
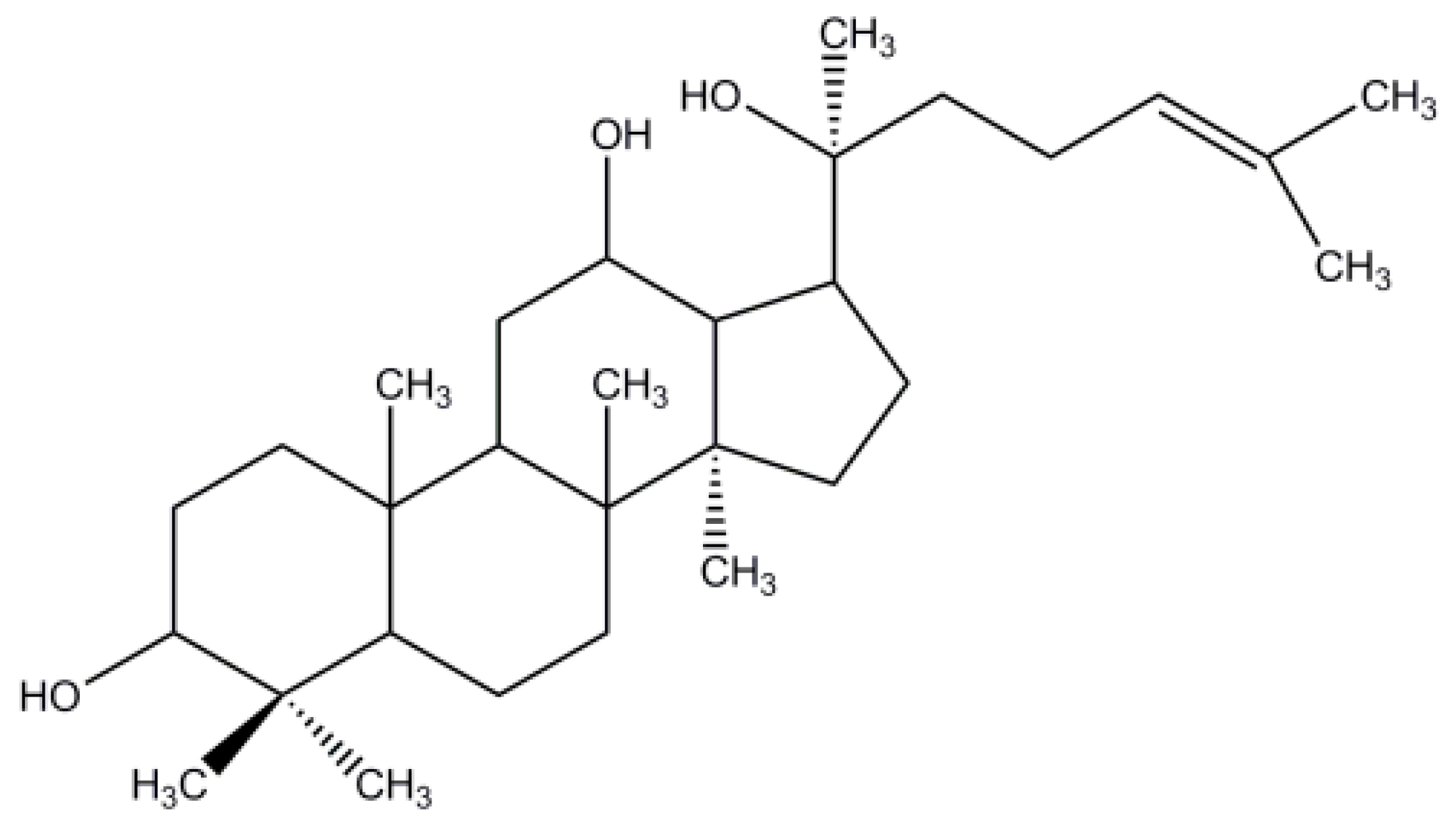
Characterization, Molecular Docking, and In Vitro Dissolution Studies of Solid Dispersions of 20(S)-Protopanaxadiol

 ,
,
Abstract
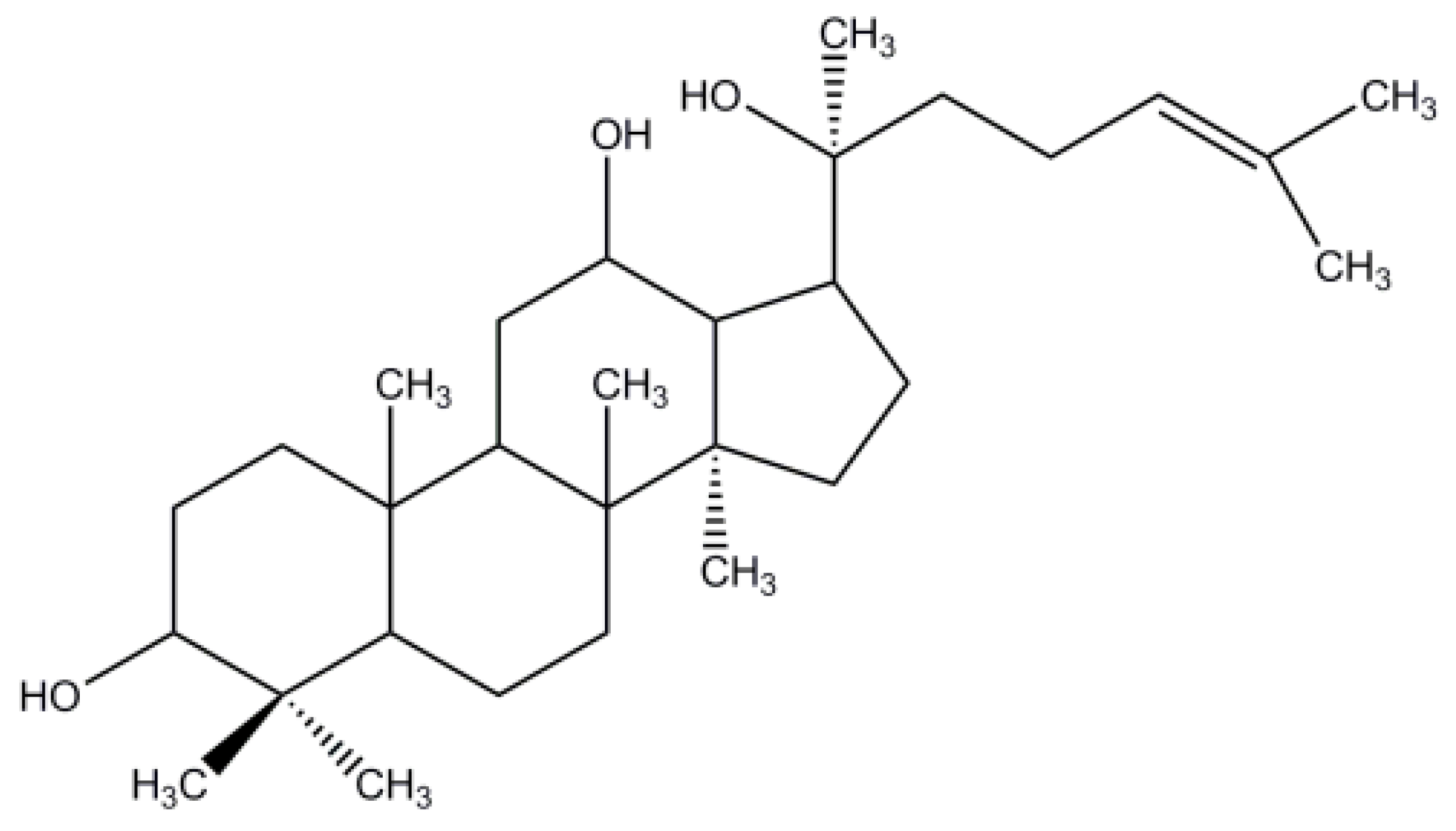
:1. Introduction
2. Results and Discussion
2.1. Drug Loading Efficiency and Solubility Measurement
2.2. Characterization
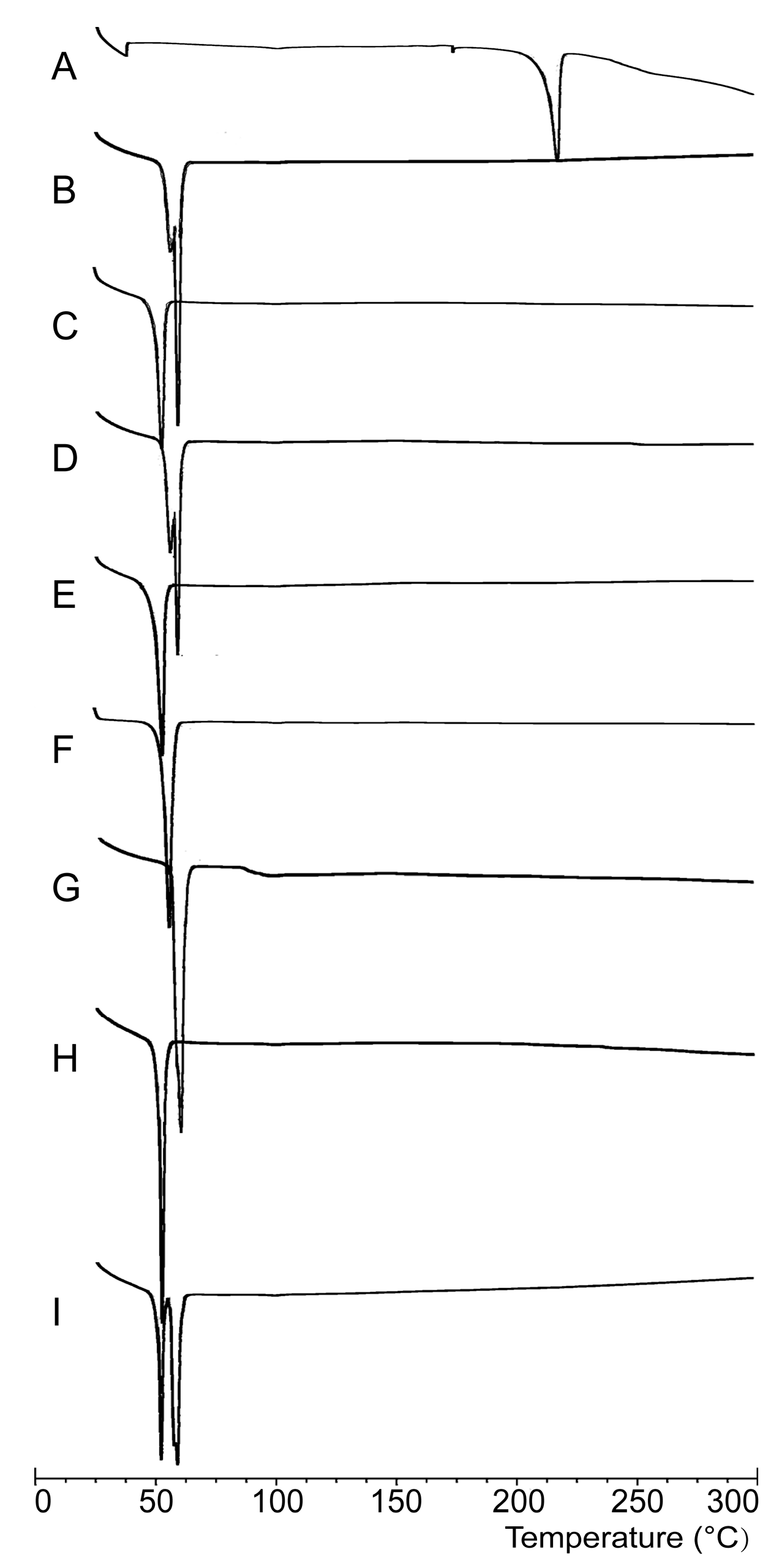
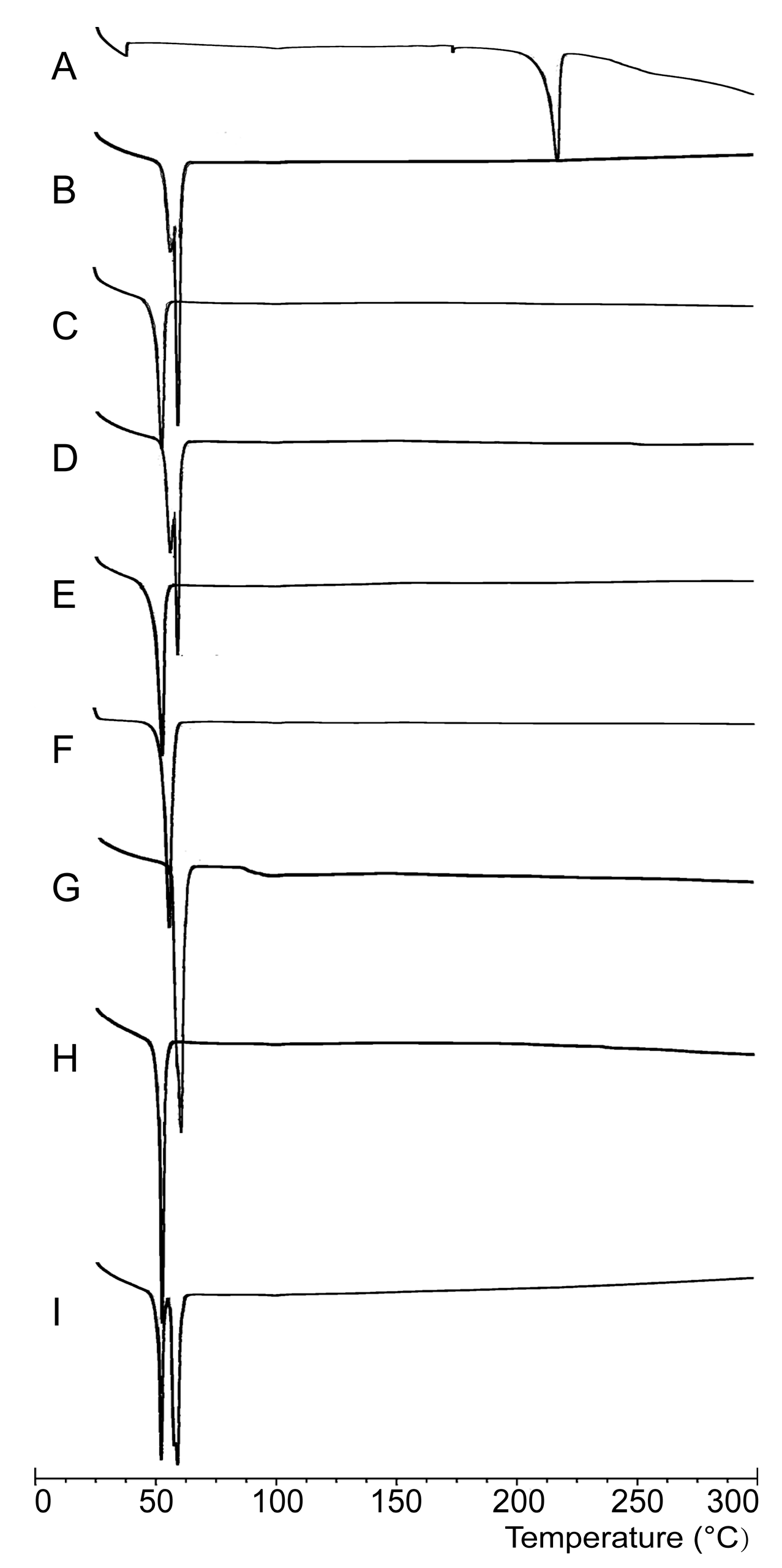
2.2.1. DSC Analysis
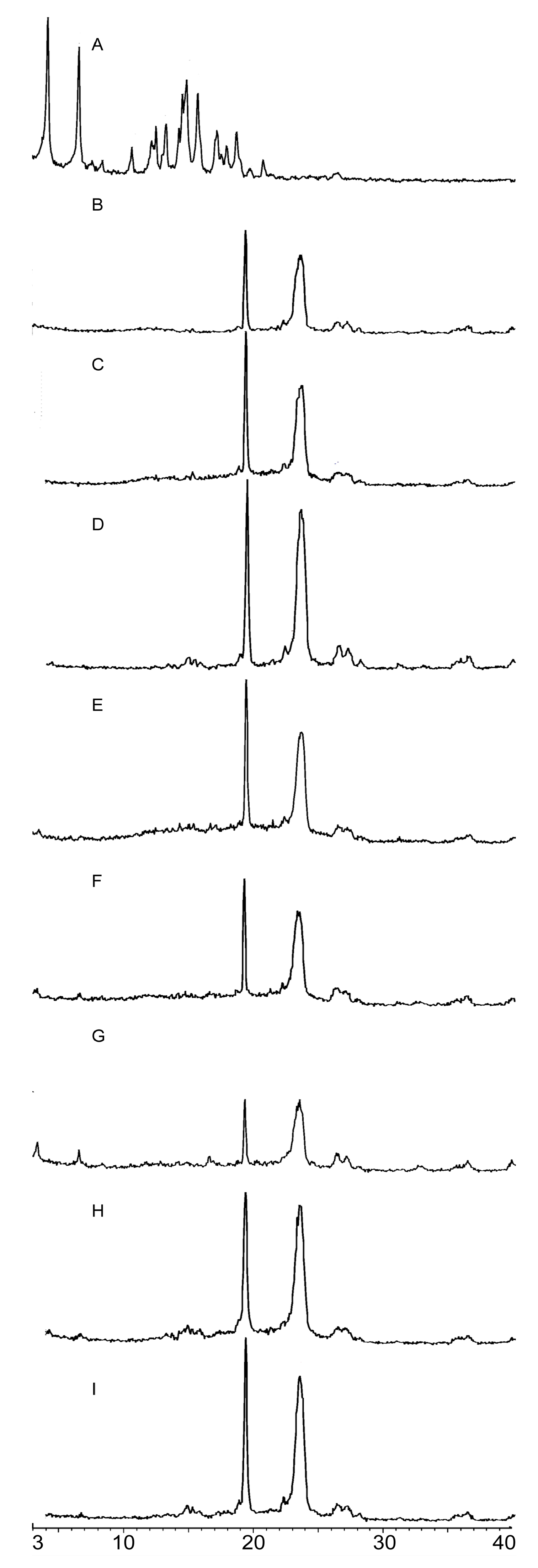
2.2.2. PXRD Analysis
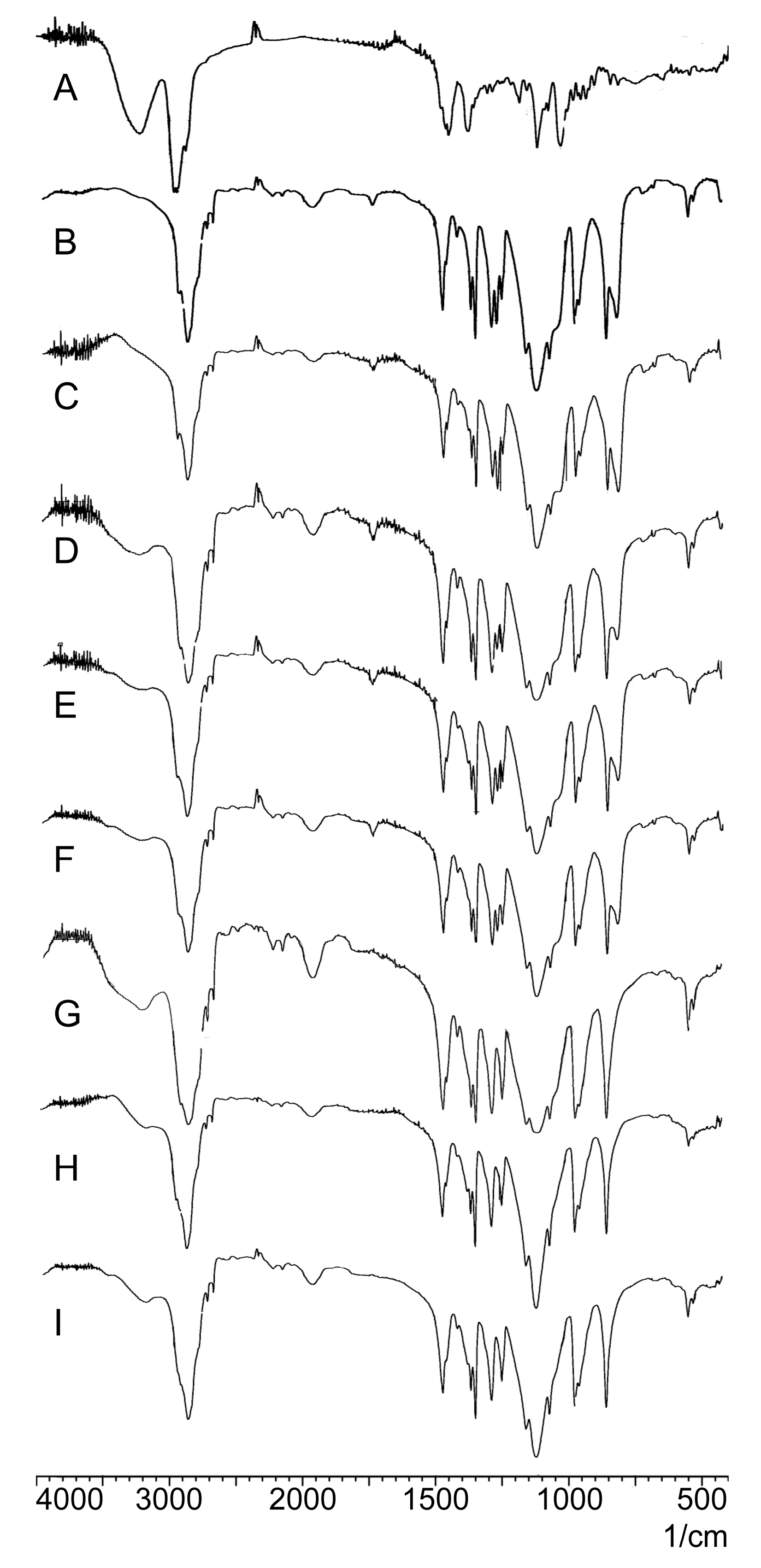
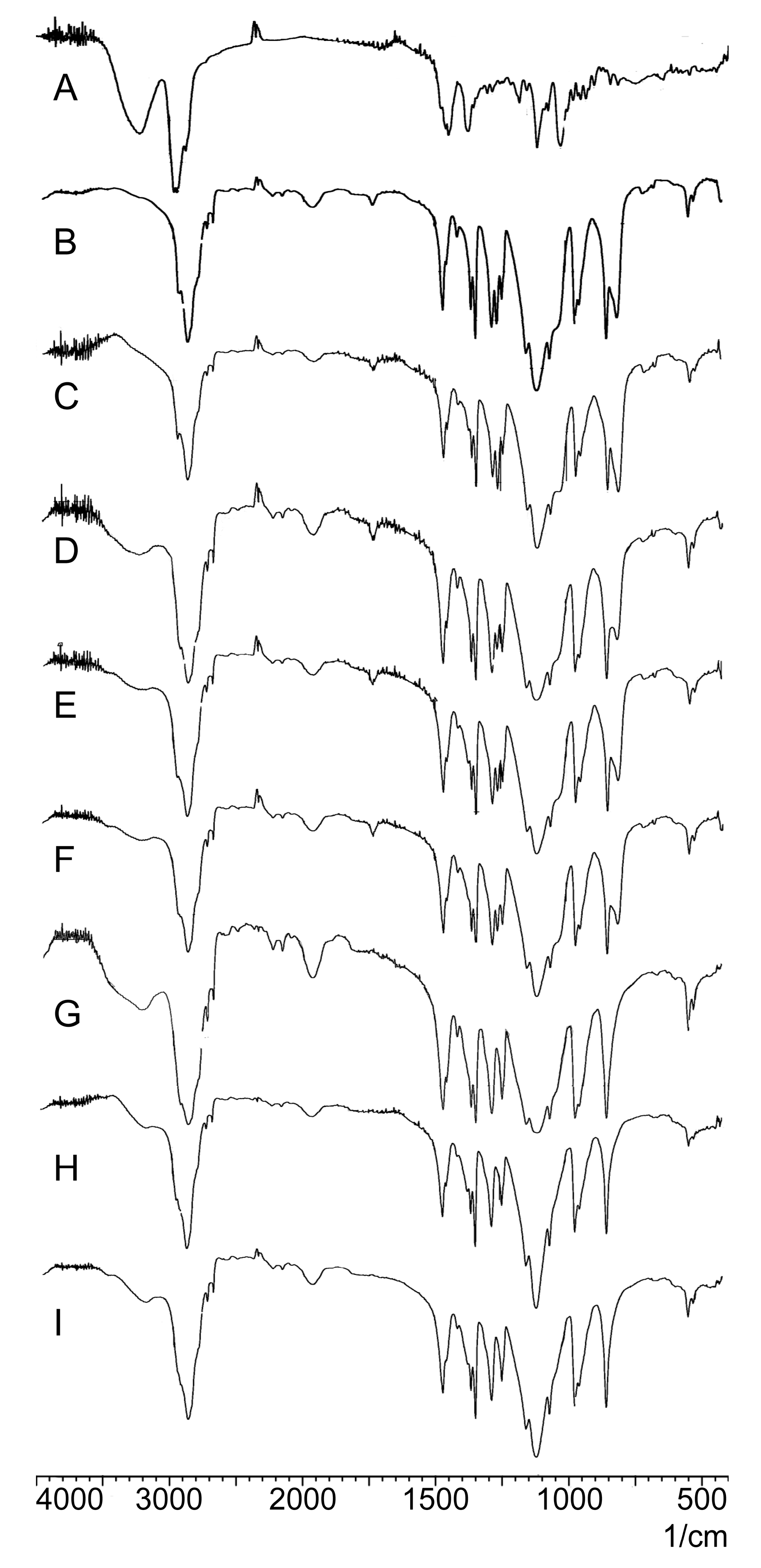
2.2.3. FTIR Analysis
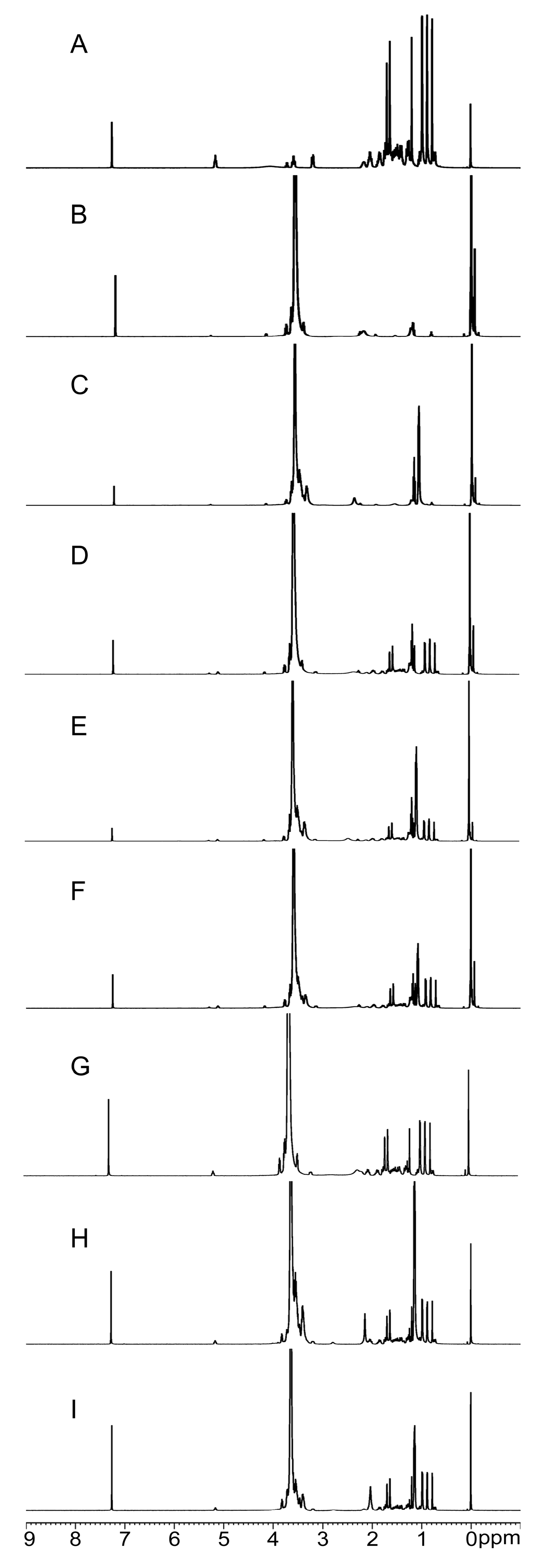
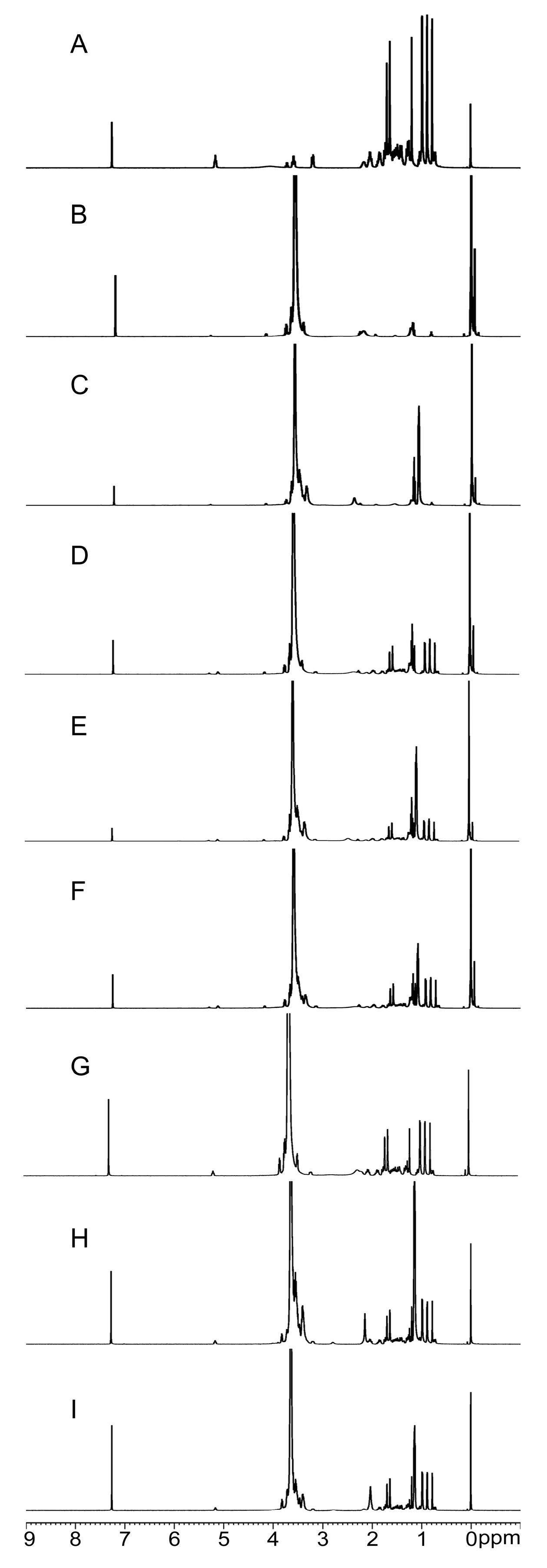
2.2.4. NMR Analysis
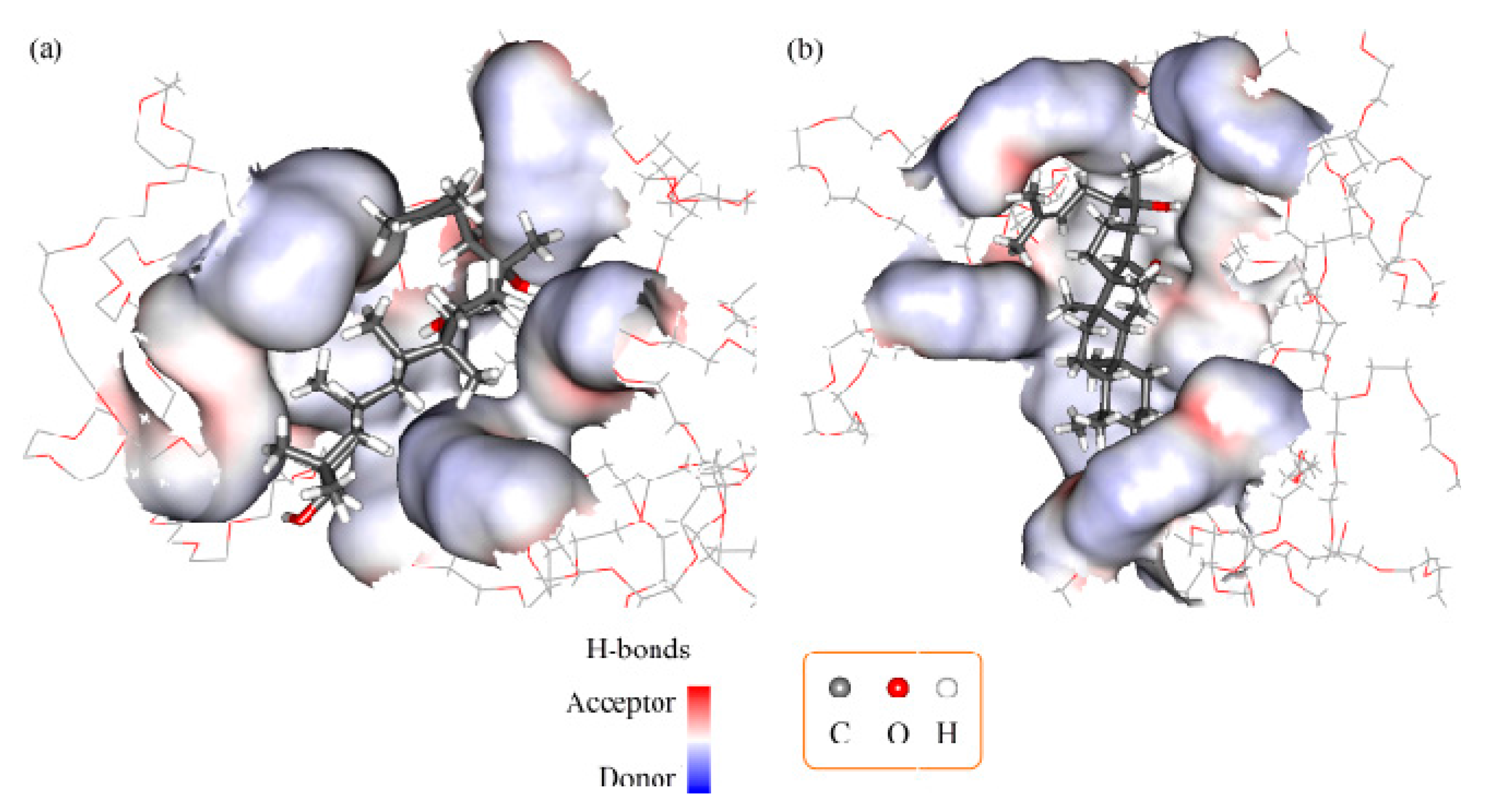
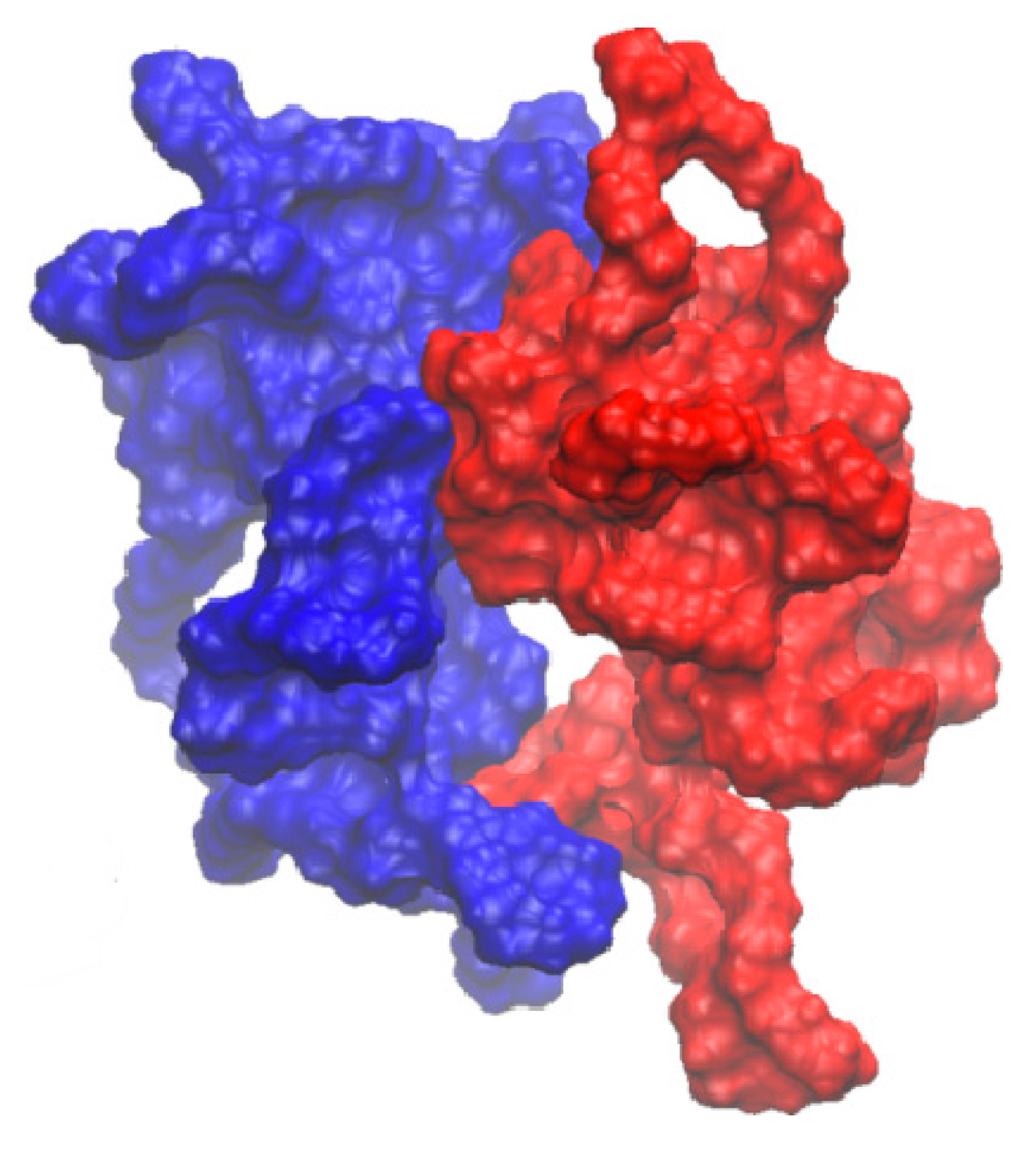
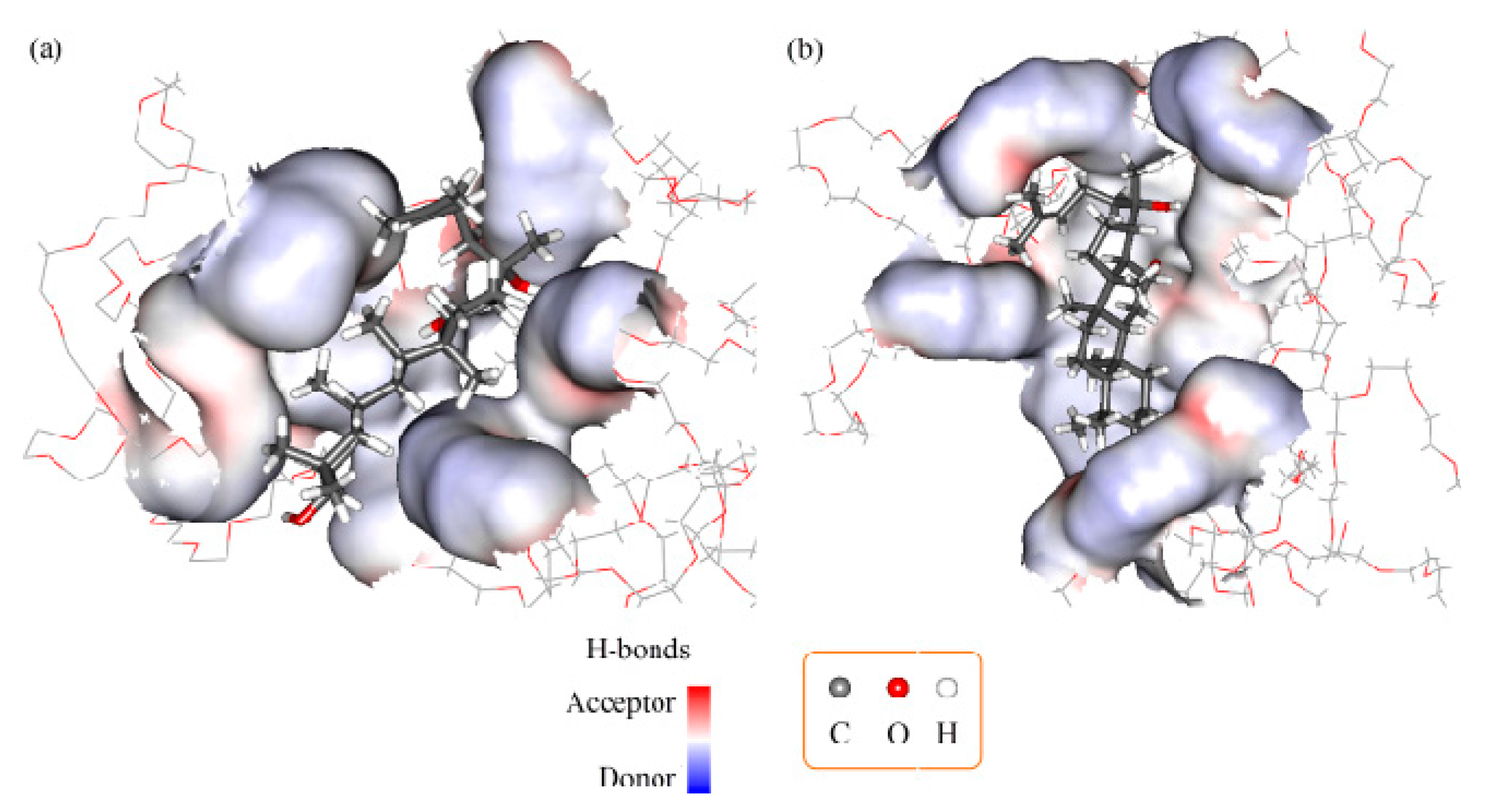
2.3. Molecular Docking and Dynamics Calculation
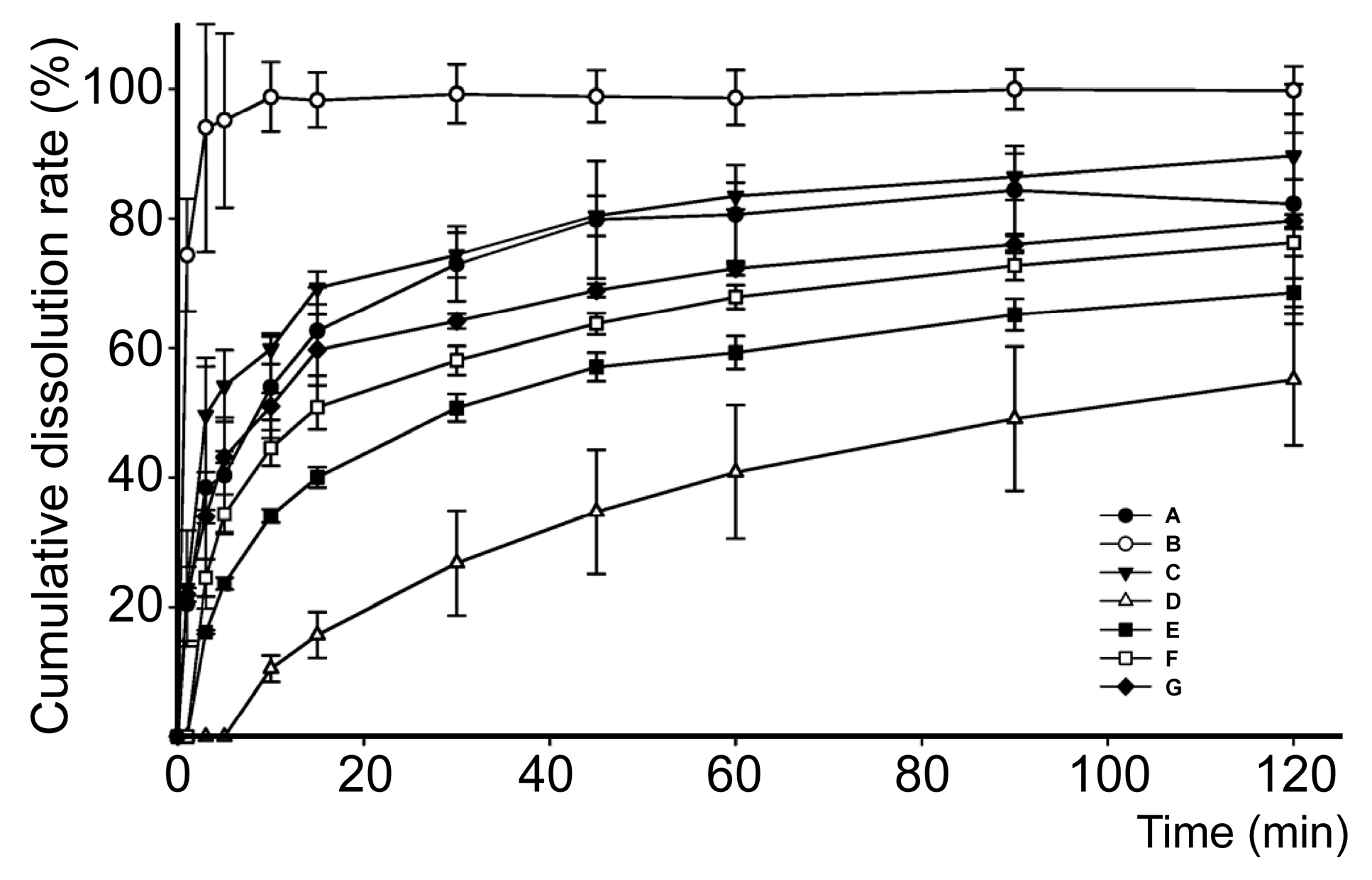
2.4. In Vitro Dissolution Study
3. Materials and Methods
3.1. Materials
3.2. Formulation Design
3.3. SD Preparation
3.4. Physical Mixture Preparation
3.5. Determination of Drug Loading Efficiency
3.6. Characterization of SDs
3.6.1. DSC Analysis
3.6.2. PXRD Analysis
3.6.3. FTIR Analysis
3.6.4. Nuclear Magnetic Resonance
3.7. Molecular Docking and Dynamics Calculation
3.8. Solubility Measurements
3.9. In Vitro Dissolution Study
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Helms, S. Cancer Prevention and Therapeutics: Panax Ginseng. Altern. Med. Rev. 2004, 9, 259–274. [Google Scholar] [PubMed]
- Xiang, Y.Z.; Shang, H.C.; Gao, X.M.; Zhang, B.L. A comparison of the ancient use of ginseng in traditional Chinese medicine with modern pharmacological experiments and clinical trials. Phytother. Res. 2008, 22, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.F.; Chiou, W.F.; Zhang, J.T. Comparison of the pharmacological effects of Panax ginseng and Panax quinquefolium. Acta Pharmacol. Sin. 2008, 29, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, Y.; Rayburn, E.R.; Hill, D.L.; Wang, H.; Zhang, R. In vitro anti-cancer activity and structure-activity relationships of natural products isolated from fruits of Panax ginseng. Cancer Chemother. Pharmacol. 2007, 59, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Radad, K.; Gille, G.; Liu, L.; Rausch, W.-D. Use of Ginseng in Medicine with Emphasis on Neurodegenerative Disorders. J. Pharmacol. Sci. 2006, 100, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Anoja, S.A.; Ji, A.W.; Yuan, C.-S. Ginseng pharmacology - Multiple constituents and multiple actions. Biochem. Pharmacol. 1999, 58, 1685–1693. [Google Scholar]
