
Exploring Flow Procedures for Diazonium Formation




Abstract
:
1. Introduction
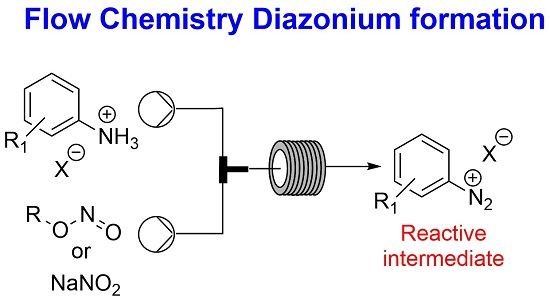
2. Results and Discussion
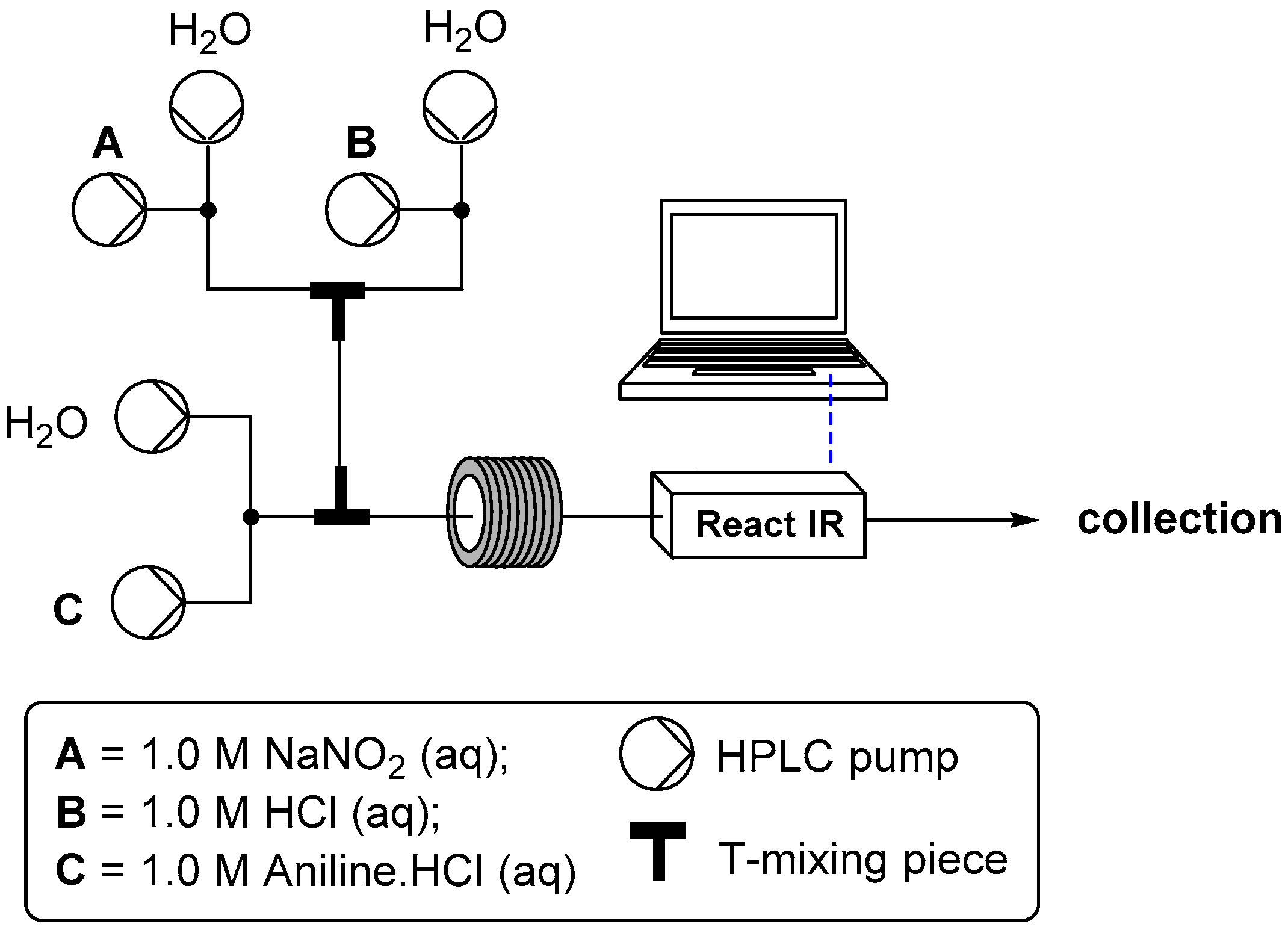
2.1. Formation of Aryl Diazonium Species under Aqueous Conditions
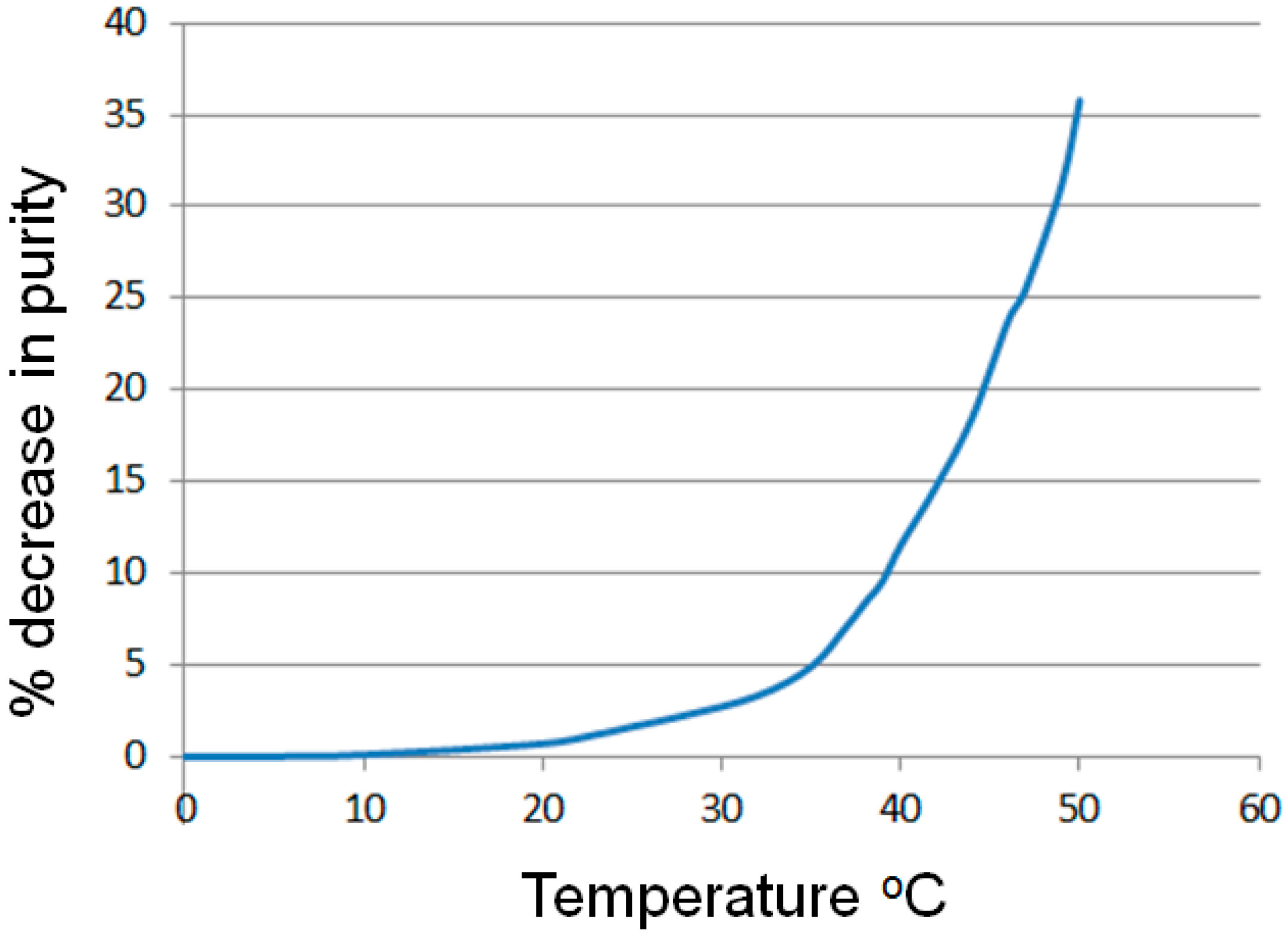
2.1.1. Temperature Dependence
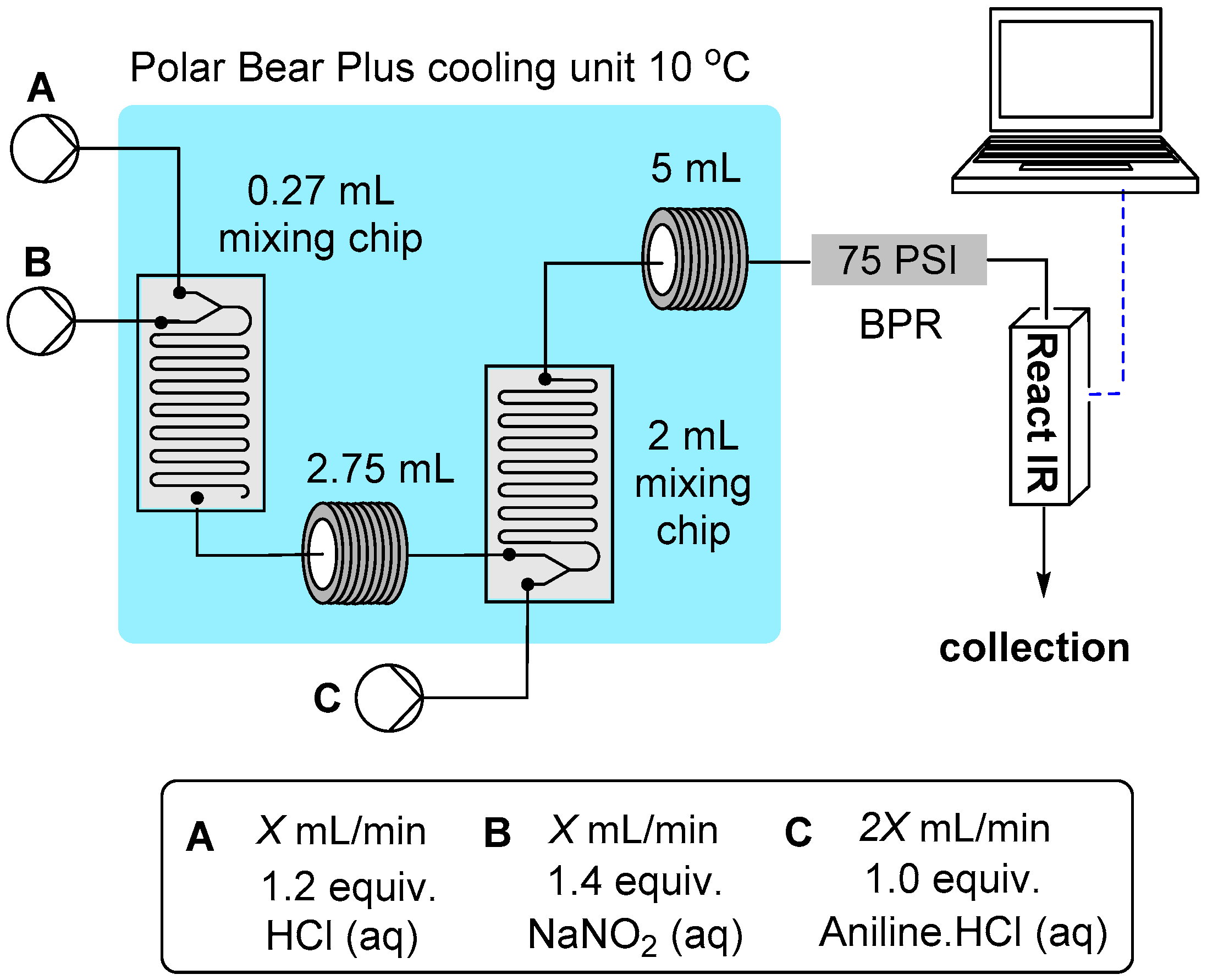
2.1.2. Flow Rate Analysis
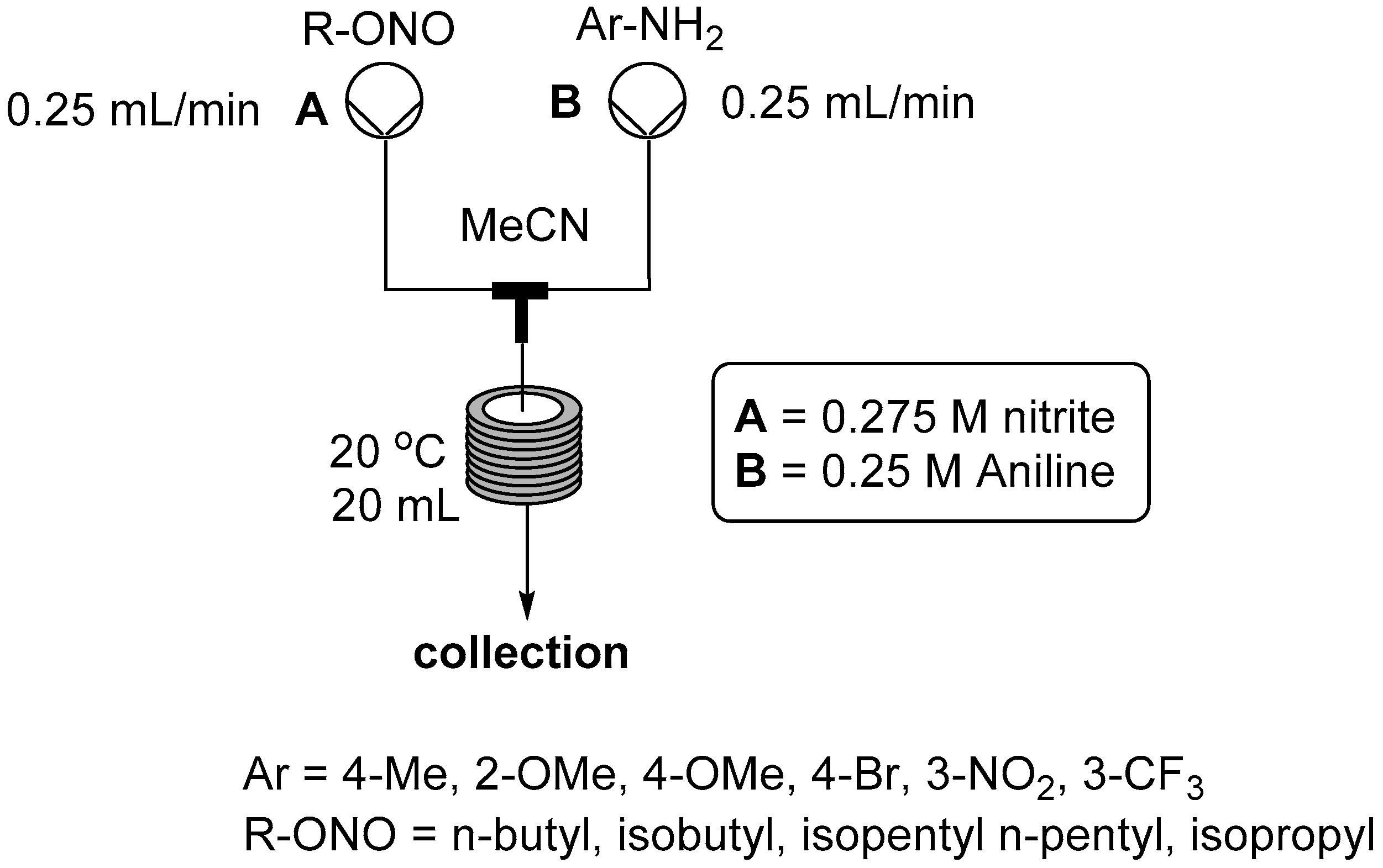
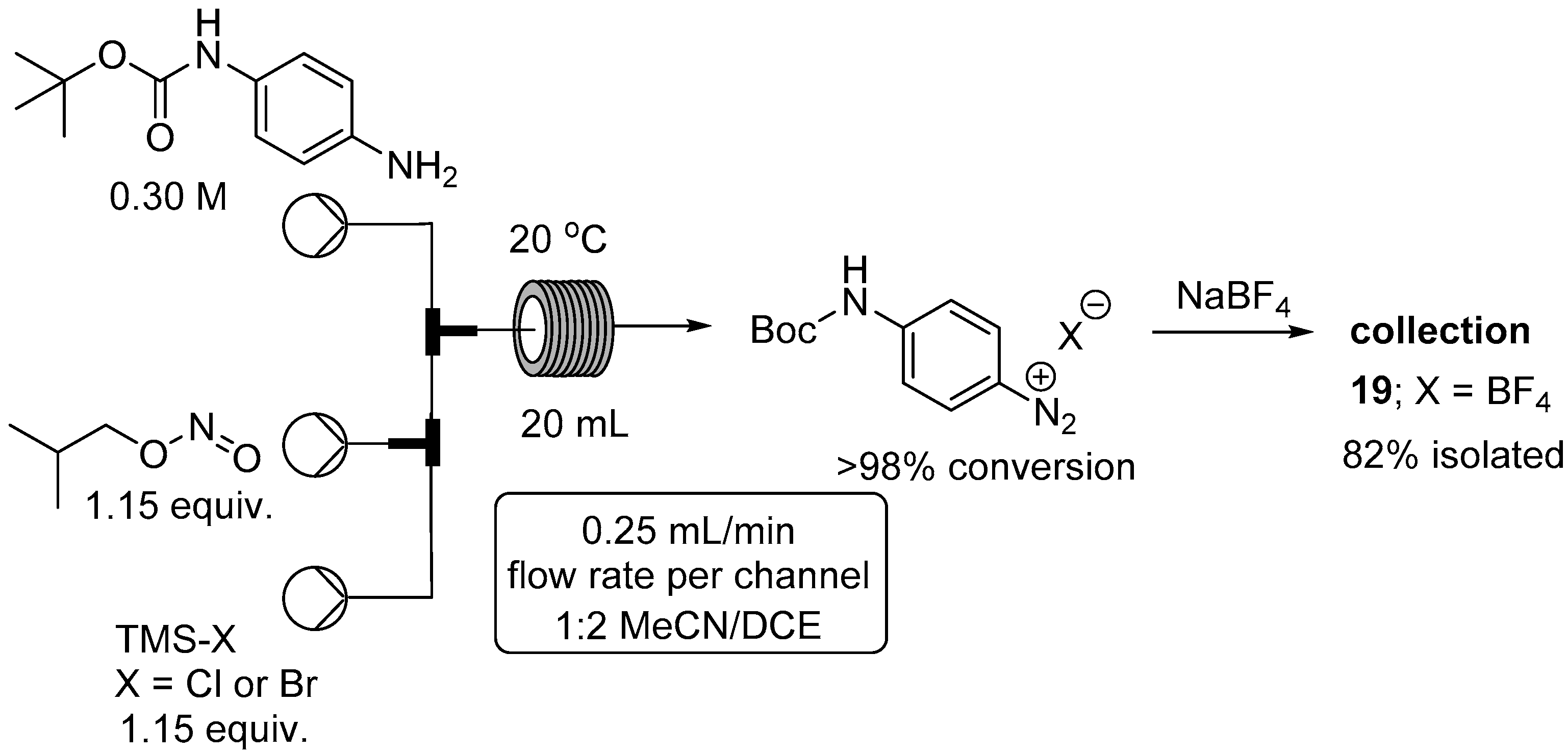
2.2. Formation of Aryl Diazonium Species under Organic Non Aqueous Conditions
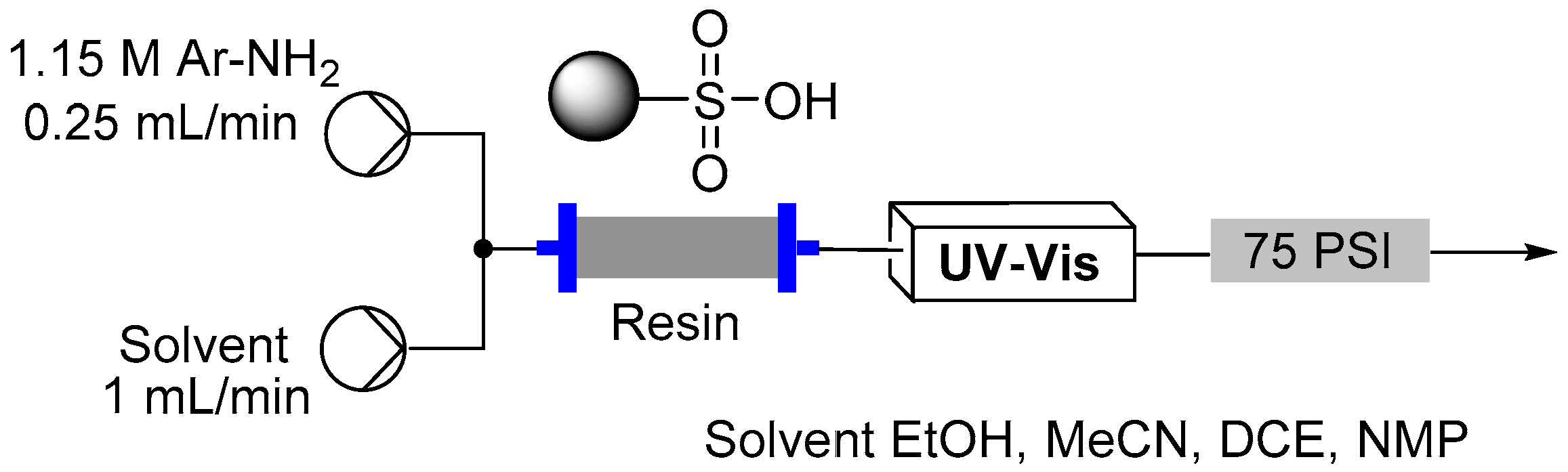
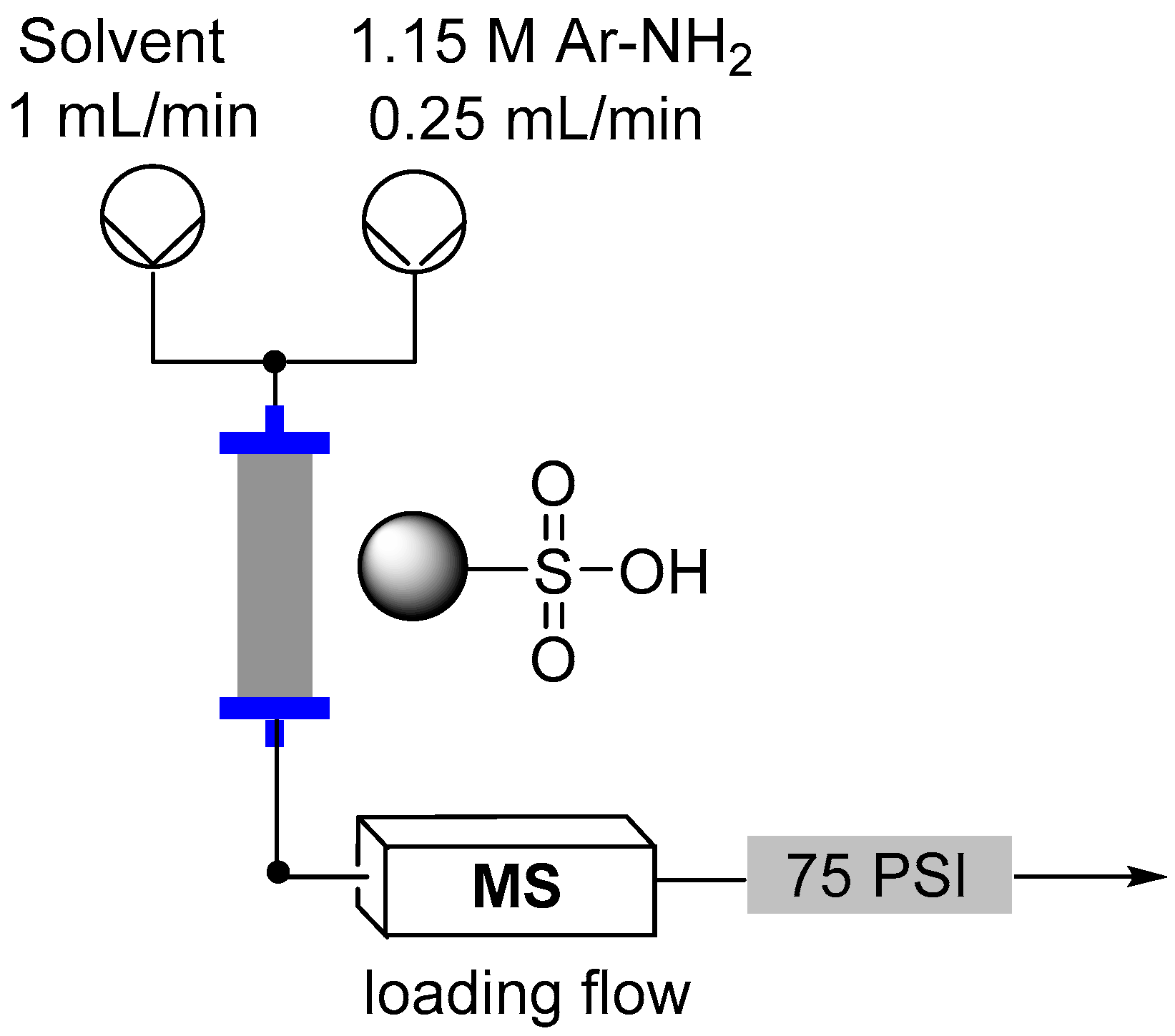
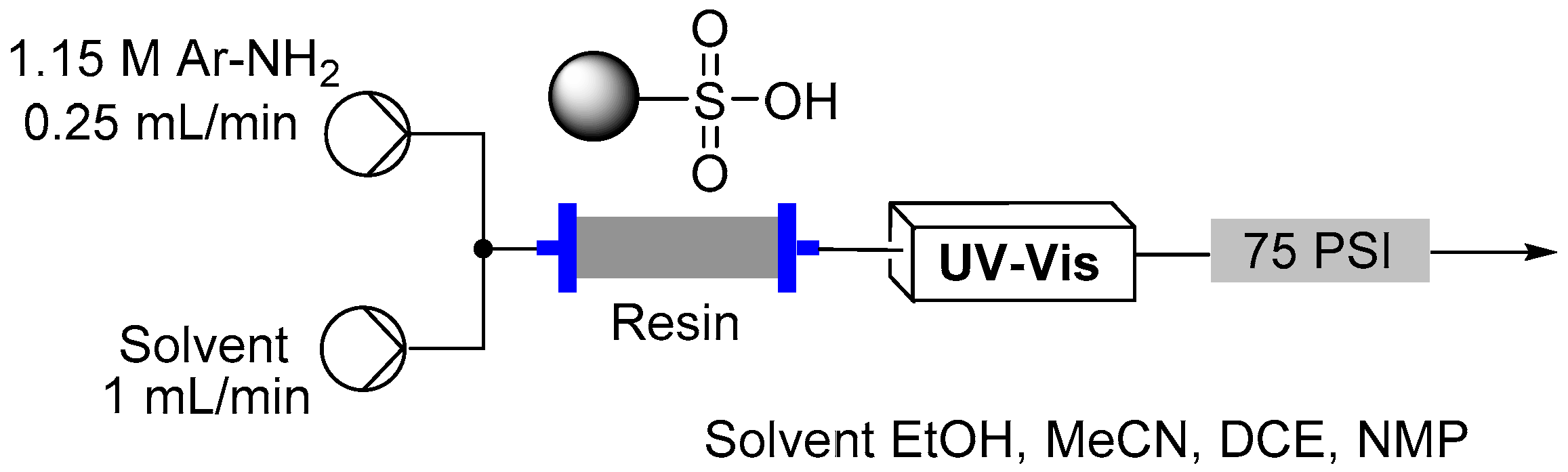
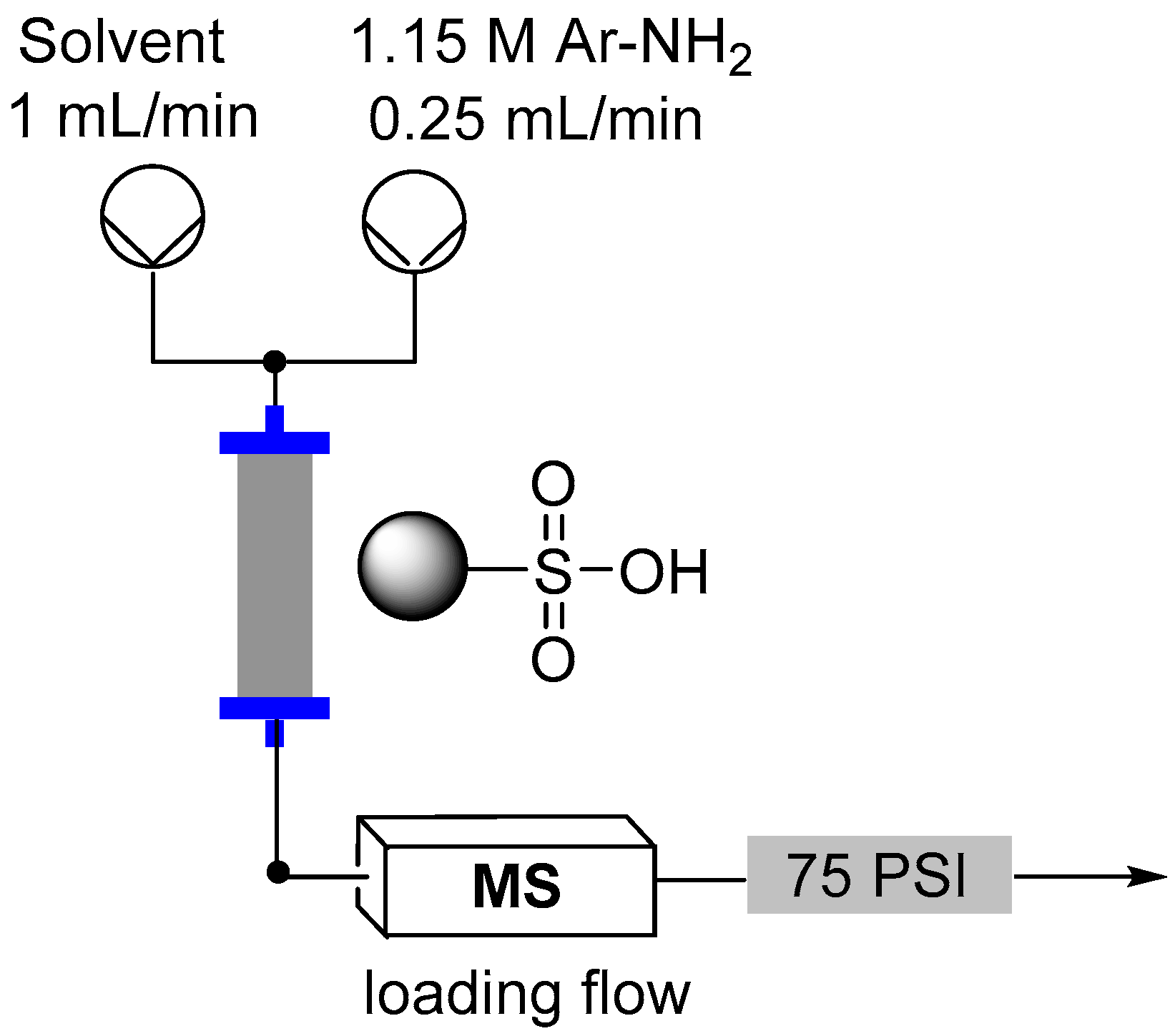
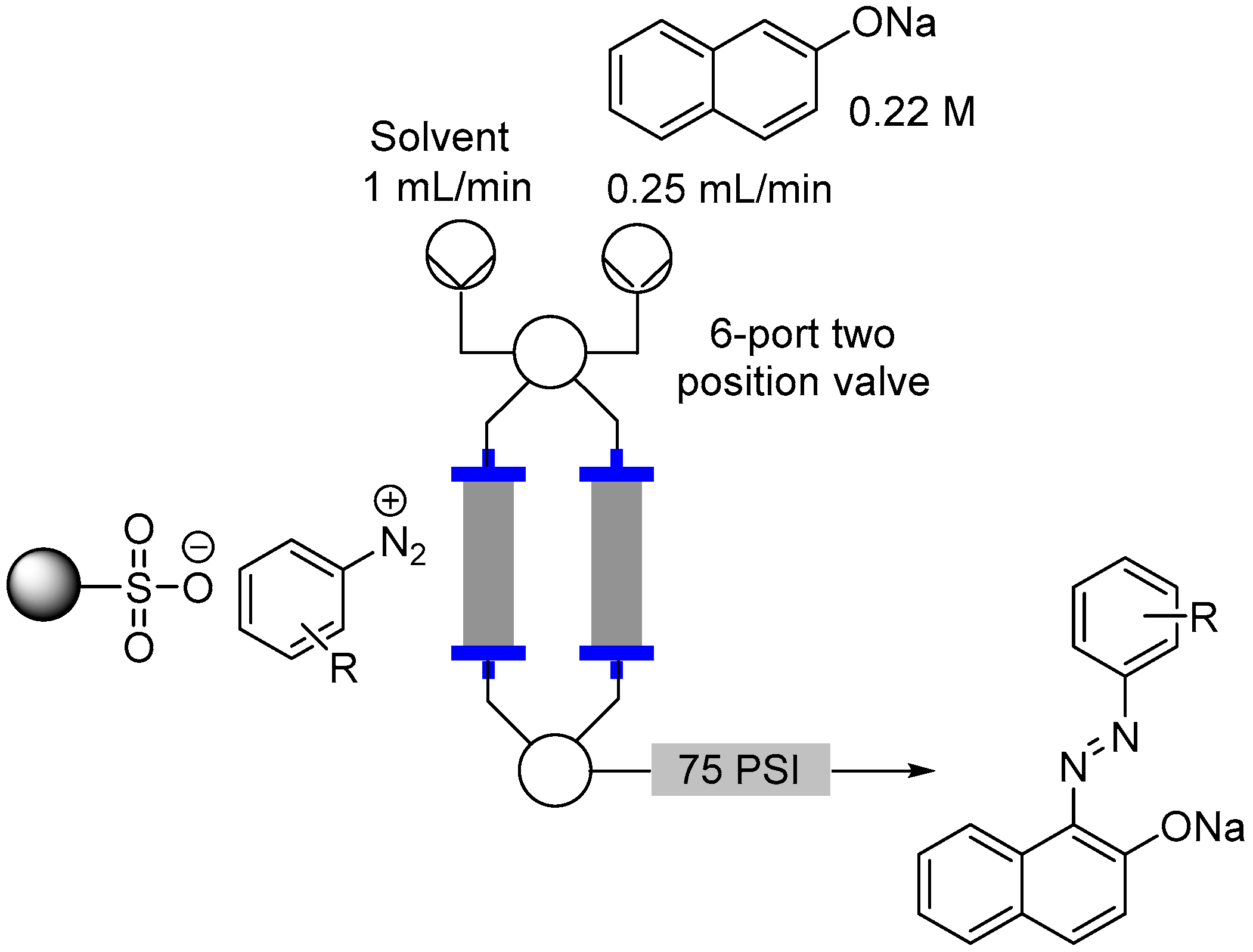
2.3. Formation of Aryl Diazonium Species Using Solid Phase Techniques
3. Experimental Section
3.1. General Information
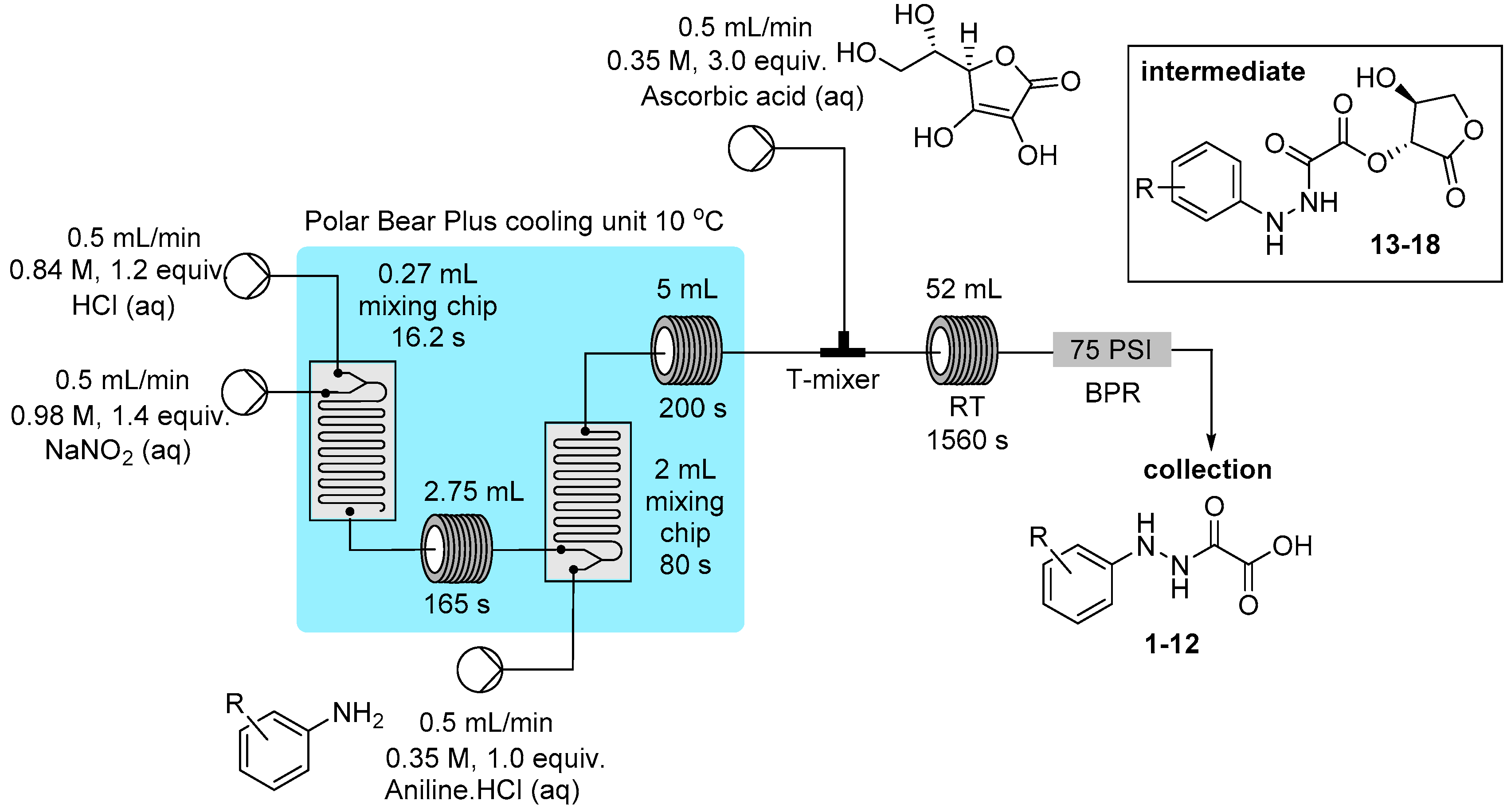
3.2. Reactor Configuration for the Synthesis of Hydrazine Derivatives from Diazonium Salts 1–18
3.3. Product Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Yoshida, J.; Takahashi, Y.; Nagaki, A. Flash chemistry: Flow chemistry that cannot be done in batch. Chem. Commun. 2013, 49, 9896–9904. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, B.; Kappe, C.O. Forbidden Chemistries go flow in API. Chim. Oggi Chem. Today 2015, 33, 18–25. [Google Scholar]
- Baxendale, I.R. The integration of flow reactors into synthetic organic chemistry. J. Chem. Technol. Biotechnol. 2013, 88, 519–552. [Google Scholar] [CrossRef]
- Shang, M.; Noël, T.; Wang, Q.; Su, Y.; Miyabayashi, K.; Hessel, V.; Hasebe, S. 2- and 3-Stage temperature ramping for the direct synthesis of adipic acid in micro-flow packed-bed reactors. Chem. Eng. J. 2015, 260, 454–462. [Google Scholar] [CrossRef]
