-mediated drug efflux by fumitremorgin-type indolyl diketopiperazines. Bioorg. Med. Chem. Lett. 2001, 11, 29–32. [Google Scholar] [CrossRef]
- Weidner, L.D.; Zoghbi, S.S.; Lu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Mulder, J.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The inhibitor Ko143 is not specific for ABCG2. J. Pharmacol. Exp. Ther. 2015, 354, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Szolomajer-Csikos, O.; Beery, E.; Kosa, L.; Rajnai, Z.; Jani, M.; Hetenyi, A.; Jakab, K.T.; Krajcsi, P.; Toth, G.K. Synthesis and ABCG2 inhibitory activity of novel fumitremorgin c analogs—Specificity and structure activity correlations. Med. Chem. 2013, 9, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Hino, T.; Kawate, T.; Nakagawa, M. A synthesis of so-called fumitremorgin c. Tetrahedron 1989, 45, 1941–1944. [Google Scholar] [CrossRef]
- Jiang, D.; Xu, Z.; Jia, Y. Mg(CLO4)2-catalyzed intramolecular allylic amination: Application to the total synthesis of demethoxyfumitremorgin C. Tetrahedron 2012, 68, 4225–4232. [Google Scholar] [CrossRef]
- Li, Y.; Hayman, E.; Plesescu, M.; Prakash, S.R. Synthesis of potent bcrp inhibitor—Ko143. Tetrahedron Lett. 2008, 49, 1480–1483. [Google Scholar] [CrossRef]
- Kanoh, K.; Kohno, S.; Asari, T.; Harada, T.; Katada, J.; Muramatsu, M.; Kawashima, H.; Sekiya, H.; Uno, I. (−)-phenylahistin: A new mammalian cell cycle inhibitor produced by aspergillus ustus. Bioorg. Med. Chem. Lett. 1997, 7, 2847–2852. [Google Scholar] [CrossRef]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T.C. Npi-2358 is a tubulin-depolymerizing agent: In vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.; Aren, O.; Millward, M.; Mainwaring, P.; Mita, A.; Mita, M.; Bazhenova, L.; Blum, R.; Polikoff, J.; Gadgeel, S.; et al. Abstract c30: Phase 1/2 study of the vascular disrupting agent (VDA) plinabulin (NPI-2358) combined with docetaxel in patients with non-small cell lung cancer (NSCLC). Mol. Cancer Ther. 2009, 8, C30. [Google Scholar] [CrossRef]
- Stratmann, T.; Burgoyne, D.L.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Hapalosin, a cyanobacterial cyclic depsipeptide with multidrug-resistance reversing activity. J. Org. Chem. 1995, 60, 2950–2950. [Google Scholar] [CrossRef]
- Smith, C.D. New compounds from cyanobacteria to circumvent mdr. Drug News Perspect. 1995, 8, 423–425. [Google Scholar]
- Dinh, T.Q.; Smith, C.D.; Du, X.; Armstrong, R.W. Design, synthesis, and evaluation of the multidrug resistance-reversing activity of d-glucose mimetics of hapalosin. J. Med. Chem. 1998, 41, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Ohmori, K.; Nishiyama, S.; Yamamura, S.; Nakamura, K.; Houk, K.N.; Okamoto, K. Chemical study on hapalosin, a cyclic depsipeptide possessing multidrug resistance reversing activities: Synthesis, structure and biological activity. Tetrahedron 1996, 52, 14723–14734. [Google Scholar] [CrossRef]
- Dinh, T.Q.; Smith, C.D.; Armstrong, R.W. Analogs incorporating trans-4-hydroxy-l-proline that reverse multidrug resistance better than hapalosin. J. Org. Chem. 1997, 62, 790–791. [Google Scholar] [CrossRef]
- Kashihara, N.; To-e, S.; Nakamura, K.; Umezawa, K.; Yamamura, S.; Nishiyama, S. Synthesis and biological activities of hapalosin derivatives with modification at the c12 position. Bioorg. Med. Chem. Lett. 2000, 10, 101–103. [Google Scholar] [CrossRef]
