
Natural Products as Chemopreventive Agents by Potential Inhibition of the Kinase Domain in ErbB Receptors


Abstract
:1. Introduction
2. Results and Discussion
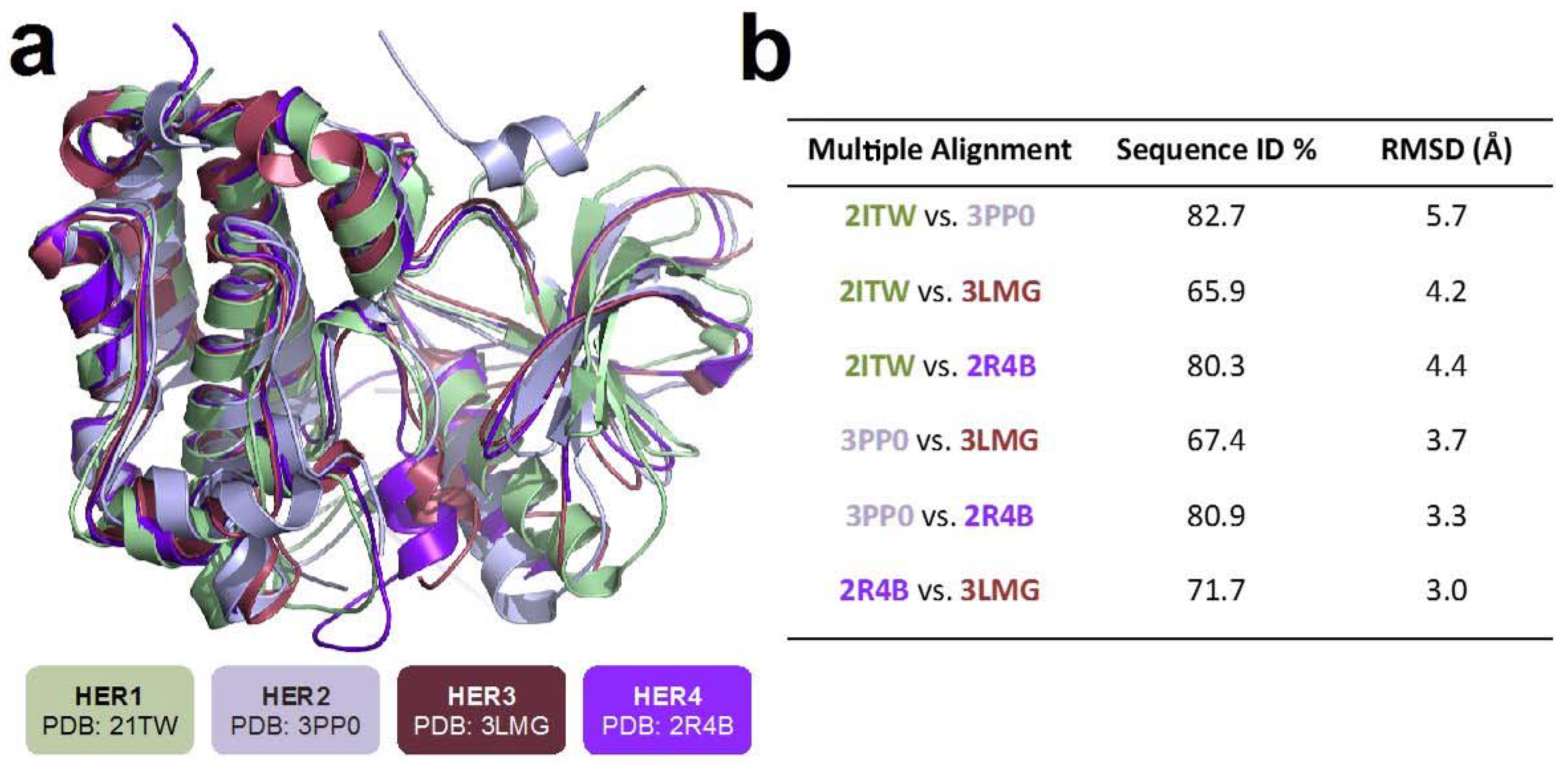
2.1. Structural Comparison of HER Receptors
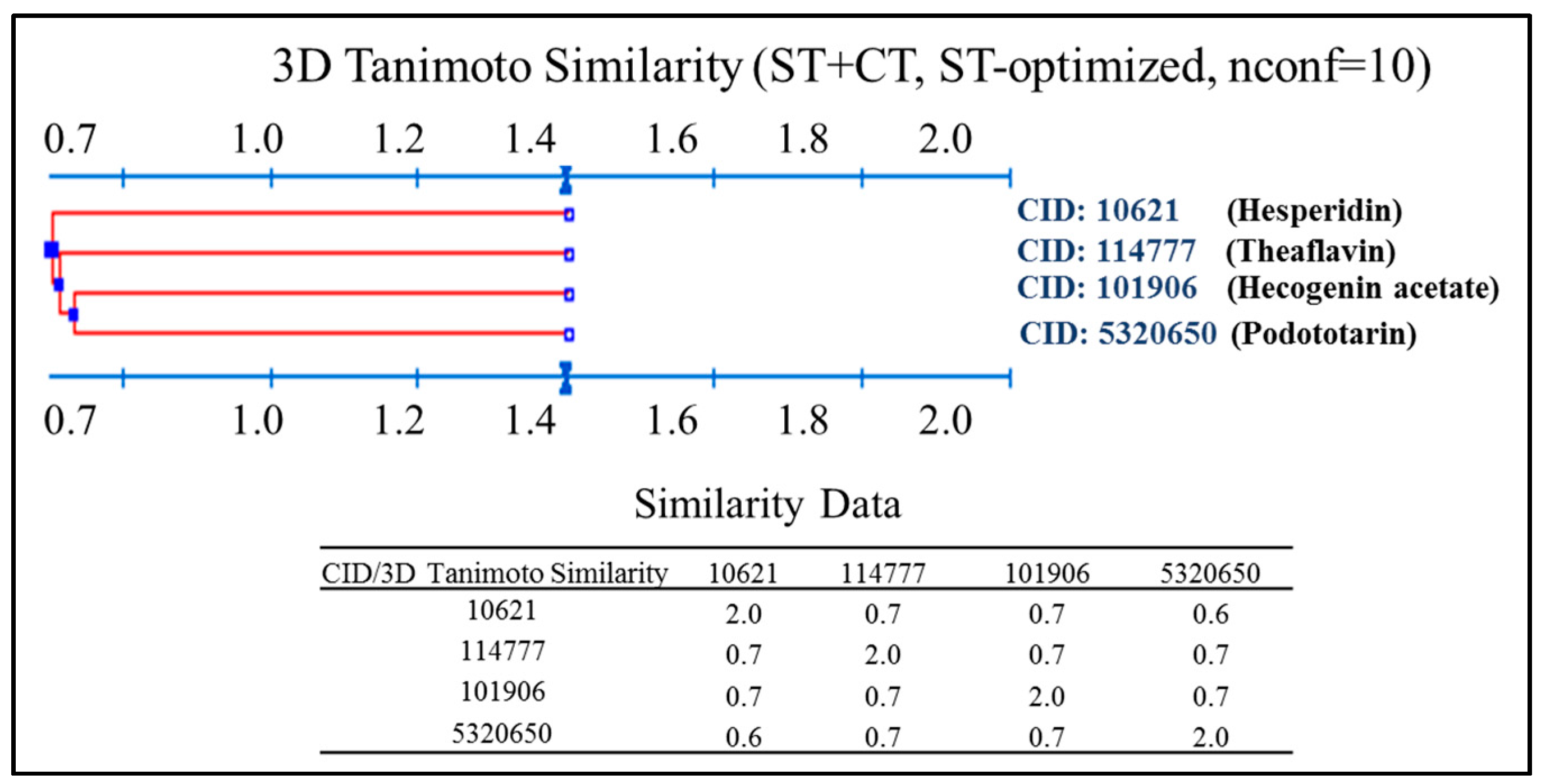
2.2. Virtual Screening for Identification of Potential Inhibitors of HER Kinase Domain
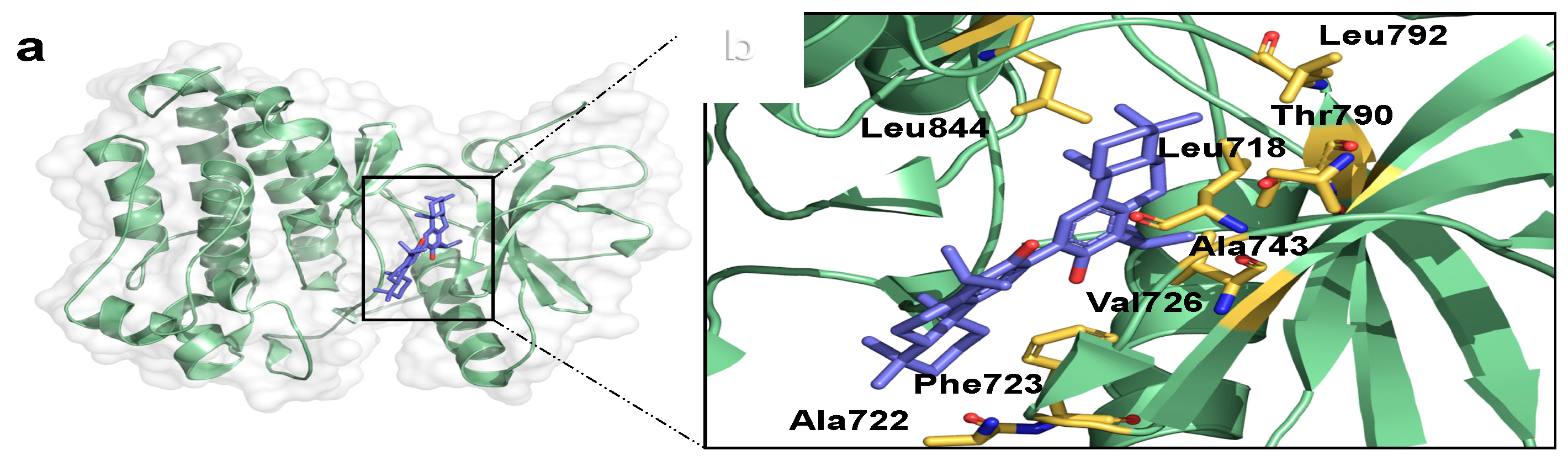
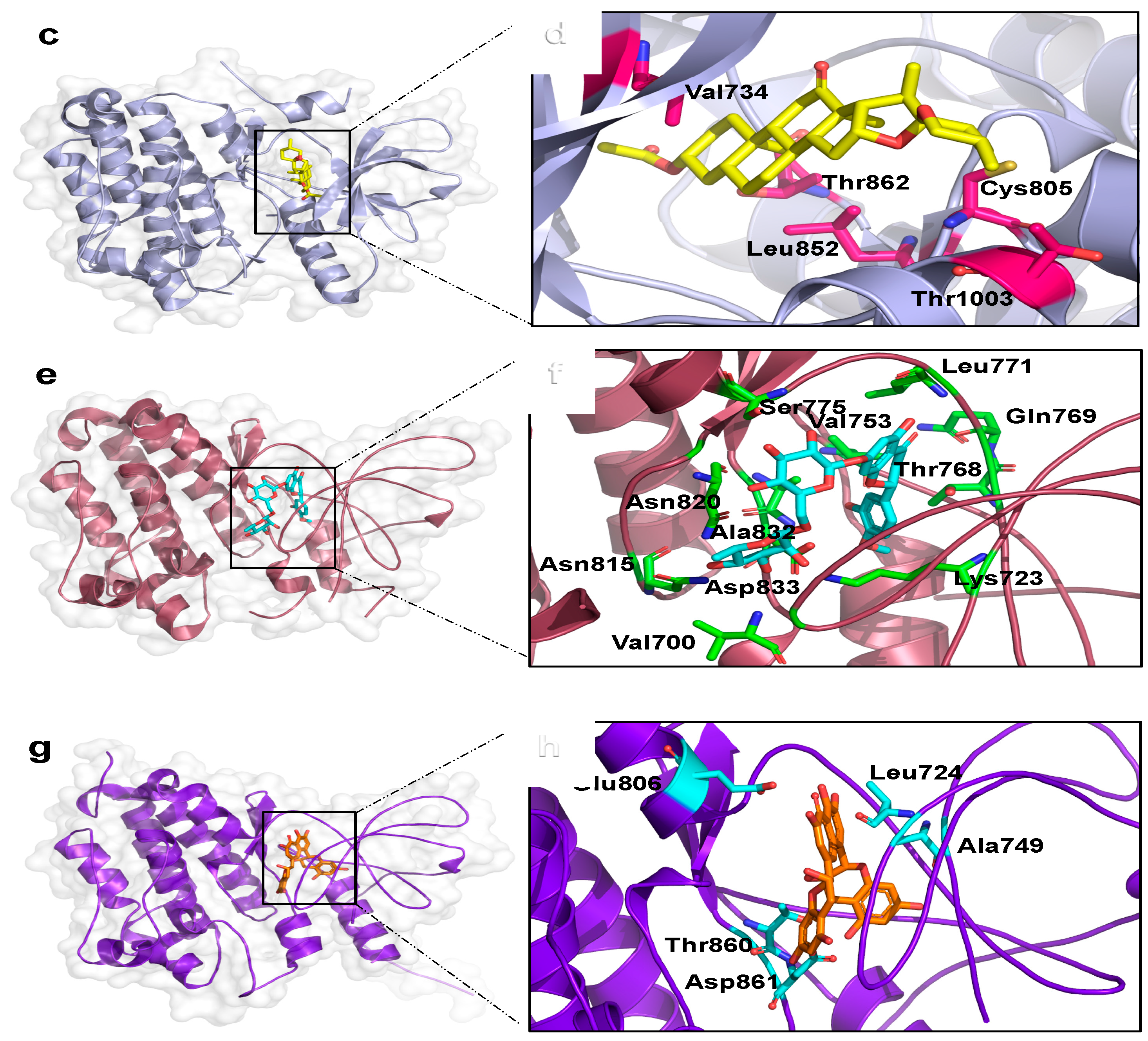
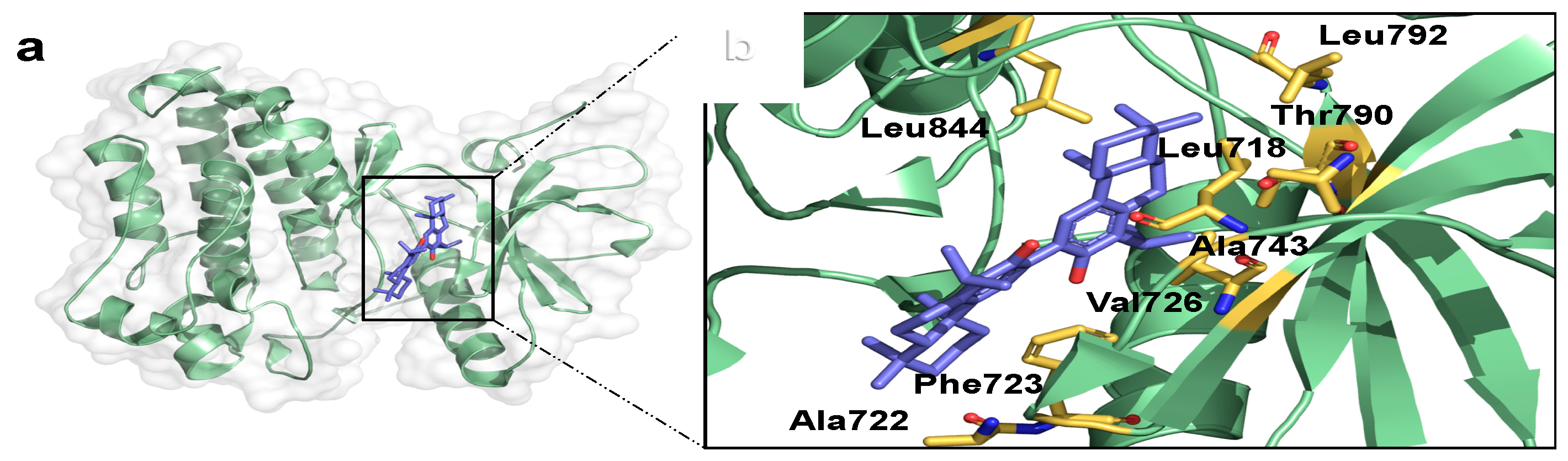
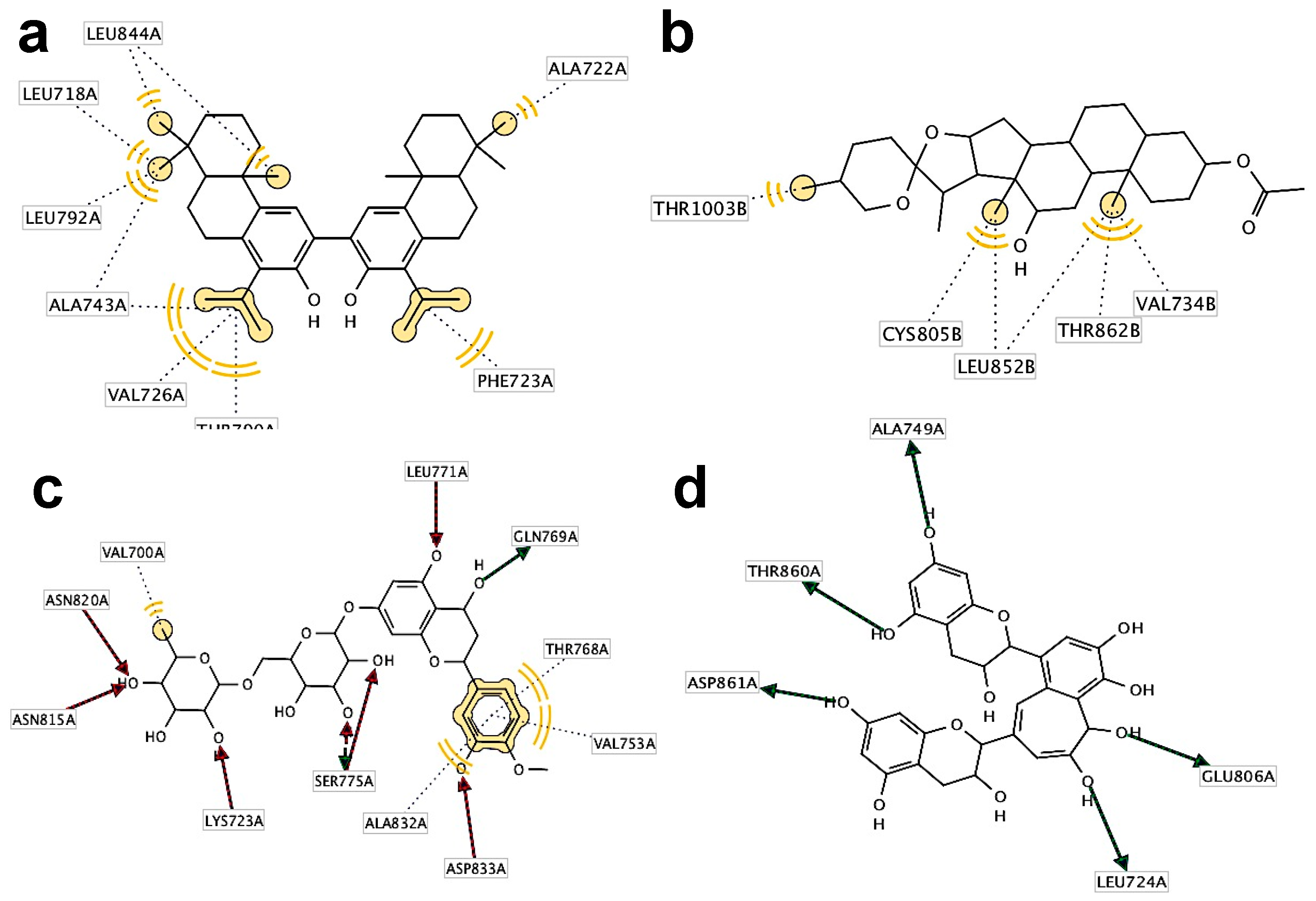
2.3. Binding Mode Analysis and Interacting Residues for NPs on HER Kinase Domain Binding Site
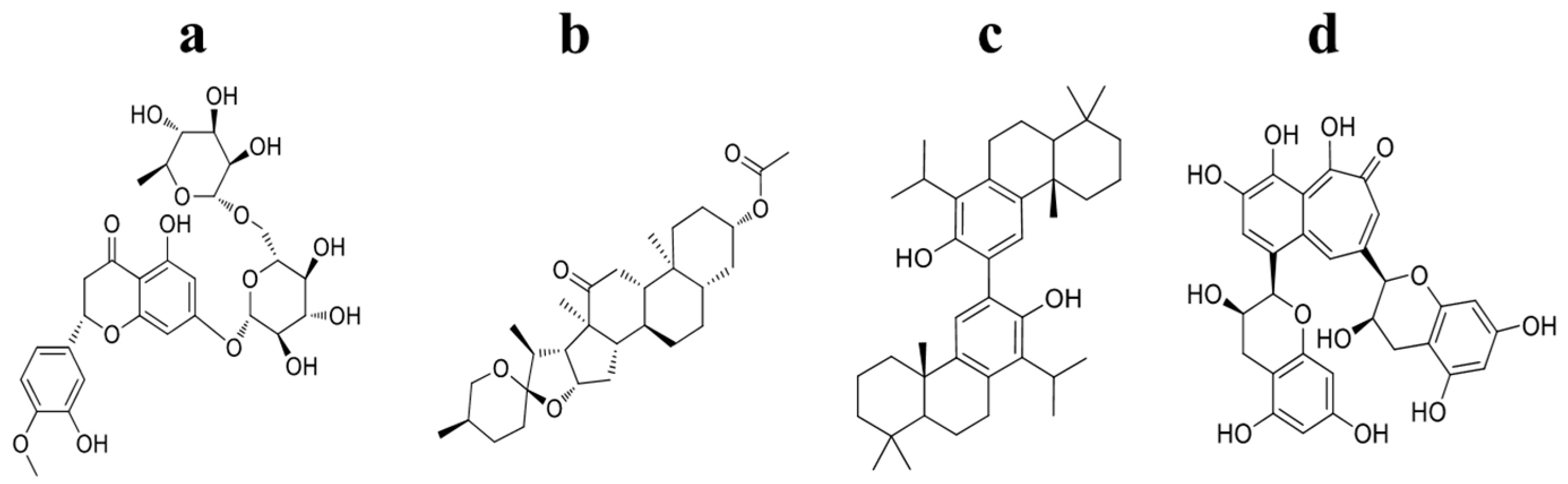
2.4. Information Regarding the Selected NPs Is Described in the Following Section
2.5. Prediction of ADME-TOX Properties for Promisory Compounds Using FAF-Drugs3 Server
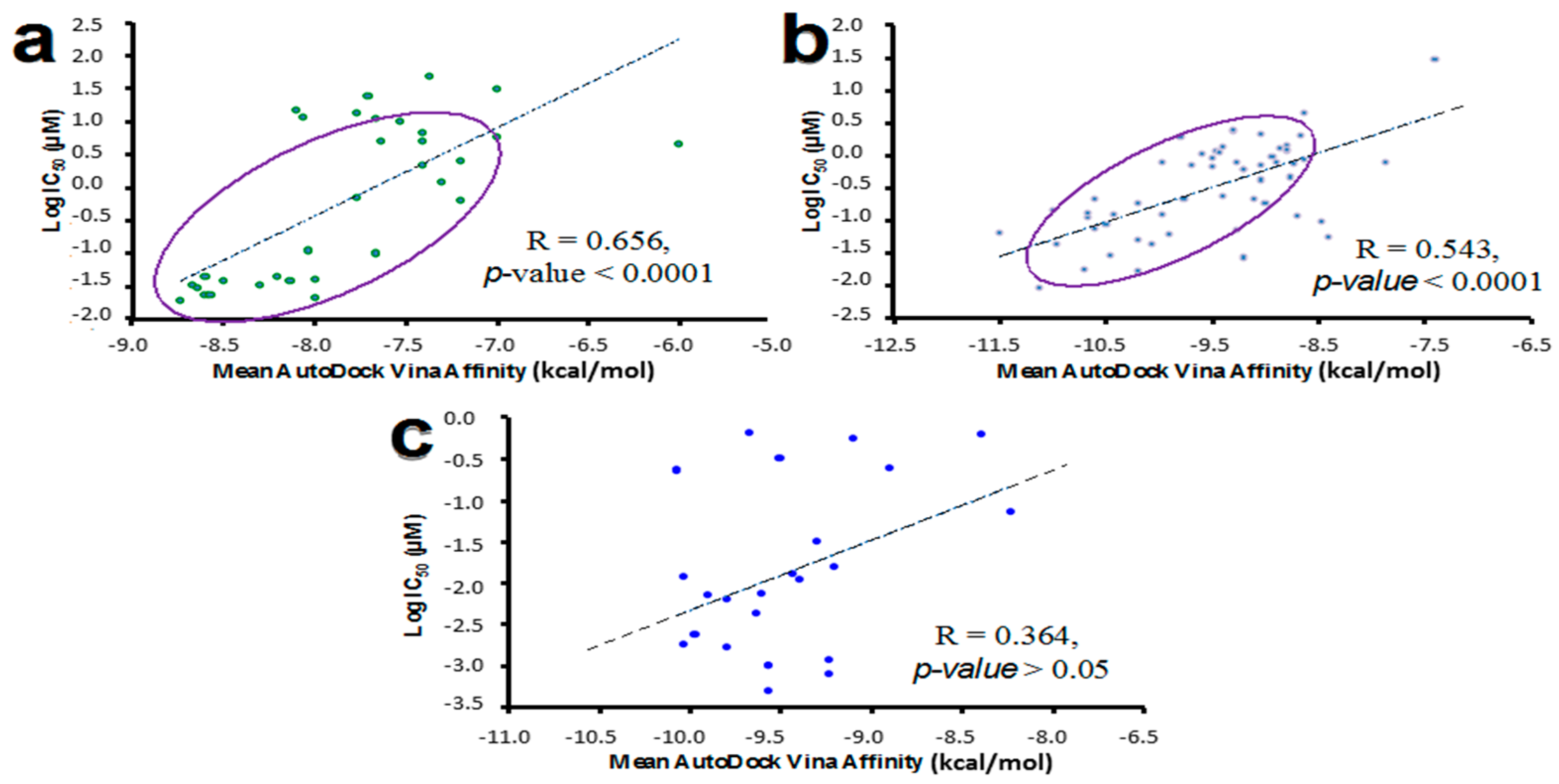
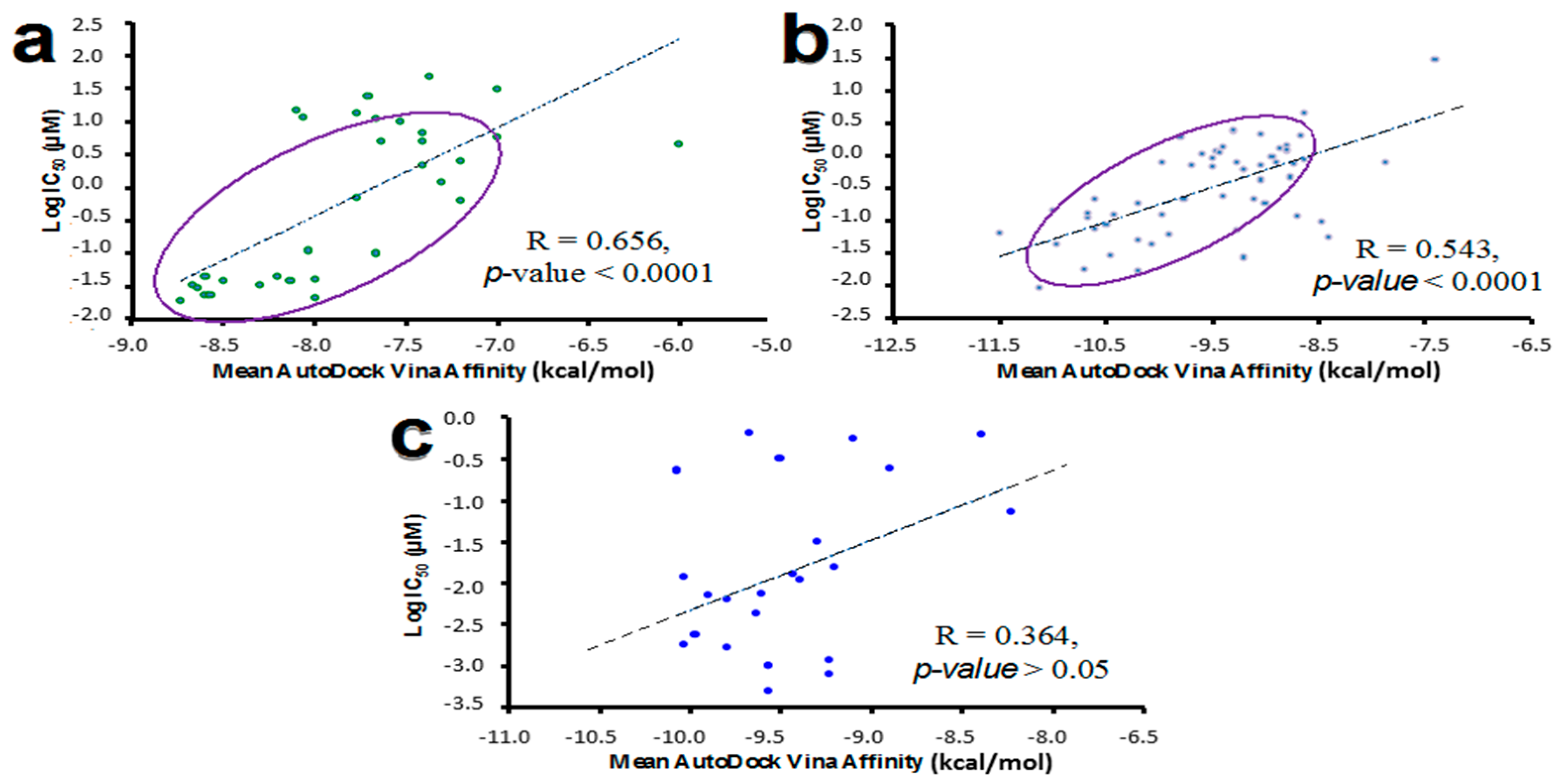
2.6. Molecular Docking Validation Using Reported Biological Data for Known HER Inhibitors
3. Materials and Methods
3.1. Virtual Screening by Molecular Docking with AutoDock Vina
3.1.1. Preparation and Selection of Crystallographic Structures of HER Kinase Domains for Molecular Docking with NPs
3.1.2. Preparation of NP Ligand Library (NatProd Collection)
3.1.3. Docking Calculations of NPs on HER Receptors
3.2. Identification of Main Interaction of Selected NPs on HER Kinase Domain
3.3. Computational Prediction of ADME-TOX Properties Using FAF-Drugs3
3.4. Molecular Docking Validation Using Reported Biological Data for HER Inhibitors