- Snead, D.R.; Jamison, T.F. A Three-Minute Synthesis and Purification of Ibuprofen: Pushing the Limits of Continuous-Flow Processing. Angew. Chem. Int. Ed. 2015, 54, 983–987. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, S.A.M.W.; Leliveld, J.R.; Becker, R.; Delville, M.M.E.; Nieuwland, P.J.; Koch, K.; Rutjes, F.P.J.T. Continuous Flow Production of Thermally Unstable Intermediates in a Microreactor with Inline IR-Analysis: Controlled Vilsmeier–Haack Formylation of Electron-Rich Arenes. Org. Process Res. Dev. 2012, 16, 934–938. [Google Scholar] [CrossRef]
- Hafner, A.; Ley, S.V. Generation of Reactive Ketenes under Flow Conditions through Zinc-Mediated Dehalogenation. Synlett 2015, 26, 1470–1474. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Process Res. Dev. 2016, 20, 2–25. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R. The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry. Beilstein J. Org. Chem. 2015, 11, 1194–1219. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology—A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef] [PubMed]
- Denčić, I.; Ott, D.; Kralisch, D.; Noël, T.; Meuldijk, J.; de Croon, M.; Hessel, V.; Laribi, Y.; Perrichon, P. Continuous Processing in the Manufacture of Active Pharmaceutical Ingredients and Finished Dosage Forms: An Industry Perspective. Org. Process Res. Dev. 2014, 18, 1326–1338. [Google Scholar] [CrossRef]
- Newman, S.G.; Jensen, K.F. The role of flow in green chemistry and engineering. Green Chem. 2013, 15, 1456–1472. [Google Scholar] [CrossRef]