- Kennedy, O.D.; Scholey, B.A. Ginseng: Potential for the enhancement of cognitive performance and mood. Pharmacol. Biochem. Behav. 2003, 75, 687–700. [Google Scholar] [CrossRef]
- Endale, M.; Im, E.J.; Lee, J.Y.; Kim, S.D.; Yayeh, T.; Song, Y.B.; Kwak, Y.S.; Kim, C.; Kim, S.H.; Roh, S.S.; et al. Korean Red Ginseng Saponin Fraction Rich in Ginsenoside-Rb1, Rc and Rb2 Attenuates the Severity of Mouse Collagen-Induced Arthritis. Mediat. Inflamm. 2014, 2014, 748964:1–748964:14. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Z.; Hu, Y.; Wu, W.Y.; Ye, M.; Guo, D.A. Saponins in the genus Panax L. (Araliaceae): A systematic review of their chemical diversity. Phytochemistry 2014, 106, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Choi, H.; Kim, N.; Kim, D. Anxiolytic-like Effects of Ginsenosides Rg3 and Rh2 from Red Ginseng in the Elevated Plus-Maze Model. Planta Med. 2009, 75, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yao, Q.; Chen, C. Ginseng Compounds: An Update on their Molecular Mechanisms and Medical Applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as Anticancer Agents: In vitro and in vivo Activities, Structure–Activity Relationships, and Molecular Mechanisms of Action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Bai, L.P.; Wong, V.K.; Zhou, H.; Wang, J.R.; Liu, Y.; Jiang, Z.H.; Liu, L. The In Vitro Structure-Related Anti-Cancer Activity of Ginsenosides and Their Derivatives. Molecules 2011, 16, 10619–10630. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ye, M.; Guo, H.; Tian, Y.; Zhang, J.; Zhou, J.; Hu, Y.; Guo, D. Biotransformation of 20(S)-protopanaxadiol by Mucor spinosus. Phytochemistry 2009, 70, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, X.M.; Wang, Q.J.; Zhang, D.L.; Fang, Z.M.; Wang, C.Y.; Wang, Z.; Sun, B.S.; Wu, H.; Sung, C.K. Enzymatic preparation of 20(S, R)-protopanaxadiol by transformation of 20(S, R)-Rg3 from black ginseng. Phytochemistry 2010, 71, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.H.; Yeom, S.J.; Park, C.S.; Lee, K.W.; Oh, D.K. Production of aglycon protopanaxadiol via compound K by a thermostable β-glycosidase from Pyrococcus furiosus. Appl. Microbiol. Biotechnol. 2011, 89, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, C.; Lu, M.; Sun, F.; Fu, Y.; Jin, F. Purification and characterization of new special ginsenosidase hydrolyzing multi-glycisides of protopanaxadiol ginsenosides, ginsenosidase type I. Chem. Pharm. Bull. 2007, 55, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Sung, J.-H.; Lee, S.-J.; Moon, C.-K.; Lee, B.-H. Antitumor activity of a novel ginseng saponin metabolite in human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer Lett. 1999, 144, 39–43. [Google Scholar] [CrossRef]