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martinez-Useros, J.; Garcia-Foncillas, J. Review: The challenge of blocking a wider family members of EGFR against head and neck squamous cell carcinomas. Oral Oncol. 2015, 51, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Bilgin, E.; Karabulut, S.; Duranyildiz, D. Clinical significance of serum epidermal growth factor receptor (EGFR) levels in patients with breast cancer. Cytokine 2015, 71, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. EGFR INHIBITORS: Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 903–913. [Google Scholar] [CrossRef] [PubMed]
- RoskoskiRobert, J. Invited Review: ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol. Res. 2014, 87, 42–59. [Google Scholar]
- Gonzales, A.J.; Fry, D.W. G1 cell cycle arrest due to the inhibition of erbB family receptor tyrosine kinases does not require the retinoblastoma protein. Exp. Cell Res. 2005, 303, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Gil-Benso, R.; Megías, J.; Muñoz-Hidalgo, L.; San-Miguel, T.; Callaghan, R.C.; González-Darder, J.M.; López-Ginés, C.; Cerdá-Nicolás, M.J. Alteration of major vault protein in human glioblastoma and its relation with EGFR and PTEN status. Neuroscience 2015, 297, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Mooso, B.A.; Vinall, R.L.; Mudryj, M.; Yap, S.A.; deVere White, R.W.; Ghosh, P.M. Review Article: The Role of EGFR Family Inhibitors in Muscle Invasive Bladder Cancer: A Review of Clinical Data and Molecular Evidence. J. Urol. 2015, 193, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ge, Y.; Wang, C.; Huang, S.; Shu, X.; Liu, K.; Zhou, Y.; Ma, X. Challenges and Perspectives on the Development of Small-Molecule EGFR Inhibitors against T790M-Mediated Resistance in Non-Small-Cell Lung Cancer. J. Med. Chem. 2016, 59, 6580–6594. [Google Scholar] [CrossRef] [PubMed]