- Dinh, T.Q.; Du, X.; Smith, C.D.; Armstrong, R.W. Synthesis, conformational analysis, and evaluation of the multidrug resistance-reversing activity of the triamide and proline analogs of hapalosin. J. Org. Chem. 1997, 62, 6773–6783. [Google Scholar] [CrossRef]
- Dinh, T.Q.; Du, X.H.; Armstrong, R.W. Synthesis and conformational analysis of the multidrug resistance-reversing agent hapalosin and its non-n-methyl analog. J. Org. Chem. 1996, 61, 6606–6616. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, K.; Okuno, T.; Nishiyama, S.; Yamamura, S. Synthetic study on hapalosin, a cyclic depsipeptide possessing multidrug resistance reversing activities. Tetrahedron Lett. 1996, 37, 3467–3470. [Google Scholar] [CrossRef]
- Hermann, C.; Giammasi, C.; Geyer, A.; Maier, M.E. Syntheses of hapalosin analogs by solid-phase assembly of acyclic precursors. Tetrahedron 2001, 57, 8999–9010. [Google Scholar] [CrossRef]
- Hermann, C.; Pais, G.C.G.; Geyer, A.; Kuhnert, S.M.; Maier, M.E. Total synthesis of hapalosin and two ring expanded analogs. Tetrahedron 2000, 56, 8461–8471. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; García, J.M.; González, A.; Pazos, R.; Odriozola, J.M.; Bañuelos, P.; Tello, M.; Linden, A. A practical total synthesis of hapalosin, a 12-membered cyclic depsipeptide with multidrug resistance-reversing activity, by employing improved segment coupling and macrolactonization†. J. Org. Chem. 2004, 69, 4126–4134. [Google Scholar] [CrossRef] [PubMed]
- Pais, G.C.G.; Maier, M.E. Efficient synthesis of the γ-amino-β-hydroxy acid subunit of hapalosin. J. Org. Chem. 1999, 64, 4551–4554. [Google Scholar] [CrossRef]
- Maier, M.E.; Hermann, C. Synthesis of the γ-amino-β-hydroxy acid of hapalosin via an asymmetric dihydroxylation route. Tetrahedron 2000, 56, 557–561. [Google Scholar] [CrossRef]
- Kumar, H.; Reddy, A.S.; Yadav, J.S.; Reddy, B.V.S. A highly stereoselective formal synthesis of hapalosin. Synlett 2013, 24, 1415–1419. [Google Scholar]
- Parkes, K.E.B.; Bushnell, D.J.; Crackett, P.H.; Dunsdon, S.J.; Freeman, A.C.; Gunn, M.P.; Hopkins, R.A.; Lambert, R.W.; Martin, J.A. Studies toward the large-scale synthesis of the hiv proteinase inhibitor ro 31-8959. J. Org. Chem. 1994, 59, 3656–3664. [Google Scholar] [CrossRef]
- McDonald, L.A.; Christopher Swersey, J.; Ireland, C.M.; Carroll, A.R.; Coll, J.C.; Bowden, B.F.; Fairchild, C.R.; Cornell, L. Botryllamides A–D, new brominated tyrosine derivatives from styelid ascidians of the genus botryllus. Tetrahedron 1995, 51, 5237–5244. [Google Scholar] [CrossRef]
- Rao, M.R.; Faulknert, D.J. Botryllamides E–H, four new tyrosine derivatives from the ascidian botrylloides tyreum. J. Nat. Prod. 2004, 67, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Henrich, C.J.; Robey, R.W.; Takada, K.; Bokesch, H.R.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; McMahon, J.B.; Gustafson, K.R. Botryllamides: Natural product inhibitors of ABCG2. ACS Chem. Biol. 2009, 4, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Imamura, N.; Gustafson, K.R.; Henrich, C.J. Synthesis and structure-activity relationship of botryllamides that block the ABCG2 multidrug transporter. Bioorg. Med. Chem. Lett. 2010, 20, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Cao, L.; Matsui, K.; Rachmat, R.; Akiyama, S.I.; Kobayashi, M. Kendarimide a, a novel peptide reversing P-glycoprotein-mediated multidrug resistance in tumor cells, from a marine sponge of haliclona sp. Tetrahedron 2004, 60, 7053–7059. [Google Scholar] [CrossRef]