- Hasegawa, H.; Lee, K.-S.; Nagaoka, T.; Tezuka, Y.; Uchiyama, M.; Kadota, S.; Saiki, I. Pharmacokinetics of ginsenoside deglycosylated by intestinal bacteria and its transformation to biologically active fatty acid esters. Biol. Pharm. Bull. 2000, 23, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ko, W.; Kim, J.; Sung, J.; Moon, C.; Lee, B. Induction of apoptosis by a novel intestinal metabolite of ginseng saponin via cytochrome c-mediated activation of caspase-3 protease. Biochem. Pharmacol. 2000, 60, 677–685. [Google Scholar] [CrossRef]
- Bae, E.; Han, M.; Choo, M.; Park, S.; Kim, D. Metabolism of 20(S)- and 20(R)-Ginsenoside Rg3 by Human Intestinal Bacteria and Its Relation to in Vitro Biological Activities. Biol. Pharm. Bull. 2002, 25, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhou, Q.; Hang, Y.; Bu, X.; Jia, W. Antiestrogenic effect of 20S-protopanaxadiol and its synergy with tamoxifen on breast cancer cells. Cancer 2007, 109, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, Z.; Sun, Y.; Liu, K.; Wang, Z. Ginsenoside 20(S)-Protopanaxadiol Inhibits the Proliferation and Invasion of Human Fibrosarcoma HT1080 cells. Basic Clin. Pharmacol. Toxicol. 2006, 98, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, C.; Murakami, K.; Hasegawa, H.; Murata, J.; Saiki, I. An intestinal bacterial metabolite of ginseng protopanaxadiol saponins has the ability to induce apoptosis in tumor cells. Biochem. Biophys. Res. Commun. 1998, 246, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Hang, M.H.; Chen, J.; Chen, S.L.; Wang, X.T. Preparation and study in vitro of 20(S)-protopanaxadiol pharmacosome. China J. Chin. Mater. Med. 2010, 35, 842–846. (in Chinese). [Google Scholar]
- Wang, B.; Wang, Y.; Pu, Y.; Xu, B.; Tao, J.; Zhang, T. Apparent Oil/water Partition Coefficient of 20(S)-PPD and Its Intestinal Absorption in Rats. Chin. J. Inf. Tradit. Chin. Med. 2011, 18, 50–61. [Google Scholar]
