
New Cytotoxic Seco-Type Triterpene and Labdane-Type Diterpenes from Nuxia oppositifolia

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
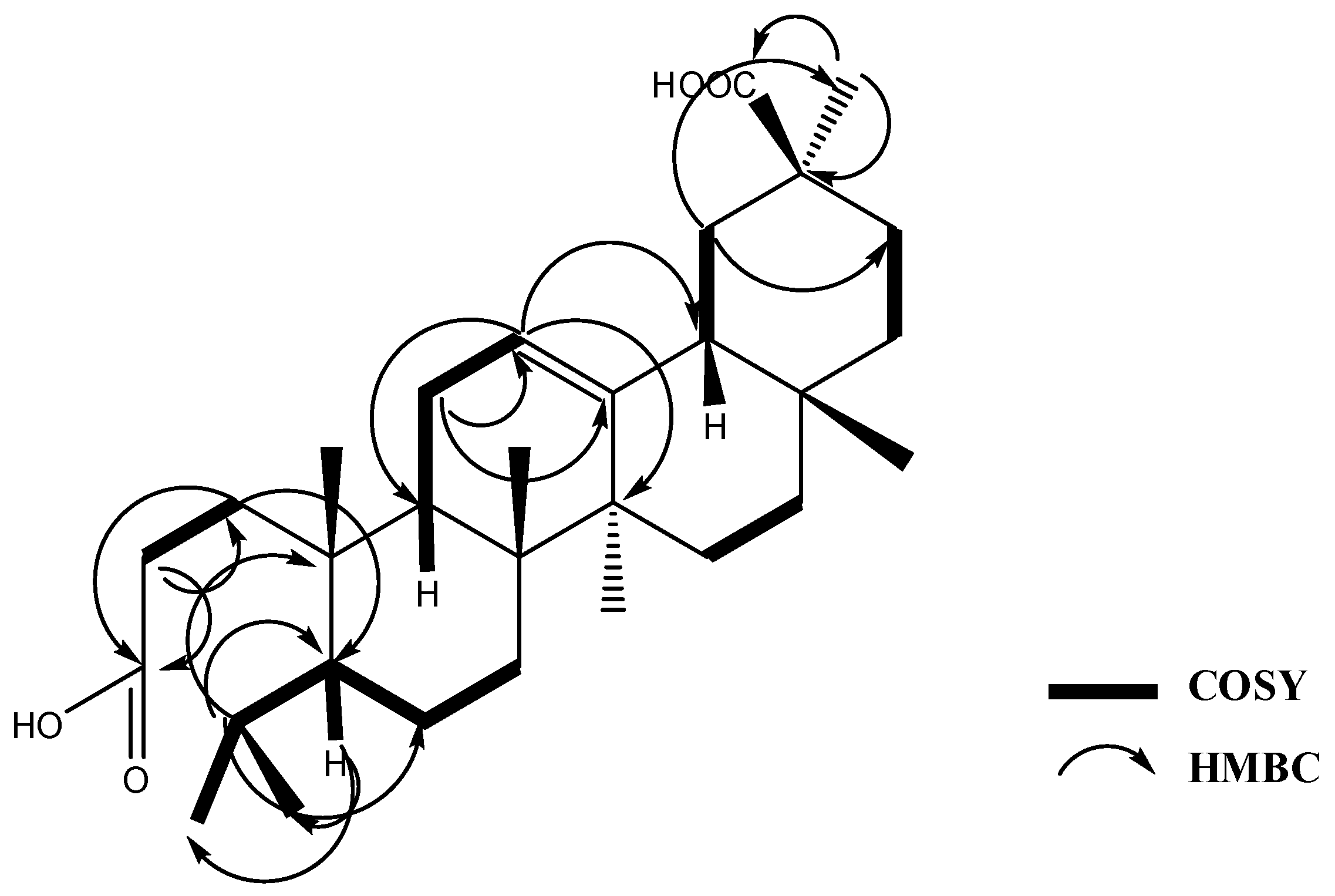
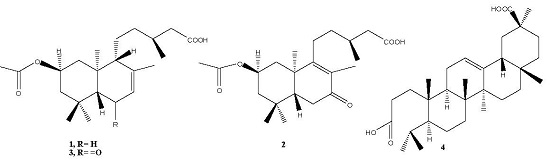
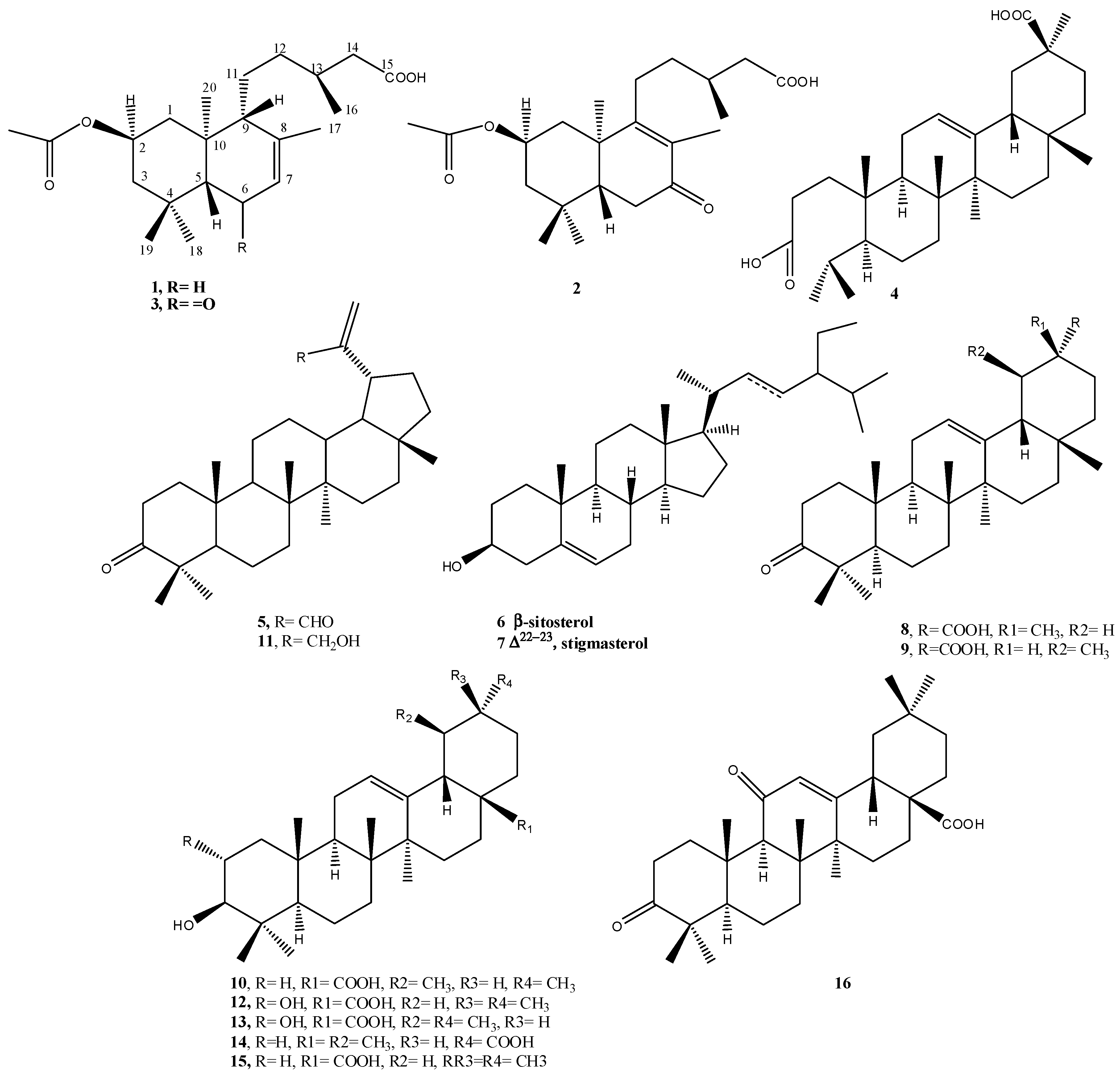