- Kotoku, N.; Cao, L.; Aoki, S.; Kobayashi, M. Absolute stereo-structure of kendarimide a, a novelmdr modulator, from a marine sponge. Heterocycles 2005, 65, 563–578. [Google Scholar]
- Fu, X.; Do, T.; Schmitz, F.J.; Andrusevich, V.; Engel, M.H. New cyclic peptides from the ascidian lissoclinum patella. J. Nat. Prod. 1998, 61, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Rasko, D.A.; Sudek, S.; Eisen, J.A.; Haygood, M.G.; Ravel, J. Patellamide a and c biosynthesis by a microcin-like pathway in prochloron didemni, the cyanobacterial symbiont of lissoclinum patella. Proc. Natl. Acad. Sci. USA 2005, 102, 7315–7320. [Google Scholar] [CrossRef] [PubMed]
- García-Reynaga, P.; VanNieuwenhze, M.S. A new total synthesis of patellamide a. Org. Lett. 2008, 10, 4621–4623. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Miyamoto, T.; Higuchi, R.; Uchiumi, T.; Kuwano, M.; Van Soest, R.W.M. Structure of a novel multidrug resistance modulator, irciniasulfonic acid, isolated from a marine sponge, ircinia sp. Tetrahedron Lett. 2001, 42, 3335–3337. [Google Scholar] [CrossRef]
- Emura, C.; Higuchi, R.; Miyamoto, T. Irciniasulfonic acid B, a novel taurine conjugated fatty acid derivative from a japanese marine sponge, Ircinia sp. Tetrahedron 2006, 62, 5682–5685. [Google Scholar] [CrossRef]
- Kim, C.K.; Song, I.H.; Park, H.Y.; Lee, Y.J.; Lee, H.S.; Sim, C.J.; Oh, D.C.; Oh, K.B.; Shin, J. Suvanine sesterterpenes and deacyl irciniasulfonic acids from a tropical coscinoderma sp. Sponge. J. Nat. Prod. 2014, 77, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Emura, C.; Higuchi, R.; Miyamoto, T. Synthetic studies on the natural multidrug resistance modulator, irciniasulfonic acid B. Chem. Lett. 2010, 39, 1002–1003. [Google Scholar] [CrossRef]
- Adrian, P.; Dobbs, A.V.; Butler, L.A.; Parker, R.J. Document first total synthesis of the irciniasulfonic acids. Synlett 2005, 4, 652–654. [Google Scholar]
- Steyn, P.S. The isolation, structure and absolute configuration of secalonic acid D, the toxic metabolite of penicillium oxalicum. Tetrahedron 1970, 26, 51–57. [Google Scholar] [CrossRef]
- Ren, H.; Tian, L.; Gu, Q.Q.; Zhu, W.M. Secalonic acid D; a cytotoxic constituent from marine lichen-derived fungus Gliocladium sp. T31. Arch. Pharm. Res. 2006, 29, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Hong, R. Secalonic acid d as a novel DNA topoisomerase I inhibitor from marine lichen-derived fungus Gliocladium sp. T31. Pharm. Biol. 2011, 49, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-P.; Tao, L.-Y.; Wang, F.; Zhang, J.-Y.; Liang, Y.-J.; Fu, L.-W. Secalonic acid d reduced the percentage of side populations by down-regulating the expression of ABCG2. Biochem. Pharmacol. 2013, 85, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Guru, S.K.; Pathania, A.S.; Kumar, S.; Ramesh, D.; Kumar, M.; Rana, S.; Kumar, A.; Malik, F.; Sharma, P.R.; Chandan, B.K.; et al. Secalonic acid-D represses HIF1A/VEGF-mediated angiogenesis by regulating the Akt/mTOR/p70S6K signaling cascade. Cancer Res. 2015, 75, 2886–2896. [Google Scholar] [CrossRef] [PubMed]