- Xie, H.T.; Wang, G.J.; Sun, J.G.; Tucker, I.; Zhao, X.C.; Xie, Y.Y.; Li, H.; Jiang, X.L.; Wang, R.; Xu, M.J.; et al. High performance liquid chromatographic-mass spectrometric determination of ginsenoside Rg3 and its metabolites in rat plasma using solid-phase extraction for pharmacokinetic studies. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2005, 818, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.; Li, D.; Zhong, D. Identification of 20(S)-Protopanaxadiol Metabolites in Human Liver Microsomes and Human Hepatocytes. Drug Metab. Dispos. Biol. Fate Chem. 2011, 39, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, X.; Wang, L.; Meng, Q.; Liu, W. Stereoselective Property of 20(S)-Protopanaxadiol Ocotillol Type Epimers Affects Its Absorption and Also the Inhibition of P-Glycoprotein. PLoS ONE 2014, 9, e98887. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.; Tran, T.T.; Park, J.B.; Lee, B.J. Controlled release systems containing solid dispersions: Strategies and mechanisms. Pharm. Res. 2011, 28, 2353–2378. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.M.; Avachat, A.M. Enhancement of solubility and permeability of candesartan cilexetil by using different pharmaceutical interventions. Curr. Drug Deliv. 2011, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Rudraraju, V.S. A systematic approach to design and prepare solid dispersions of poorly water-soluble drug. AAPS PharmSciTech 2014, 15, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.; Piao, Z.; Kim, T.; Cao, Q.; Kim, A.; Lee, B. Effect of solubilizing and microemulsifying excipients in polyethylene glycol 6000 solid dispersion on enhanced dissolution and bioavailability of ketoconazole. Arch. Pharm. Res. 2005, 28, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, H.; Buckton, G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech 2009, 10, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Joshi, V.Y. Evaluation of SLS: APG mixed surfactant systems as carrier for solid dispersion. AAPS PharmSciTech 2008, 9, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Damian, F.; Blaton, N.; Kinget, R.; Mooter, G.V.d. Physical stability of solid dispersions of the antiviral agent UC-781 with PEG 6000, Gelucire® 44/14 and PVP K30. Int. J. Pharm. 2002, 244, 87–98. [Google Scholar] [CrossRef]
- Ahuja, N.; Katare, O.P.; Singh, B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur. J. Pharm. Biopharm. 2007, 65, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Kolasinac, N.; Kachrimanis, K.; Homsek, I.; Grujic, B.; Ethuric, Z.; Ibric, S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int. J. Pharm. 2012, 436, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Chatterji, A.