- Malet-Sanz, L.; Susanne, F. Continuous Flow Synthesis. A Pharma Perspective. J. Med. Chem. 2012, 55, 4062–4098. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.G. Practical Use of Continuous Processing in Developing and Scaling Up Laboratory Processes. Org. Process Res. Dev. 2001, 5, 613–621. [Google Scholar] [CrossRef]
- Oger, N.; Le Grognec, E.; Felpin, F.-X. Handling diazonium salts in flow for organic and material chemistry. Org. Chem. Front. 2015, 2, 590–614. [Google Scholar] [CrossRef]
- Oger, N.; d’Halluin, M.; Le Grognec, E.; Felpin, F.-X. Aryldiazonium Tetrafluoroborate Salts as Green and Efficient Coupling Partners for the Suzuki–Miyaura Reaction: From Optimisation to Mole Scale. Org. Process Res. Dev. 2014, 18, 1786–1801. [Google Scholar] [CrossRef]
- Smith, C.J.; Nikbin, N.; Ley, S.V.; Lange, H.; Baxendale, I.R. A fully automated, multistep flow synthesis of 5-amino-4-cyano-1,2,3-triazoles. Org. Biomol. Chem. 2011, 9, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Browne, D.L.; Baxendale, I.R.; Ley, S.V. Piecing together the puzzle: understanding a mild, metal free reduction method for the large scale synthesis of hydrazines. Tetrahedron 2011, 67, 10296–10303. [Google Scholar] [CrossRef]
- Smith, C.J.; Nikbin, N.; Smith, C.D.; Ley, S.V.; Baxendale, I.R. Flow synthesis of organic azides and the multistep synthesis of imines and amines using a new monolithic triphenylphosphine reagent. Org. Biomol. Chem. 2011, 9, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Malet-Sanz, L.; Madrzak, J.; Ley, S.V.; Baxendale, I.R. Preparation of arylsulfonyl chlorides by chlorosulfonylation of in situ generated diazonium salts using a continuous flow reactor. Org. Biomol. Chem. 2010, 8, 5324–5332. