
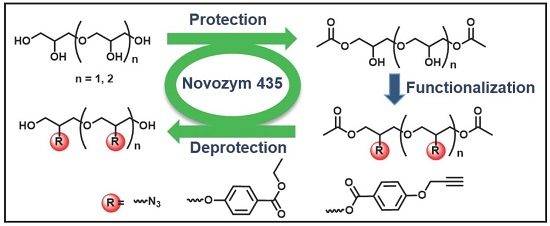
Chemo-Enzymatic Synthesis of Oligoglycerol Derivatives



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
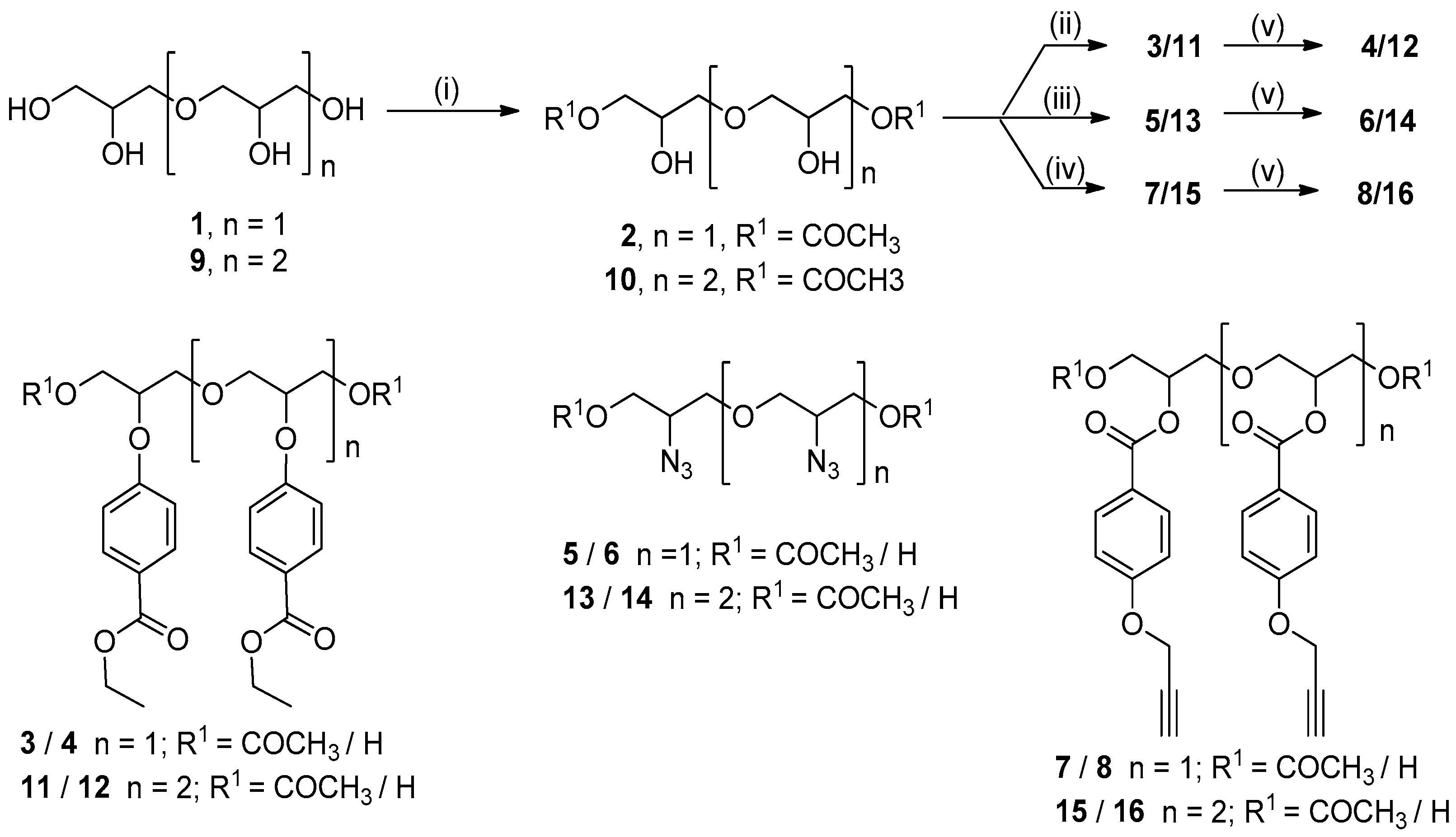
2. Results
3. Experimental Section
3.1. General Information
3.2. Synthesis and Characterization
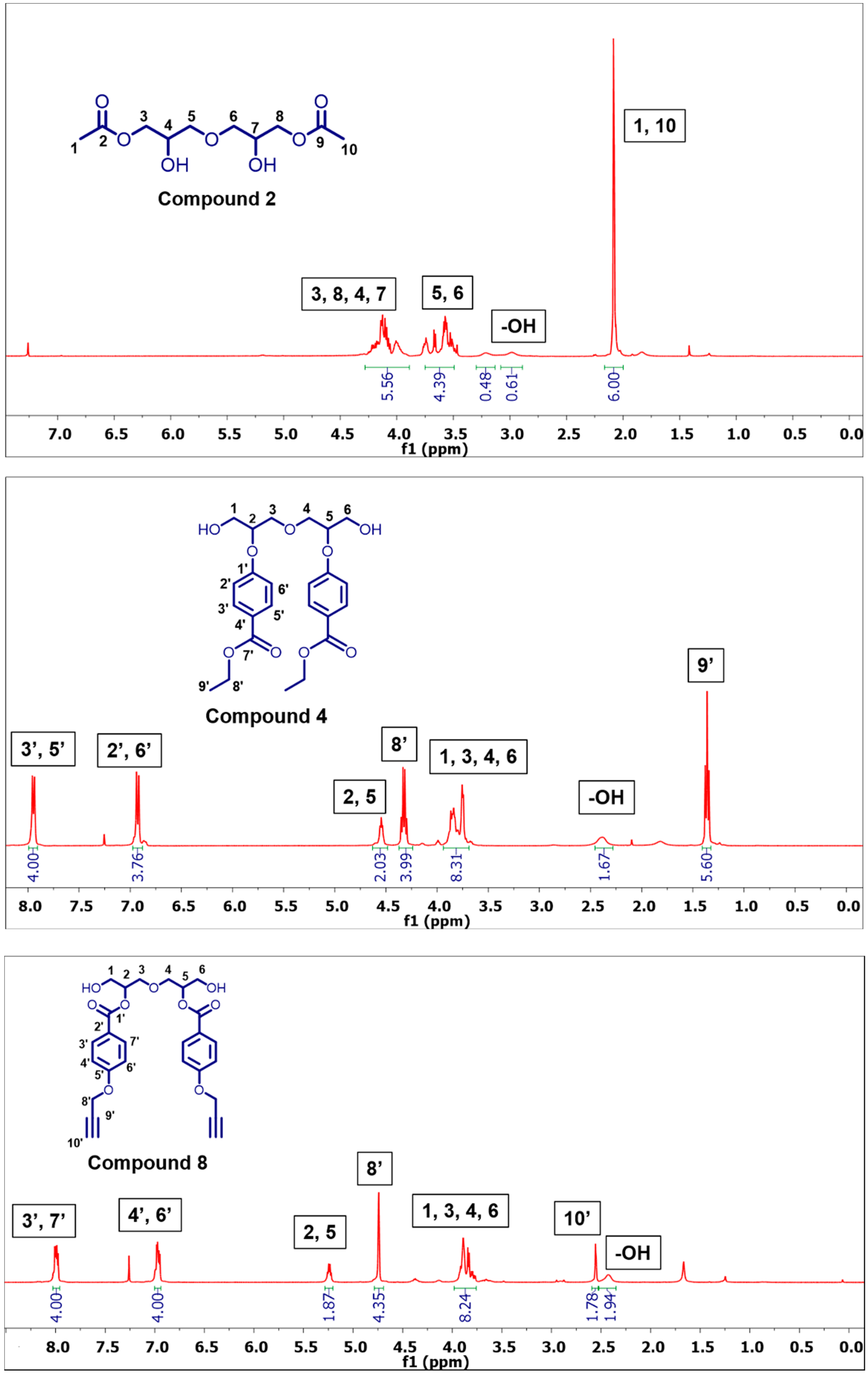
3.2.1. Synthesis of Oxybis(2-hydroxypropan-3,1-diyl) Diacetate (2)
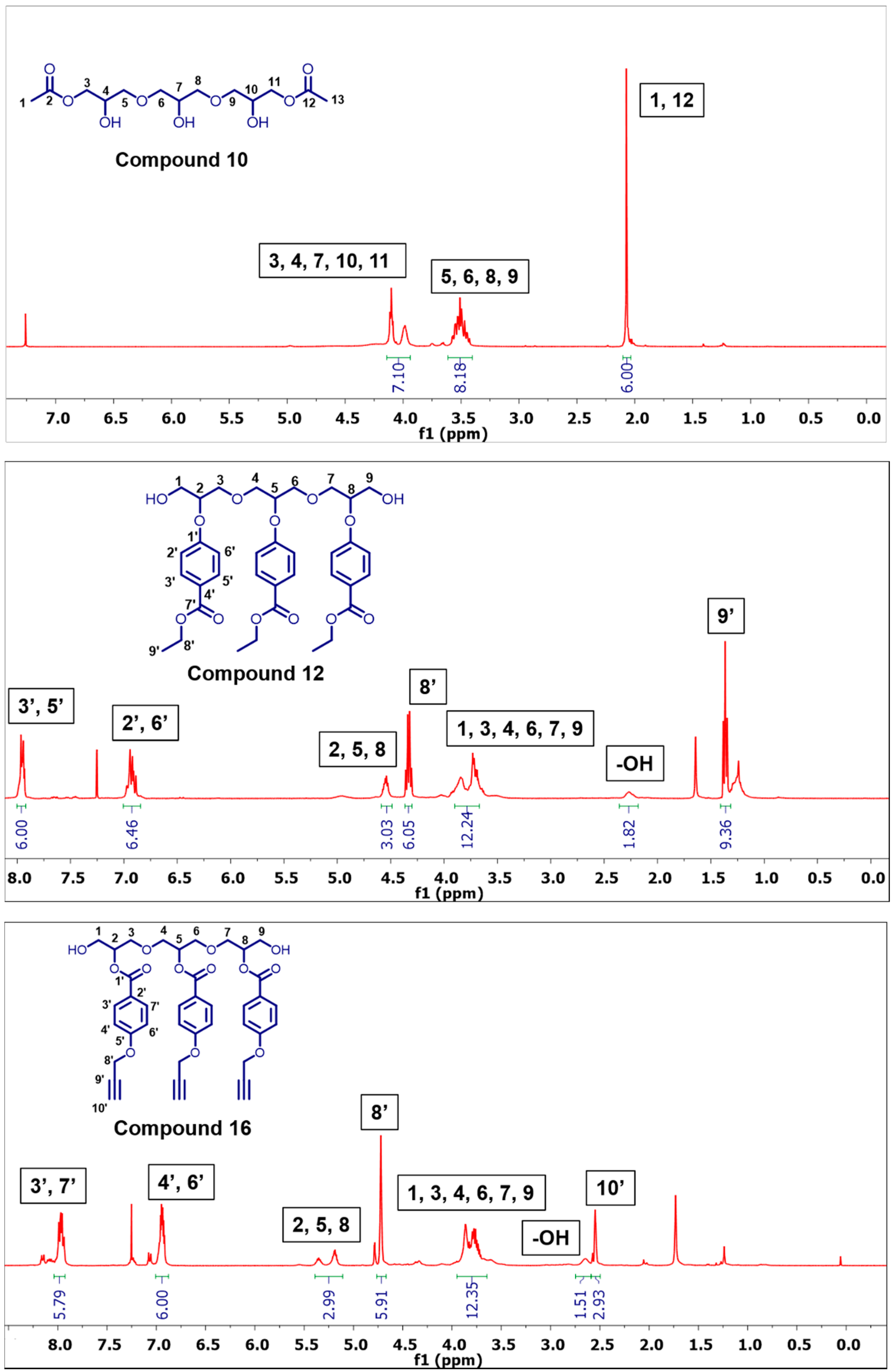
3.2.2. Synthesis of ((2-Hydroxypropane-1,3-diyl)bis(oxy))bis(2-hydroxypropane-3,1-diyl) Diacetate (10)
3.2.3. Synthesis of Diethyl 4,4′-((Oxybis(1-acetoxypropane-3,2-diyl))bis(oxy))dibenzoate (3)
3.2.4. Synthesis of Diethyl 4,4′-((9-(4-(Ethoxycarbonyl)phenoxy)-2,16-dioxo-3,7,11,15-tetraoxahepta-decane-5,13-diyl)bis(oxy))dibenzoate (11)
3.2.5. Synthesis of Oxybis(2-azidopropane-3,1-diyl) Diacetate (5)
3.2.6. Synthesis of ((2-Azidopropane-1,3-diyl)bis(oxy))bis(2-azidopropane-3,1-diyl) Diacetate (13)
3.2.7. Synthesis of Oxybis(1-acetoxypropane-3,2-diyl) bis(4-(prop-2-yn-1-yloxy)benzoate) (7)
3.2.8. Synthesis of 2,16-Dioxo-9-((4-(prop-2-yn-1-yloxy)benzoyl)oxy)-3,7,11,15-tetraoxaheptadecane-5,13-diyl bis(4-(prop-2-yn-1-yloxy)benzoate) (15)
3.2.9. Synthesis of 4,4′-((Oxybis(1-hydroxypropane-3,2-diyl))bis(oxy))dibenzoate (4)
3.2.10. Synthesis of Diethyl 4,4′-((((2-(4-(ethoxycarbonyl)phenoxy)propane-1,3-diyl)bis(oxy))bis(1-hydroxypropane-3,2-diyl))bis(oxy))dibenzoate (12)
3.2.11. Synthesis of 3,3′-Oxybis(2-azidopropan-1-ol) (6)
3.2.12. Synthesis of 3,3′-((2-Azidopropane-1,3-diyl)bis(oxy))bis(2-azidopropan-1-ol) (14)
3.2.13. Synthesis of Oxybis(1-hydroxypropane-3,2-diyl) bis(4-(prop-2-yn-1-yloxy)benzoate) (8)
3.2.14. Synthesis of ((2-((4-(Prop-2-yn-1-yloxy)benzoyl)oxy)propane-1,3-diyl)bis(oxy))bis(1-hydroxypropane-3,2-diyl) bis(4-(Prop-2-yn-1-yloxy)benzoate) (16)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DMF | N,N-dimethylformamide |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| FT-IR | Fourier transform infrared |