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Shah, N. Evaluation of solid state properties of solid dispersions prepared by hot-melt extrusion and solvent co-precipitation. Int. J. Pharm. 2008, 355, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Sahoo, J.; Murthy, P.N.; Giradkar, R.P.; Avari, J.G. Enhancement of dissolution rate of gliclazide using solid dispersions with polyethylene glycol 6000. AAPS PharmSciTech 2008, 9, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Sawicki, W.; Haber, K.; Knapik, J.; Wojnarowska, Z.; Paluch, M.; Lepek, P.; Hawelek, L.; Tajber, L. Physicochemical properties of tadalafil solid dispersions—Impact of polymer on the apparent solubility and dissolution rate of tadalafil. Eur. J. Pharm. Biopharm. 2015, 94, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.K.; Balogh, A.; Demuth, B.; Pataki, H.; Vigh, T.; Szabo, B.; Molnar, K.; Schmidt, B.T.; Horak, P.; Marosi, G.; et al. High speed electrospinning for scaled-up production of amorphous solid dispersion of itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, C.; Fu, Q.; Di, D.; Kuang, X.; Wang, C.; He, Z.; Wang, J.; Sun, J. Solvent-shift strategy to identify suitable polymers to inhibit humidity-induced solid-state crystallization of lacidipine amorphous solid dispersions. Int. J. Pharm. 2016, 503, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Craig, D. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Karavas, E.; Ktistis, G.; Xenakis, A.; Georgarakis, E. Effect of hydrogen bonding interactions on the release mechanism of felodipine from nanodispersions with polyvinylpyrrolidone. Eur. J. Pharm. Biopharm. 2006, 63, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Muhrer, G.; Meier, U.; Fusaro, F.; Albano, S.; Mazzotti, M. Use of compressed gas precipitation to enhance the dissolution behavior of a poorly water-soluble drug: Generation of drug microparticles and drug-polymer solid dispersions. Int. J. Pharm. 2006, 308, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Moschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Ha, E.S.; Baek, I.H.; Kim, M.S.; Cho, C.W.; Hwang, S.J. Enhanced supersaturation and oral absorption of sirolimus using an amorphous solid dispersion based on Eudragit® E. Molecules 2015, 20, 9496–9509. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.G.; Zhu, L.M.; Branford-White, C.J.; Yang, J.H.; Wang, X.; Li, Y.; Qian, W. Solid dispersions in the form of electrospun core-sheath nanofibers. Int. J. Nanomed. 2011, 6, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Chen, J.; Wang, Y.; Chen, S.; Wang, X. Determination of 20(S)-protopanaxadiol in rat plasma by LC-MS/MS and its application to the pharmacokinetic study: A comparative study of its solution and two oral formulations. J. Anal. Chem. 2013, 68, 730–735. [Google Scholar] [CrossRef]