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xie, X.; Dong, H.; Liu, J.; Su, W. Continuous-Flow Process for the Synthesis of m-Nitrothioanisole. Org. Process Res. Dev. 2016, 20, 774–779. [Google Scholar] [CrossRef]
- Deadman, B.J.; Collins, S.G.; Maguire, A.R. Taming Hazardous Chemistry in Flow: The Continuous Processing of Diazo and Diazonium Compounds. Chem. Eur. J. 2015, 21, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.N.; Battilocchio, C.; Lou, S.-B.; Hawkins, J.M.; Ley, S.V. Flow chemistry as a discovery tool to access sp2–sp3 cross-coupling reactions via diazo compounds. Chem. Sci. 2015, 6, 1120–1125. [Google Scholar] [CrossRef]
- Yu, Z.; Tong, G.; Xie, X.; Zhou, P.; Lv, Y.; Su, W. Continuous-Flow Process for the Synthesis of 2-Ethylphenylhydrazine Hydrochloride. Org. Process Res. Dev. 2015, 19, 892–896. [Google Scholar] [CrossRef]
- Nalivela, K.S.; Tilley, M.; McGuire, M.A.; Organ, M.G. Multicomponent, Flow Diazotization/Mizoroki–Heck Coupling Protocol: Dispelling Myths about Working with Diazonium Salts. Chem. Eur. J. 2014, 20, 6603–6607. [Google Scholar] [CrossRef] [PubMed]
- Oger, N.; Le Grognec, E.; Felpin, F.-X. Continuous-Flow Heck–Matsuda Reaction: Homogeneous versus Heterogeneous Palladium Catalysts. J. Org. Chem. 2014, 79, 8255–8262. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lv, Y.; Yu, C.; Su, W. Continuous flow reactor for Balz–Schiemann reaction: A new procedure for the preparation of aromatic fluorides. Tetrahedron Lett. 2013, 54, 1261–1263. [Google Scholar] [CrossRef]