| HRMS | high-resolution mass spectrometry |
| IR | infrared |
| MeOH | methanol |
| NMR | nuclear magnetic resonance |
| pKa | acid dissociation constant |
| ppm | parts per million |
| PVA | polyvinyl acetate |
| Q-TOF LCMS | quadrupoletime of flight liquid chromatography mass spectrometry |
| RB | round-bottom |
| rpm | round per min |
| SAR | structure-activity relationship |
| THF | tetrahydrofuran |
| TLC | thin layer chromatography |
| TMS | tetramethylsilane |
References
- Katryniok, B.; Kimura, H.; Skrzynska, E.; Girardon, J.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: perspective for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Cintas, P.; Tagliapietra, S.; Gaudino, E.C.; Palmisano, G.; Cravotto, G. Glycerol: A solvent and a building block of choice for microwave and ultrasound irradiation procedure. Green Chem. 2014, 16, 1056–1065. [Google Scholar] [CrossRef]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilization of renewable ressources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- García, J.; Marín, H.; Pires, E. Glycerol based solvents: Synthesis, properties and applications. Green Chem. 2014, 16, 1007–1033. [Google Scholar] [CrossRef]
- Diaz-Alvarez, A.E.; Francos, J.; Lastra-Barreira, B.; Crochet, P.; Cadierno, V. Glycerol and derived solvents: New sustainable reaction media for organic synthesis. Chem. Commun. 2011, 47, 6208–6227. [Google Scholar] [CrossRef] [PubMed]
- Len, C.; Merlot, A.S.; Postel, D.; Ronco, G.; Villa, P.; Goubert, C.; Jeufrault, E.; Mathon, B.; Simon, H. Synthesis and antifungal activity of novel bis (dithiocarbamate) derivatives of glycerol. J. Agric. Food Chem. 1996, 44, 2856–2858. [Google Scholar] [CrossRef]