- Han, M.; Chen, J.; Chen, S.; Wang, X. Development of a UPLC-ESI-MSMS Assay for 20(S)-Protopanaxadiol and Pharmacokinetic Application of its Two Formulations in Rats. Anal. Sci. 2010, 26, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, Z.H.; Li, S.L.; Sun, E.; Tan, X.B.; Song, J.; Jia, X.B. A nanostructured liquid crystalline formulation of 20(S)-protopanaxadiol with improved oral absorption. Fitoterapia 2013, 84, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Musende, A.G.; Eberding, A.; Wood, C.A.; Adomat, H.; Fazli, L.; Hurtado-Coll, A.; Jia, W.; Bally, M.B.; Tomlinson Guns, E.S. A novel oral dosage formulation of the ginsenoside aglycone protopanaxadiol exhibits therapeutic activity against a hormone-insensitive model of prostate cancer. Anti-Cancer Drugs 2012, 23, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pu, Y.; Xu, B.; Tao, J.; Wang, Y.; Zhang, T.; Wu, P. Self-Microemulsifying Drug Delivery System Improved Oral Bioavailability of 20(S)-protopanaxadiol: From preparation to evaluation. Chem. Pharm. Bull. 2015, 63, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, Y.; Jang, J.; Hur, B.; Chi, S. Solid dispersions containing 20-O-β-d-Glucopyranosyl-20(S)-protopanaxadiol. EP2510924 (A1), 18 May 2012. [Google Scholar]
- Eloy, J.O.; Marchetti, J.M. Solid dispersions containing ursolic acid in Poloxamer 407 and PEG 6000: A comparative study of fusion and solvent methods. Powder Technol. 2014, 253, 98–106. [Google Scholar] [CrossRef]
- Kreidel, R.N.; Duque, M.D.; Serra, C.H.R.; Velasco, M.V.R.; Baby, A.R.; Kaneko, T.M.; Consiglieri, V.O. Dissolution Enhancement and Characterization of Nimodipine Solid Dispersions with Poloxamer 407 or PEG 6000. J. Dispers. Sci. Technol. 2012, 33, 1354–1359. [Google Scholar] [CrossRef]
- Newa, M.; Bhandari, K.H.; Li, D.X.; Kwon, T.H.; Kim, J.A.; Yoo, B.K.; Woo, J.S.; Lyoo, W.S.; Yong, C.S.; Choi, H.G. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int. J. Pharm. 2007, 343, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Newa, M.; Bhandari, K.H.; Oh, D.H.; Kim, Y.R.; Sung, J.H.; Kim, J.O.; Woo, J.S.; Choi, H.G.; Yong, C.S. Enhanced dissolution of ibuprofen using solid dispersion with poloxamer 407. Arch. Pharm. Res. 2008, 31, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Kakumanu, V.K.; Bansal, A.K. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J. Pharm. Sci. 2006, 95, 1641–1665. [Google Scholar] [CrossRef] [PubMed]