- Chen, M.; Buchwald, S.L. Continuous-Flow Synthesis of 1-Substituted Benzotriazoles from Chloronitrobenzenes and Amines in a C-N Bond Formation/Hydrogenation/Diazotization/Cyclization Sequence. Angew. Chem. Int. Ed. 2013, 52, 4247–4250. [Google Scholar] [CrossRef] [PubMed]
- Delville, M.M.E.; van Hest, J.C.M.; Rutjes, F.P.J.T. Ethyl diazoacetate synthesis in flow. Beilstein J. Org. Chem. 2013, 9, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Widlicka, D.; Boucher, S.; Hayward, C.; Lucas, J.; Murray, J.C.; O’Neil, B.T.; Pfisterer, D.; Samp, L.; van Alsten, J.; et al. Telescoped Flow Process for the Syntheses of N-Aryl Pyrazoles. Org. Process Res. Dev. 2012, 16, 2031–2035. [Google Scholar] [CrossRef]
- Yu, Z.; Lv, Y.; Yu, C. A Continuous Kilogram-Scale Process for the Manufacture of o-Difluorobenzene. Org. Process Res. Dev. 2012, 16, 1669–1672. [Google Scholar] [CrossRef]
- Hu, D.X.; O’Brien, M.; Ley, S.V. Continuous Multiple Liquid–Liquid Separation: Diazotization of Amino Acids in Flow. Org. Lett. 2012, 14, 4246–4249. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.V.; Zemyatova, S.V. Flow-injection spectrophotometry of nitrites based on the diazotization reactions of azine dyes. J. Anal. Chem. 2007, 62, 637–639. [Google Scholar] [CrossRef]
- Behringer, H.; Karrenbauer, K. Continuous Diazotization of Amines. U.S. Patent 4268437, 19 May 1981. [Google Scholar]
- Wootton, R.C.R.; Fortt, R.; de Mello, A.J. On-chip generation and reaction of unstable intermediates—Monolithic nanoreactors for diazonium chemistry: Azo dyes. Lab. Chip 2002, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Guenther, P.M.; Moeller, F.; Henkel, T.; Koehler, J.M.; Groβ, G.A. Formation of Monomeric and Novolak Azo Dyes in Nanofluid Segments by Use of a Double Injector Chip Reactor. Chem. Eng. Technol. 2005, 28, 520–527. [Google Scholar] [CrossRef]