- Uribe, P.; Gonzalez, S. Review: Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol. Res. Pract. 2011, 207, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yuan, H.; Li, Y.; Han, Y. Review: The role of HER3 in gastric cancer. Biomed. Pharmacother. 2014, 68, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Tripathy, D. Co-targeting estrogen receptor and HER2 pathways in breast cancer. Breast 2014, 23, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Ju, J.; Yang, W.; Lee, K.; Nam, K.; Shin, I. EGFR negates the proliferative effect of oncogenic HER2 in MDA-MB-231 cells. Arch. Biochem. Biophys. 2015, 575, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Mahato, R.; Cheng, K. Review: The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Kol, A.; van Scheltinga, A.G.T.; Timmer-Bosscha, H.; Lamberts, L.E.; Bensch, F.; de Vries, E.G.E.; Schröder, C.P. HER3, serious partner in crime: therapeutic approaches and potential biomarkers for effect of HER3-targeting. Pharmacol. Ther. 2014, 143, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Yarden, Y. Structure and function of epigen, the last EGFR ligand. Semin. Cell Dev. Biol. 2014, 28, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Settleman, J.; Reshkin, S.J.; Azzariti, A.; Bellizzi, A.; Paradiso, A. The complexity of targeting EGFR signalling in cancer: From expression to turnover. Biochim. Biophys. Acta (BBA) Rev. Cancer 2006, 1766, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Mudjupa, C.; Abdelhamed, S.; Refaat, A.; Yokoyama, S.; Saiki, I.; Vajragupta, O. Original Research Article: Lead compound bearing caffeic scaffold induces EGFR suppression in solid tumor cancer cells. J. Appl. Biomed. 2015, 13, 305–317. [Google Scholar] [CrossRef]
- Kempf, E.; Lacroix, L.; Soria, J.-C. First Reported Case of Unexpected Response to an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in the I744M Uncommon EGFR Mutation. Clin. Lung Cancer 2015, 16, e259–e261. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Serebriiskii, I.G.; DunbrackRoland, L.J.; Robinson, M.K.; Burtness, B.; Golemis, E.A. Protein-intrinsic and signaling network-based sources of resistance to EGFR- and ErbB family-targeted therapies in head and neck cancer. Drug Resist. Updates 2011, 14, 260–279. [Google Scholar] [CrossRef]
- Remon, J.; Morán, T.; Majem, M.; Reguart, N.; Dalmau, E.; Márquez-Medina, D.; Lianes, P. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: A new era begins. Cancer Treat. Rev. 2014, 40, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.G.; Vallée, A.; Théoleyre, S. EGFR T790M resistance mutation in non small-cell lung carcinoma. Clin. Chim. Acta 2015, 444, 81–85. [Google Scholar] [CrossRef]
- Yim-Im, W.; Sawatdichaikul, O.; Semsri, S.; Horata, N.; Mokmak, W.; Tongsima, S.; Suksamrarn, A.; Choowongkomon, K. Computational analyses of curcuminoid analogs against kinase domain of HER. BMC Bioinform. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Cambie, R.C.; Madden, R.J.; Parnell, J.C. Chemistry of the Podocarpaceae. XXVIII. Constituents of some Podocarpus and other species. Aust. J. Chem. 1971, 24, 217–221. [Google Scholar] [CrossRef]
- Gama, K.B.; Quintans, J.S.S.; Antoniolli, A.R.; Quintans-Júnior, L.J.; Santana, W.A.; Branco, A.; Soares, M.B.P.; Villarreal, C.F. Evidence for the involvement of descending pain-inhibitory mechanisms in the antinociceptive effect of hecogenin acetate. J. Nat. Prod. 2013, 76, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Rajavel, T.; Nabavi, S.F.; Setzer, W.N.; Ahmadi, A.; Mansouri, K.; Nabavi, S.M. Hesperidin: A promising anticancer agent from nature. Ind. Crop. Prod. 2015, 76, 582–589. [Google Scholar] [CrossRef]