- Len, C.; Postel, D.; Ronco, G.; Villa, P.; Goubert, C.; Jeufrault, E.; Mathon, B.; Simon, H. Synthesis of carbamic esters derivatives of itols: Antifungal activity against various crop diseases. J. Agric. Food Chem. 1997, 45, 3–6. [Google Scholar] [CrossRef]
- Rafin, C.; Veignie, E.; Sancholle, M.; Postel, D.; Len, C.; Villa, P.; Ronco, G. Synthesis and antifungal activity of novel bisdithiocarbamate derivatives of carbohydrates against Fusarium oxysporum f. sp. Lini. J. Agric. Food Chem. 2000, 48, 5283–5287. [Google Scholar] [CrossRef] [PubMed]
- DeSousa, R.; Thurier, C.; Len, C.; Pouilloux, Y.; Barrault, J.; Jerome, F. Regioselective functionalization of glycerol with a dithiocarbamate moiety: An environmentally friendly route to safer fungicides. Green Chem. 2011, 13, 1129–1132. [Google Scholar] [CrossRef]
- Saggadi, H.; Luart, D.; Thiebault, N.; Polaert, I.; Estel, L.; Len, C. Toward the synthesis of 6-hydroxyquinoline starting from glycerol via improved microwave-assisted modified Skraup reaction. Catal. Commun. 2014, 44, 15–18. [Google Scholar] [CrossRef]
- Len, C.; Luque, R. Continuous flow transformations of glycerol to valuable products: An overview. Sustain. Chem. Process. 2014, 2. [Google Scholar] [CrossRef]
- Saggadi, H.; Luart, D.; Thiebault, N.; Polaert, I.; Estel, L.; Len, C. Quinoline and phenanthroline preparation starting from glycerol via improved microwave-assisted modified Skraup reaction. RSC Adv. 2014, 4, 21456–21464. [Google Scholar] [CrossRef]
- Saggadi, H.; Polaert, I.; Luart, D.; Len, C.; Estel, L. Microwaves under pressure for the continuous production of quinolone from glycerol. Catal. Today 2015, 255, 66–74. [Google Scholar] [CrossRef]