- Fortt, R.; Wootton, R.C.R.; de Mello, A.J. Continuous-Flow Generation of Anhydrous Diazonium Species: Monolithic Microfluidic Reactors for the Chemistry of Unstable Intermediates. Org. Process Res. Dev. 2003, 7, 762–768. [Google Scholar] [CrossRef]
- Mettler Toledo (http://uk.mt.com) See: Carter, C.C.; Lange, H.; Ley, S.V.; Baxendale, I.R.; Wittkamp, B.; Goode, J.G.; Gaunt, N.L. ReactIR Flow Cell: A New Analytical Tool for Continuous Flow Chemical Processing. Org. Process Res. Dev. 2010, 14, 393–402. [Google Scholar] .
- Carter, C.C.; Lange, H.; Ley, S.V.; Baxendale, I.R. The Continuous Flow Synthesis of Butane-2,3-Diacetal Protected Building Blocks Using Microreactors. Org. Biomol. Chem. 2010, 8, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Either the commercial hydrochloride salt was bought or the salt was prepared by the addition of one equivalent of concentrated hydrochloric acid (37%) to the aqueous solution and the mixture sonicated until the aniline fully dissolved.
- Characterized by cleaner visual reaction streams comprising of non-brown homogenous mixtures, the elimination of solution cloudiness and significant ppt or oil formation at higher concentrations. By analysis less by-product formation {diazo dye formation <2% N=N signal at ~2250}.
- Westcott, B.; Enemark, J. Inorganic Electronic Structure and Spectroscopy; Soloman, E., Lever, A., Eds.; Wiley: New York, NY, USA, 1999; Volume II. [Google Scholar] It should be noted that longer interludes will lead to loss of active NO via diffusion through the PFA tubing.