- Wezeman, T.; Masters, K.S.; Bräse, S. Double trouble—The art of synthesis of chiral dimeric natural products. Angew. Chem. Int. Ed. 2014, 53, 4524–4526. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, H.; Uffman, E.W.; Guo, L.; Zhang, Y.; Yang, X.; Birman, V.B. Kinetic resolution of secondary alcohols using amidine-based catalysts. J. Org. Chem. 2012, 77, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Porco, J.A., Jr. Total syntheses of secalonic acids a and d. Angew. Chem. Int. Ed. 2014, 53, 3107–3110. [Google Scholar] [CrossRef] [PubMed]
- Nising, C.F.; Schmid, U.K.; Nieger, M.; Bräse, S. A new protocol for the one-pot synthesis of symmetrical biaryls. J. Org. Chem. 2004, 69, 6830–6833. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Wang, S.Y.; Shen, H.; Yao, X.F.; Zhang, F.L.; Lai, D. The marine-derived fungal metabolite, terrein, inhibits cell proliferation and induces cell cycle arrest in human ovarian cancer cells. Int. J. Mol. Med. 2014, 34, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Cho, H.J.; Lee, H.K.; Lee, W.H.; Park, E.S.; Youn, S.W.; Park, K.C. Terrein, a fungal metabolite, inhibits the epidermal proliferation of skin equivalents. J. Dermatol. Sci. 2007, 46, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, D.S.; Kim, W.G.; Ryoo, I.J.; Lee, D.H.; Huh, C.H.; Youn, S.W.; Yoo, I.D.; Park, K.C. Terrein: A new melanogenesis inhibitor and its mechanism. Cell. Mol. Life Sci. 2004, 61, 2878–2885. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.Y.; Shen, C.N.; Lin, L.H.; Yang, Y.L.; Han, H.Y.; Chen, J.W.; Kuo, S.C.; Wu, S.H.; Liaw, C.C. Asperjinone, a nor-neolignan, and terrein, a suppressor of ABCG2-expressing breast cancer cells, from thermophilic aspergillus terreus. J. Nat. Prod. 2012, 75, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Mandai, H.; Omori, K.; Yamamoto, D.; Tsumura, T.; Murota, K.; Yamamoto, S.; Mitsudo, K.; Ibaragi, S.; Sasaki, A.; Maeda, H.; et al. Synthetic (+)-terrein suppresses interleukin-6/soluble interleukin-6 receptor induced-secretion of vascular endothelial growth factor in human gingival fibroblasts. Bioorg. Med. Chem. 2014, 22, 5338–5344. [Google Scholar] [CrossRef] [PubMed]
- Khalil, Z.G.; Huang, X.-C.; Raju, R.; Piggott, A.M.; Capon, R.J. Shornephine a: Structure, chemical stability, and p-glycoprotein inhibitory properties of a rare diketomorpholine from an australian marine-derived aspergillus sp. J. Org. Chem. 2014, 79, 8700–8705. [Google Scholar] [CrossRef] [PubMed]

















































© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, S.; Sousa, E.; Kijjoa, A.; Pinto, M.M.M. Marine Natural Products as Models to Circumvent Multidrug Resistance. Molecules 2016, 21, 892. https://doi.org/10.3390/molecules21070892
Long S, Sousa E, Kijjoa A, Pinto MMM. Marine Natural Products as Models to Circumvent Multidrug Resistance. Molecules. 2016; 21(7):892. https://doi.org/10.3390/molecules21070892
Chicago/Turabian StyleLong, Solida, Emília Sousa, Anake Kijjoa, and Madalena M. M. Pinto. 2016. "Marine Natural Products as Models to Circumvent Multidrug Resistance" Molecules 21, no. 7: 892. https://doi.org/10.3390/molecules21070892
APA StyleLong, S., Sousa, E., Kijjoa, A., & Pinto, M. M. M. (2016). Marine Natural Products as Models to Circumvent Multidrug Resistance. Molecules, 21(7), 892. https://doi.org/10.3390/molecules21070892