- Borba, P.A.; Pinotti, M.; de Campos, C.E.; Pezzini, B.R.; Stulzer, H.K. Sodium alginate as a potential carrier in solid dispersion formulations to enhance dissolution rate and apparent water solubility of BCS II drugs. Carbohydr. Polym. 2016, 137, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, S.; Blaton, N.; Kinget, R.; Mooter, G.V.D. Mechanism of increased dissolution of diazepam and temazepam from polyethylene glycol 6000 solid dispersions. Int. J. Pharm. 2002, 249, 45–58. [Google Scholar] [CrossRef]
- Damian, F.; Blaton, N.; Naesens, L.; Balzarini, J.; Kinget, R.; Augustijns, P.; Mooter, G.V.D. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur. J. Pharm. Sci. 2000, 10, 311–322. [Google Scholar] [CrossRef]
- Guedes, F.L.; de Oliveira, B.G.; Hernandes, M.Z.; De Simone, C.A.; Veiga, F.J.; de Lima Mdo, C.; Pitta, I.R.; Galdino, S.L.; Neto, P.J. Solid dispersions of imidazolidinedione by PEG and PVP polymers with potential antischistosomal activities. AAPS PharmSciTech 2011, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.J.; Zhang, Z.H.; Jin, X.; Hu, Q.; Chen, X.Y.; Jia, X.B. A novel drug-phospholipid complex enriched with micelles: Preparation and evaluation in vitro and in vivo. Int. J. Nanomed. 2013, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Onoue, S.; Kojo, Y.; Suzuki, H.; Yuminoki, K.; Kou, K.; Kawabata, Y.; Yamauchi, Y.; Hashimoto, N.; Yamada, S. Development of novel solid dispersion of tranilast using amphiphilic block copolymer for improved oral bioavailability. Int. J. Pharm. 2013, 452, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital lectronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Lin, Q.; Ling, L.Q.; Guo, L.; Gong, T.; Sun, X.; Zhang, Z.R. Intestinal absorption characteristics of imperialine: In vitro and in situ assessments. Acta Pharm. Sin. 2015, 36, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Kong, Y.; Sui, H.; Feng, J.; Zhu, R.; Wang, W. Enhanced oral bioavailability of glycyrrhetinic acid via nanocrystal formulation. Drug Deliv. Transl. Res. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zulal, N.A.; Lakshmi, P.K. Enhancement of Solubility and Bioavailability of Candesartan Cilexetil using Natural P-Glycoprotein Inhibitors. Trop. J. Pharm. Res. 2015, 14, 21. [Google Scholar] [CrossRef]