- Wiseman, F.L. Monitoring the Rate of Solvolytic Decomposition of Benzenediazonium Tetrafluoroborate in Aqueous Media Using a pH Electrode. J. Chem. Educ. 2005, 82, 1841. [Google Scholar] [CrossRef]
- Taylor, J.E.; Feltis, T.J. The Hydrolytic Decomposition of Diazonium Salts. I. The Determination of Very Precise Rates in Very Dilute Solutions. J. Am. Chem. Soc. 1952, 74, 1331–1333. [Google Scholar] [CrossRef]
- Canning, P.S.J.; McCrudden, K.; Masnill, H.; Sexton, B. Rates and mechanisms of the thermal solvolytic decomposition of arenediazonium ions. J. Chem. Soc. Perkin Trans. 1999, 2, 2735–2740. [Google Scholar] [CrossRef]
- Smith, B.D. The Investigation of the Decomposition of Diazonium Salts in Aqueous Solution; Cox, J.R., Jr., Ed.; Georgia Institute of Technology: Atlanta, GA, USA, 1966. [Google Scholar]
- Broxton, T.J.; McLeish, M.J. Reactions of aryl diazonium salts and arylazo alkyl ethers. VI. A comparison of the available methods for the measurement of the rate of ionization of (Z)-arylazo alkyl ethers in alcoholic solvents. Aust. J. Chem. 1982, 35, 319–329. [Google Scholar] [CrossRef]
- Moelwyn-Hugh, E.A.; Johnson, P. The kinetics of the decomposition of benzene diazonium chloride in water. Trans. Faraday Soc. 1940, 36, 948–956. [Google Scholar] [CrossRef]
- Hegarty, A.F. Kinetics and mechanisms of reactions involving diazonium and diazo groups. In Diazonium and Diazo Groups; Patai, S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 1978; Volume 2. [Google Scholar]
- Polar Bear Plus Reactor Available from Cambridge Reactor Design. Available online: http://www.cambridgereactordesign.com/polarbearplus/index.html (accessed on 13 July 2016).
- Uniqsis Glass Static Mixer-Reactor Chips. Available online: http://www.uniqsis.com/ (accessed on 13 July 2016).
- Poh, J.-S.; Browne, D.L.; Ley, S.V. A multistep continuous flow synthesis machine for the preparation of pyrazoles via a metal-free amine-redox process. React. Chem. Eng. 2016, 1, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, C.P.; Hellier, P.; Pettman, A.; Watkinson, S. Second-Generation Process Research towards Eletriptan: A Fischer Indole Approach. Org. Process Res. Dev. 2011, 15, 98–103. [Google Scholar] [CrossRef]
- Norris, T.; Bezze, C.; Franz, S.Z.; Stivanello, M. Heavy-Metal-Free Reduction Methodology for Large-Scale Synthesis Applicable to Heterocyclic and Arylhydrazines. Org. Process Res. Dev. 2009, 13, 354–357. [Google Scholar] [CrossRef]
- Doyle, M.P.; Nesloney, C.L.; Shanklin, M.S.; Marsh, C.A.; Brown, K.C. Formation and characterization of 3-O-arenediazoascorbic acids. New stable diazo ethers. J. Org. Chem. 1989, 54, 3785–3789. [Google Scholar] [CrossRef]
- Weiss, R.; Wagner, K.-G. Notizen. Die Erzeugung von Nitrosylsalzen in wasserfreien organischen Medien. Chem. Ber. 1984, 117, 1973–1976. [Google Scholar] [CrossRef]
- Colas, C.; Goeldner, M. An Efficient Procedure for the Synthesis of Crystalline Aryldiazonium Trifluoroacetates—Synthetic Applications. Eur. J. Org. Chem. 1999, 1357–1366. [Google Scholar] [CrossRef]
- Mallia, C.J.; Baxendale, I.R. The Use of Gases in Flow Synthesis. Org. Process Res. Dev. 2016, 20, 327–360. [Google Scholar] [CrossRef]
- Koos, P.; Gross, U.; Polyzos, A.; O’Brien, M.; Baxendale, I.R.; Ley, S.V. Teflon AF-2400 mediated gas–liquid contact in continuous flow methoxycarbonylations and in-line FTIR measurement of CO concentration. Org. Biomol. Chem. 2011, 9, 6903–6908. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Taylor, N.; Polyzos, A.; Baxendale, I.R.; Ley, S.V. Hydrogenation in flow: Homogeneous and heterogeneous catalysis using Teflon AF-2400 to effect gas–liquid contact at elevated pressure. Chem. Sci. 2011, 2, 1250–1257. [Google Scholar] [CrossRef]
- O’Brien, M.; Baxendale, I.R.; Ley, S.V. Flow Ozonolysis Using a Semipermeable Teflon AF-2400 Membrane To Effect Gas−Liquid Contact. Org. Lett. 2010, 12, 1596–1598. [Google Scholar] [CrossRef] [PubMed]
- Baxendale, I.R.; Mallia, C.J.; Brocken, L. Flow chemistry approaches directed at improving chemical synthesis. Green Process Synth. 2013, 2, 211–230. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R.; Ley, S.V. The flow synthesis of heterocycles for natural product and medicinal chemistry applications. Mol. Divers. 2011, 15, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Chaturbhuj, G.U.; Akamanchi, K.G. Copper catalyzed Gomberg–Buchmann–Hey reaction using aryldiazonium tosylate. Tetrahedron Lett. 2011, 52, 4950–4953. [Google Scholar] [CrossRef]
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A New, One-Step, Effective Protocol for the Iodination of Aromatic and Heterocyclic Compounds via Aprotic Diazotization of Amines. Synthesis 2007, 81–84. [Google Scholar] [CrossRef]
- Lu, Y.-T.; Arai, C.; Ge, J.-F.; Ren, W.-S.; Kaiser, M.; Wittlin, S.; Brun, R.; Lu, J.-M.; Ihara, M. Synthesis and in vitro antiprotozoal activities of water-soluble, inexpensive phenothiazinium chlorides. Dyes Pigment. 2011, 91, 44–48. [Google Scholar] [CrossRef]
- Zarei, A.; Khazdooz, L.; Pirisedigh, A.; Hajipour, A.R.; Seyedjamali, H.; Aghaei, H. Aryldiazonium silica sulfates as efficient reagents for Heck-type arylation reactions under mild conditions. Tetrahedron Lett. 2011, 52, 4554–4557. [Google Scholar] [CrossRef]
- Filimonov, V.D.; Trusova, M.; Postnikov, P.; Krasnokutskaya, E.A.; Lee, Y.M.; Hwamg, H.Y.; Kim, H.; Chi, K.-W. Unusually Stable, Versatile, and Pure Arenediazonium Tosylates: Their Preparation, Structures, and Synthetic Applicability. Org. Lett. 2008, 10, 3961–3964. [Google Scholar] [CrossRef] [PubMed]
- Caldarelli, C.; Baxendale, I.R.; Ley, S.V. Clean and efficient synthesis of azo dyes using polymer-supported reagents. Green Chem. 2000, 43–45. [Google Scholar] [CrossRef]
- MP-TsOH Sulfonic Acid Resin Is Available from Biotage AB (Part Number 800463). Available online: http://www.biotage.com (accessed on 13 July 2016).
- SiliaBond® Propylsulfonic Acid (SCX-2) and SiliaBond® Tosic Acid (SCX). Available online: http://www.silicycle.com (accessed on 13 July 2016).
- Nafion® NR50, CAS Number 31175–20–9 (loading 0.8 meq/g) is available from Sigma Aldrich. (accessed on 13 July 2016).
- This approach had been used to great effect in the synthesis of coumarin-8-carbaldehydes. Zak, J.; Ron, D.; Riva, E.; Harding, H.P.; Cross, B.C.S.; Baxendale, I.R. Establishing a Flow Process to Coumarin-8-Carbaldehydes as Important Synthetic Scaffolds. Chem. Eur. J. 2012, 32, 9901–9910. [Google Scholar] .