- Zhou, C.H.; Beltramini, J.N.; Fana, Y.X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Richter, M. Oligomerization of glycerol: A critical review. Eur. J. Lipid Sci. Technol. 2011, 113, 100–117. [Google Scholar] [CrossRef]
- Shi, Y.; Dayoub, W.; Chen, G.R.; Lemaire, M. Selective synthesis of 1-O-alkyl glycerol and diglycerol ethers by reductive alkylation of alcohols. Green Chem. 2010, 12, 2189–2195. [Google Scholar] [CrossRef]
- Cassel, S.; Debaig, C.; Benvegnu, T.; Chaimbault, P.; Lafosse, M.; Plusquellec, D.; Rollin, P. Original synthesis of linear, branched and cyclic oligoglycerol standards. Eur. J. Org. Chem. 2001, 875–896. [Google Scholar] [CrossRef]
- Sutter, M.; Dayoub, W.; Métay, E.; Raoul, Y.; Lemaire, M. 1-O-Alkyl (di)glycerol ethers synthesis from methyl esters and triglycerides by two pathways: catalytic reductive alkylation and transesterification/reduction. Green Chem. 2013, 15, 786–797. [Google Scholar] [CrossRef]
- Katryniok, B.; Paul, S.; Baca, V.; Reye, P. Glycerol dehydration to acrolein in the context of new uses of glycerol. Green Chem. 2010, 12, 2079–2098. [Google Scholar] [CrossRef]
- Urata, K. Unique block molecules based on glycerol skeleton as C-3 building blocks for liquid crystals formation by self-assembly and their future potential for the “nano-chemistry”. Eur. J. Lipid Sci. Technol. 2003, 105, 542–556. [Google Scholar] [CrossRef]
- Sonnati, M.O.; Amigoni, S.; Taffin de Givenchy, E.P.; Darmanin, T.; Chouletb, O.; Guittard, F. Glycerol carbonate as a versatile building block for tomorrow: Synthesis, reactivity, properties and applications. Green Chem. 2013, 15, 283–306. [Google Scholar] [CrossRef]
- Kirk, O.; Christensen, M.W. Lipases from Candida antartica: Unique biocatalysts from a unique origin. Org. Proc. Res. Dev. 2002, 6, 446–451. [Google Scholar] [CrossRef]