- Kong, L.; Qi, X.; Huang, S.; Chen, S.; Wu, Y.; Zhao, L. Theaflavins inhibit pathogenic properties of P. gingivalis and MMPs production in P. gingivalis-stimulated human gingival fibroblasts. Arch. Oral Biol. 2015, 60, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.J.; Ray, S.; Sathe, G.J.; Gajbhiye, A.; Prasad, T.S.K.; Rapole, S.; Panda, D.; Srivastava, S. A comprehensive proteomic analysis of totarol induced alterations in Bacillus subtilis by multipronged quantitative proteomics. J. Proteom. 2015, 114, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, J.; Somensi, N.; Kunzler, A.; Girardi, C.S.; de Bittencourt Pasquali, M.A.; Ramos, V.M.; Simoes-Pires, A.; Quintans-Junior, L.J.; Branco, A.; Moreira, J.C.F.; et al. Hecogenin acetate inhibits reactive oxygen species production and induces cell cycle arrest and senescence in the A549 human lung cancer cell line. Anticancer Agents Med. Chem. 2014, 14, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Omar, H.A.; Mohamed, W.R.; Arafa, E.-S.A.; Shehata, B.A.; El Sherbiny, G.A.; Arab, H.H.; Elgendy, A.N.A.M. Original research article: Hesperidin alleviates cisplatin-induced hepatotoxicity in rats without inhibiting its antitumor activity. Pharmacol. Rep. 2016, 68, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Bigoniya, P.; Singh, K. Original article: Ulcer protective potential of standardized hesperidin, a citrus flavonoid isolated from Citrus sinensis. Rev. Bras. Farmacogn. 2014, 24, 330–340. [Google Scholar] [CrossRef]
- Filho, C.B.; Del Fabbro, L.; de Gomes, M.G.; Goes, A.T.R.; Souza, L.C.; Boeira, S.P.; Jesse, C.R. Kappa-opioid receptors mediate the antidepressant-like activity of hesperidin in the mouse forced swimming test. Eur. J. Pharmacol. 