- Browne, D.L.; Wright, S.; Deadman, B.; Dunnage, S.; Baxendale, I.R.; Turner, R.; Ley, S.V. Continuous flow reaction monitoring using an on-line miniature mass spectrometer. Rapid Commun. Mass Spectrom. 2012, 26, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N.; Akien, G.R.; Savage, R.J.D.; Stanetty, C.; Baxendale, I.R.; Blacker, A.J.; Taylor, B.A.; Woodward, R.L.; Meadows, R.E.; Bourne, R.A. Online quantitative mass spectrometry for the rapid adaptive optimisation of automated flow reactors. React. Chem. Eng. 2016, 1, 96–100. [Google Scholar] [CrossRef]
- Guthrie, J.P. Hydrolysis of esters of oxy acids: pKa values for strong acids; Brønsted relationship for attack of water at methyl; free energies of hydrolysis of esters of oxy acids; and a linear relationship between free energy of hydrolysis and pKa holding over a range of 20 pK units. Can. J. Chem. 1978, 56, 2342–2354. [Google Scholar]
- Eckert, F.; Leito, I.; Kaljurand, I.; Kütt, A.; Klamt, A.; Diedenhofen, M. Prediction of acidity in acetonitrile solution with COSMO-RS. J. Comput. Chem. 2009, 30, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Kütt, A.; Ivo Leito, I.; Kaljurand, I.; Sooväli, L.; Vlasov, V.M.; Yagupolskii, L.M.; Koppel, I.A. A Comprehensive Self-Consistent Spectrophotometric Acidity Scale of Neutral Brønsted Acids in Acetonitrile. J. Org. Chem. 2006, 71, 2829–2838. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Leroux, Y.R.; Hapiot, P.; Downard, A.J. Amine-Terminated Monolayers on Carbon: Preparation, Characterization, and Coupling Reactions. Langmuir 2015, 31, 5071–5077. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, R.A.; Tatlow, J.C. The reactions of certain nitrogen-containing compounds derived from benzotrifluoride. J. Chem. Soc. 1953, 1994–1998. [Google Scholar] [CrossRef]
- Chen, S.; Tsao, M.-L. Genetic Incorporation of a 2-Naphthol Group into Proteins for Site-Specific Azo Coupling. Bioconjugate Chem. 2013, 24, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Tanaka, N.; Morisawa, T.; Nishikuri, M.; Kaji, A. Studies of Diazosulfides. I. A Kinetic Study of the Reaction of Diaryldiazosulfides with β-Naphthol in Alkaline Ethanol. Bull. Chem. Soc. Jpn. 1970, 43, 908–914. [Google Scholar] [CrossRef]
- Mohamed, S.K.; Gomaa, M.A.-M.; Nour El-Din, A.M. Reaction of Triazene 1-Oxides: Novel Synthesis of Solid Arenediazonium Chlorides. J. Chem. Res. 1997, 166–167. [Google Scholar] [CrossRef]
- Barbero, M.; Crisma, M.; Degani, I.; Fochi, R.; Perracino, P. New Dry Arenediazonium Salts, Stabilized to an Exceptionally High Degree by the Anion of o-Benzenedisulfonimide. Synthesis 1998, 8, 1171–1175. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, N.; Zhu, X.; Chen, X.; Zhang, Z. Cyclic Side-Chain Phenylazo Naphthalene Polymers: Enhanced Fluorescence Emission and Surface Relief Grating Formation. Macromol. Rapid Commun. 2012, 33, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Rahimizadeh, M.; Eshghi, H.; Shiri, A.; Ghadamyari, Z.; Matin, M.M.; Oroojalian, F.; Pordeli, P. Fe(HSO4)3 as an Efficient Catalyst for Diazotization and Diazo Coupling Reactions. J. Korean Chem. Soc. 2012, 56, 716–719. [Google Scholar] [CrossRef]
- Tedder, J.M.; Theaker, G. The direct introduction of the diazonium group into aromatic nuclei. Part II. Diazonium salts from aromatic sulphonic acids, carboxylic acids, and nitro-compounds prepared by use of mercuric ions as catalyst. J. Chem. Soc. 1957, 4008–4012. [Google Scholar] [CrossRef]
- Churkina, L.N.; Belyaev, E.Y.; Kazak, Y.Y. Synthesis of Dyes from Aromatic C-Nitroso-N-hydroxytriazenes. Russ. J. Org. Chem. 2001, 37, 680–682. [Google Scholar] [CrossRef]
- Zarchi, M.A.K.; Karimi, M. Diazotization of anilines and diazo coupling with a coupling component mediated by a polymer-supported sodium nitrite and a polymeric acid. J. Appl. Polym. Sci. 2012, 123, 2762–2767. [Google Scholar] [CrossRef]
- Gorelik, M.V.; Lomzakova, V.I.; Khamidova, E.A.; Shteiman, V.Y.; Kuznetsova, M.G.; Andrievsky, A.M. Regioselective Bromination of Anilines in the Presence of Nitrosonium Hydrogensulfate in Concentrated Sulfuric Acid. Mendeleev Comm. 1995, 5, 65–66. [Google Scholar] [CrossRef]
- Chin, A.; Hung, M.-H.; Stock, L.M. Reactions of benzenediazonium ions with adenine and its derivatives. J. Org. Chem. 1981, 46, 2203–2207. [Google Scholar] [CrossRef]
- Keirstead, K.F. The Reduction of Aromatic Nitro Compounds Magnesium and Methyl alcohol. Can. J. Chem. 1953, 31, 1064–1077. [Google Scholar] [CrossRef]
- Valizadeh, H.; Amiri, M.; Hosseinzadeh, F. Nanoparticles of organosilane-based nitrite ionic liquid immobilized on silica for the diazotization of aniline derivatives and subsequent synthesis of azo dyes. Dyes Pigments 2012, 92, 1308–1313. [Google Scholar] [CrossRef]
- Di Donna, L.; Maiuolo, L.; Mazzotti, F.; de Luca, D.; Sindona, G. Assay of Sudan I Contamination of Foodstuff by Atmospheric Pressure Chemical Ionization Tandem Mass Spectrometry and Isotope Dilution. Anal. Chem. 2004, 76, 5104–5108. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1–12, 20–21, 23–35 are available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product A | Aniline Substrate | Yield (%) |
|---|---|---|
| 1 | 2-Br | 79 |
| 2 | 3-Br | 80 |
| 3 | 4-Br | 94 |
| 4 | 2-Cl | 68 |
| 5 | 3-Cl | 83 |
| 6 | 4-Cl | 90 |
| 7 | 2-NO2 | 89 |
| 8 | 3-NO2 | 92 |
| 9 | 4-NO2 | 90 |
| 10 | 2-OMe | 67 |
| 11 | 3-Me | 72 |
| 12 | 4-Me | 77 |
| Supported Acid | Theoretical Loading (mmol/g) | Measured Loading (mmol/g) |
|---|---|---|
| MP-TOSH | 4.45 | 3.55 A/3.84 B |
| Si-SA (SCX) | 0.63 | 0.61/0.62 C/0.60 D |
| Si-SA (SCX2) | 0.54 | 0.53/0.53 C/0.53 D |
| Nafion NR50 | 0.80 | 0.37 A/0.54 B/0.77 E |
| Nafion NR50F | 0.80 | 0.66 |
| Product | Aniline Substrate | Loading Time (min) | Processed Aniline (mmol) A | Loading Efficiency (%) B | Diazo Dye Formation mmol, (%) C |
|---|---|---|---|---|---|
| 20 | 3-F | 32.0 | 16.0 | 60 | 14.2, 89 |
| 21 | 3-CF3 | 29.6 | 14.8 | 55 | 12.7, 86 |
| 22 | 4-CN | 20.0 | 10.0 | 37 | 9.3, 93 |
| 23 | 3-OMe | 47.0 | 23.5 | 87 | 21.3, 91 |
| 24 | 4-OMe | 51.4 | 25.7 | 95 | 22.9, 89 |
| 25 | 2-F,4-CN | 26.8 | 13.4 | 50 | 11.6, 87 |
| 26 | 2,4-F | 24.4 | 12.2 | 46 | 11.0, 90 |
| 27 | 2-Cl,5-OPh | 33.8 | 16.9 | 63 | nd D |
| 28 | 3-Cl | 33.2 | 16.6 | 62 | 14.3, 86 |
| 29 | 3-Me | 41.8 | 20.9 | 77 | 18.1, 87 |
| 30 | 2-Me,5-NO2 | 18.6 | 8.8 | 33 | 5.9, 67 |
| 31 | 2-NO2 | 14.2 | 7.1 | 26 | 5.8, 82 |
| 32 | 4-NO2 | 12.8 | 6.4 | 24 | 4.4, 69 |
| 32 | 4-NO2 | 13.0 | 6.5 | 25 | 5.1, 78 E |
| 33 | 4-Br | 25.2 | 12.6 | 47 | 11.3, 90 |
| 34 | H | 37.2 | 18.6 | 89 | 16.7, 90 |
| 35 | 4-Cl | 26.0 | 13.0 | 58 | 12.2, 94 |
| 36 | Dioxol-5yl | 48.4 | 24.2 | 81 | 20.2, 83 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, T.; Baxendale, I.R.; Baumann, M. Exploring Flow Procedures for Diazonium Formation. Molecules 2016, 21, 918. https://doi.org/10.3390/molecules21070918
Hu T, Baxendale IR, Baumann M. Exploring Flow Procedures for Diazonium Formation. Molecules. 2016; 21(7):918. https://doi.org/10.3390/molecules21070918
Chicago/Turabian StyleHu, Te, Ian R. Baxendale, and Marcus Baumann. 2016. "Exploring Flow Procedures for Diazonium Formation" Molecules 21, no. 7: 918. https://doi.org/10.3390/molecules21070918
APA StyleHu, T., Baxendale, I. R., & Baumann, M. (2016). Exploring Flow Procedures for Diazonium Formation. Molecules, 21(7), 918. https://doi.org/10.3390/molecules21070918