- Anderson, E.M.; Larsson, K.M.; Krik, O. One biocatalyst-many applications: The use of Candida antartica B-lipase in organic synthesis. Biocatal. Biotransform. 1998, 16, 181–204. [Google Scholar] [CrossRef]
- Krik, O.; Bjorkling, F.; Godtfredsen, S.E.; Larsen, T.O. Fatty acid specificity in lipase-catalyzed synthesis of glucoside esters. Biocatalysis 1992, 6, 127–134. [Google Scholar] [CrossRef]
- Gupta, S.; Jalal, S.; Kumar, S.; Haag, R.; Sharma, S.K. A simple and convenient chemoenzymatic approach for the synthesis of valuable triacylglycerol-based dendritic building blocks. Indian J. Chem. 2012, 51B, 1376–1387. [Google Scholar]
- Gupta, S.; Schade, B.; Kumar, S.; Böttcher, C.; Sharma, S.K.; Haag, R. Dendronized multiamphiphilic polymers as non-ionic nanotransporters for biomedical applications: Synthesis, aggregation behavior and transport properties. Small 2013, 9, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Gupta, S.; Böttcher, C.; Khandare, J.; Sharma, S.K.; Haag, R. Dendronized multifunctional amphiphilic polymers as efficient nanocarriers for biomedical applications. Macromol. Rapid Commun. 2015, 36, 254–251. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Billamboz, M.; Leonard, E.; Len, C.; Böttcher, C.; Prasad, A.K.; Haag, R.; Sharma, S.K. Self-assembly, photoresponsive behavior and transport potential of azobenzene grafted dendronized polymeric amphiphiles. RSC Adv. 2015, 5, 48301–48310. [Google Scholar] [CrossRef]
- Kumar, S.; Achazi, K.; Böttcher, C.; Licha, K.; Haag, R.; Sharma, S.K. Encapsulation and cellular internalization of cyanine dye using amphiphilic dendronized polymers. Eur. Polym. J. 2015, 69, 416–428. [Google Scholar] [CrossRef]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Legros, V.; Vanhaverbecke, C.; Souard, F.; Len, C.; Desire, J. β-Cyclodextrin-glycerol dimers: Synthesis and NMR conformational analysis. Eur. J. Org. Chem. 2013, 2013, 2583–2590. [Google Scholar] [CrossRef]