2013, 698, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.C.; de Gomes, M.G.; Goes, A.T.R.; Del Fabbro, L.; Filho, C.B.; Boeira, S.P.; Jesse, C.R. Evidence for the involvement of the serotonergic 5-HT1A receptors in the antidepressant-like effect caused by hesperidin in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 40, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, A.; Hasan, S.K.; Nafees, S.; Rashid, S.; Saidullah, B.; Sultana, S. Chemopreventive efficacy of hesperidin against chemically induced nephrotoxicity and renal carcinogenesis via amelioration of oxidative stress and modulation of multiple molecular pathways. Exp. Mol. Pathol. 2015, 99, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Zu, M.; Yang, F.; Zhou, W.; Liu, A.; Du, G.; Zheng, L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antivir. Res. 2012, 94, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Tamilselvam, K.; Radhiga, T.; Rao, S.; Essa, M.M.; Manivasagam, T. Theaflavin, a black tea polyphenol, protects nigral dopaminergic neurons against chronic MPTP/probenecid induced Parkinson’s disease. Brain Res. 2012, 1433, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, M.A.; Mulder, T.P.J.; Molhuizen, H.O.F. Theaflavins from Black Tea, Especially Theaflavin-3-gallate, Reduce the Incorporation of Cholesterol into Mixed Micelles. J. Agric. Food Chem. 2008, 56, 12031–12036. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, W.; Jia, L.; Li, B.; Chen, Y.C.; Tu, Y. Enhancement of (−)-epigallocatechin-3-gallate and theaflavin-3–3′-digallate induced apoptosis by ascorbic acid in human lung adenocarcinoma SPC-A-1 cells and esophageal carcinoma Eca-109 cells via MAPK pathways. Biochem. Biophys. Res. Commun. 2013, 438, 370–374. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.; Prince, D.; Lo, C.-Y.; Lee, L.H.; Chu, T.