- Truong, D.H.; Tran, T.H.; Ramasamy, T.; Choi, J.Y.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Preparation and characterization of solid dispersion using a novel amphiphilic copolymer to enhance dissolution and oral bioavailability of sorafenib. Powder Technol. 2015, 283, 260–265. [Google Scholar] [CrossRef]
- Won, D.H.; Kim, M.S.; Lee, S.; Park, J.S.; Hwang, S.J. Improved physicochemical characteristics of felodipine solid dispersion particles by supercritical anti-solvent precipitation process. Int. J. Pharm. 2005, 301, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Six, K.; Daems, T.; de Hoon, J.; van Hecken, A.; Depre, M.; Bouche, M.P.; Prinsen, P.; Verreck, G.; Peeters, J.; Brewster, M.E.; et al. Clinical study of solid dispersions of itraconazole prepared by hot-stage extrusion. Eur. J. Pharm. Sci. 2005, 24, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.-F.; Jia, W.; Li, S.-S.; Xu, Z.-H.; Sun, X.; Wang, X.-R.; Zhang, Y.-Y.; Xie, G.-X. A new silymarin preparation based on solid dispersion technique. Adv. Ther. 2005, 22, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Sample of the compound 20(S)-protopanaxadiol is available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | PPD (g) | Polymer (g) | Tween-80 (g) | |
|---|---|---|---|---|
| PEG6000 | F68 | |||
| F1 | 0.5 | 5.0 | - | 0.4 |
| F2 | 0.5 | - | 5.0 | 0.4 |
| F3 | 0.5 | 2.5 | 2.5 | 0.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Pu, Y.; Wang, B.; Wang, Y.; Dong, T.T.; Guo, T.; Zhang, T.; Cai, Z. Characterization, Molecular Docking, and In Vitro Dissolution Studies of Solid Dispersions of 20(S)-Protopanaxadiol. Molecules 2017, 22, 274. https://doi.org/10.3390/molecules22020274
Zhang Q, Pu Y, Wang B, Wang Y, Dong TT, Guo T, Zhang T, Cai Z. Characterization, Molecular Docking, and In Vitro Dissolution Studies of Solid Dispersions of 20(S)-Protopanaxadiol. Molecules. 2017; 22(2):274. https://doi.org/10.3390/molecules22020274
Chicago/Turabian StyleZhang, Qi, Yiqiong Pu, Bing Wang, Yuqin Wang, Tina Tingxia Dong, Tao Guo, Tong Zhang, and Zhenzhen Cai. 2017. "Characterization, Molecular Docking, and In Vitro Dissolution Studies of Solid Dispersions of 20(S)-Protopanaxadiol" Molecules 22, no. 2: 274. https://doi.org/10.3390/molecules22020274
APA StyleZhang, Q., Pu, Y., Wang, B., Wang, Y., Dong, T. T., Guo, T., Zhang, T., & Cai, Z. (2017). Characterization, Molecular Docking, and In Vitro Dissolution Studies of Solid Dispersions of 20(S)-Protopanaxadiol. Molecules, 22(2), 274. https://doi.org/10.3390/molecules22020274