- Espeel, P.; du Prez, F.E. “Click”-inspired chemistry in macromolecular science: Matching recent progress and user expectations. Macromolecules 2015, 48, 2–14. [Google Scholar] [CrossRef]
- Saku, O.; Ishida, H.; Atsumi, E.; Sugimoto, Y.; Kodaira, H.; Kato, Y.; Shirakura, S.; Nakasato, Y. Discovery of novel 5,5-diarylpentadienamides as orally available transient receptor potential vanilloid 1 (TRPV1) antagonists. J. Med. Chem. 2012, 55, 3436–3451. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Y.; Yin, T.T.; Li, X.L.; Su, M.; Xue, Y.X.; Yu, Z.P.; Liu, N.; Yin, J.; Wu, Z.Q. Chiroptical and thermotropic properties of helical styrenic polymers: effect of achiral group. Macromolecules 2014, 47, 7021–7029. [Google Scholar] [CrossRef]
- Sharma, D.; Khandelwal, A.; Sharma, R.K.; Bhatia, S.; Reddy, L.C.; Olsen, C.E.; Wengel, J.; Parmar, V.S.; Prasad, A.K. Synthesis and regioselective deacylation studies on peracylated 2′-azido arabino- and ribo-thymine nucleosides: towards 5′-O,2′-N-linked oligonucleotides. Indian J. Chem. 2009, 48B, 1712–1720. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.



© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.K.; Nguyen, R.; Galy, N.; Haag, R.; Sharma, S.K.; Len, C. Chemo-Enzymatic Synthesis of Oligoglycerol Derivatives. Molecules 2016, 21, 1038. https://doi.org/10.3390/molecules21081038
Singh AK, Nguyen R, Galy N, Haag R, Sharma SK, Len C. Chemo-Enzymatic Synthesis of Oligoglycerol Derivatives. Molecules. 2016; 21(8):1038. https://doi.org/10.3390/molecules21081038
Chicago/Turabian StyleSingh, Abhishek K., Remi Nguyen, Nicolas Galy, Rainer Haag, Sunil K. Sharma, and Christophe Len. 2016. "Chemo-Enzymatic Synthesis of Oligoglycerol Derivatives" Molecules 21, no. 8: 1038. https://doi.org/10.3390/molecules21081038