-C. Antiviral activity of theaflavin digallate against herpes simplex virus type 1. Antivir. Res. 2015, 118, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Cabarcas-Montalvo, M.; Maldonado-Rojas, W.; Montes-Grajales, D.; Bertel-Sevilla, A.; Wagner-Döbler, I.; Sztajer, H.; Reck, M.; Flechas-Alarcon, M.; Ocazionez, R.; Olivero-Verbel, J. Research paper: Discovery of antiviral molecules for dengue: In silico search and biological evaluation. Eur. J. Med. Chem. 2016, 110, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Shan, Y.; Cao, X.; Shaw, D.E.; Kuriyan, J. Structural Analysis of the Catalytically Inactive Kinase Domain of the Human EGF Receptor 3. Proc. Natl. Acad. Sci. USA 2009, 106, 21608–21613. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their useas virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Shewchuk, L.M.; Ellis, B.; Brignola, P.; Brashear, R.L.; Caferro, T.R.; Dickerson, S.H.; Dickson, H.D.; Donaldson, K.H.; Gaul, M.; et al. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Telesco, S.E.; Liu, Y.; Radhakrishnan, R.; Lemmon, M.A. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 7692–7697. [Google Scholar] [CrossRef] [PubMed]
- Powell, M.J.D. Restart procedures for the conjugate gradient method. Math. Program. 1977, 12, 241–254. [Google Scholar] [CrossRef]
- Maldonado-Rojas, W.; Olivero-Verbel, J.; Marrero-Ponce, Y. Computational fishing of new DNA methyltransferase inhibitors from natural products. J. Mol. Graph. Model. 2015, 60, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Rojas, W.; Olivero-Verbel, J. Food-related compounds that modulate expression of inducible nitric oxide synthase may act as its inhibitors. Molecules 2012, 17, 8118–8135. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, M.; Jeanmart, S. Pan Assay Interference Compounds (PAINS) and Other Promiscuous Compounds in Antifungal Research. J. Med. Chem. 2016, 59, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HER1 (PDB: 2ITW) | HER2 (PDB: 3PP0) | HER3 (PDB: 3LMG) | HER4 (PDB: 2R4B) | Natural Source | Ref. |
|---|---|---|---|---|---|---|
| Podototarin | −10.7 ± 0.0 | −9.5 ± 0.05 | −9.0 ± 0.0 | −8.8 ± 0.0 | Podocarpus spp. | [26] |
| Hecogenin acetate | −10.1 ± 0.0 | −11.2 ± 0.0 | −10.2 ± 0.0 | −10.6 ± 0.0 | Genus Agave | [27] |
| Hesperidin | −8.4 ± 0.1 | −10.5 ± 0.0 | −11.5 ± 0.0 | −9.4 ± 0.0 | Citrus Fruits | [28] |
| Theaflavin | −9.0 ± 0.0 | −10.8 ± 0.6 | −10.5 ± 0.1 | −10.7 ± 0.0 | Black teas | [29] |
| Natural Product | Molecular Descriptors a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | Log P | Log D | tPSA | Rotable Bonds | Rigid Bonds | Flexibility | HBD | HBA | Sol (mg/L) | OB (VEBER) | OB (EGAN) | |
| Podotoarin | 570.89 | 13.13 | 12.18 | 40.46 | 3 | 32 | 0.09 | 2 | 2 | 5.5 | Good | Good |
| Hecogenin acetate | 472.66 | 5.40 | 5.11 | 61.83 | 2 | 32 | 0.06 | 0 | 5 | 1125.1 | Good | Good |
| Hersperidin | 610.56 | −10.09 | −0.32 | 234.29 | 7 | 30 | 0.19 | 8 | 15 | 45,708.2 | Good | Good |
| Theaflavin | 564.49 | 2.38 | 2.78 | 217.60 | 2 | 35 | 0.05 | 9 | 12 | 3922.9 | Good | Good |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivero-Acosta, M.; Maldonado-Rojas, W.; Olivero-Verbel, J. Natural Products as Chemopreventive Agents by Potential Inhibition of the Kinase Domain in ErbB Receptors. Molecules 2017, 22, 308. https://doi.org/10.3390/molecules22020308
Olivero-Acosta M, Maldonado-Rojas W, Olivero-Verbel J. Natural Products as Chemopreventive Agents by Potential Inhibition of the Kinase Domain in ErbB Receptors. Molecules. 2017; 22(2):308. https://doi.org/10.3390/molecules22020308
Chicago/Turabian StyleOlivero-Acosta, Maria, Wilson Maldonado-Rojas, and Jesus Olivero-Verbel. 2017. "Natural Products as Chemopreventive Agents by Potential Inhibition of the Kinase Domain in ErbB Receptors" Molecules 22, no. 2: 308. https://doi.org/10.3390/molecules22020308