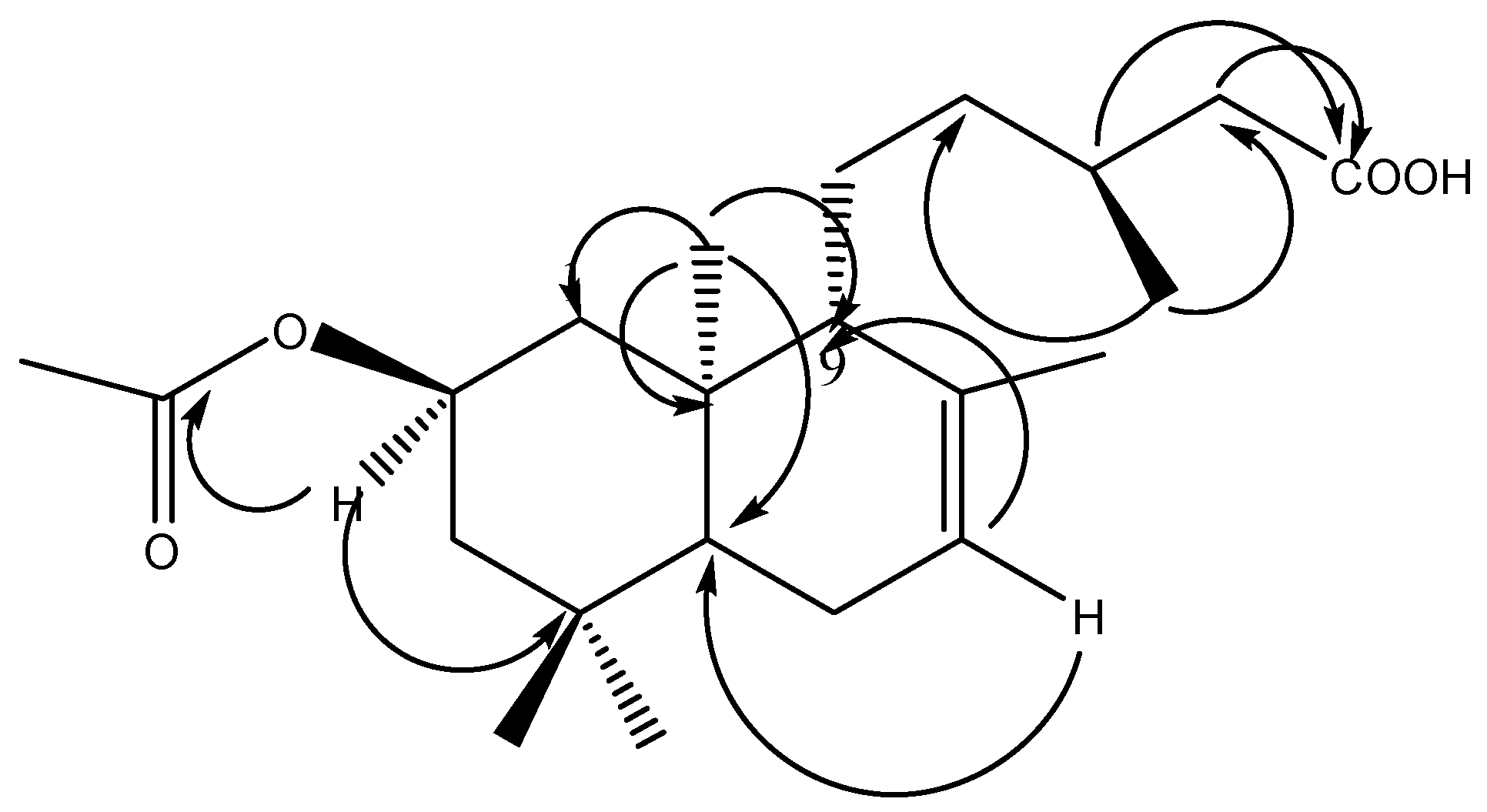
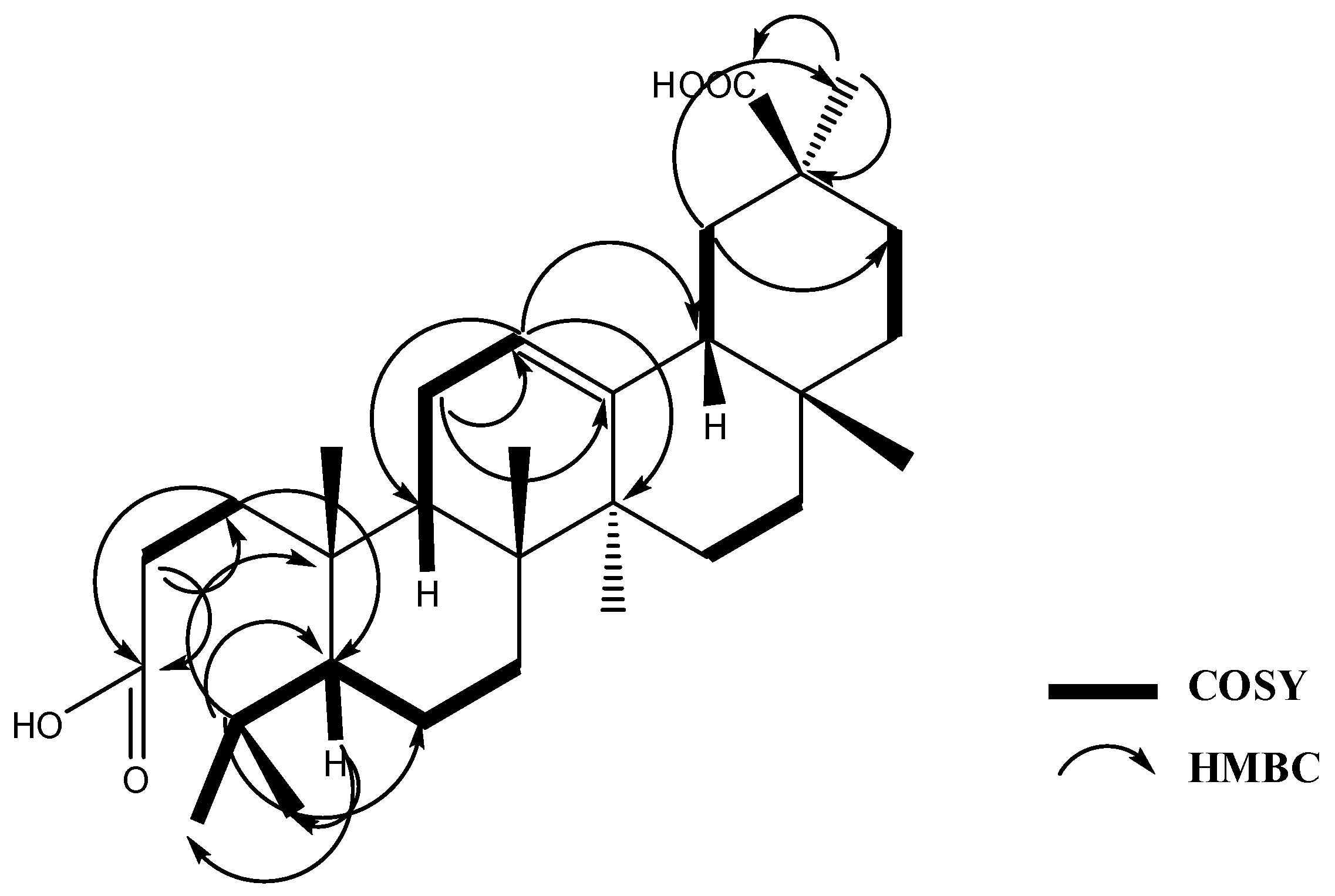
2.1. Phytochemical Study
2.2. Cytotoxicity Assay
3. Materials and Methods
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectral Data of New Compounds
3.5. Spectral Data of Known Compounds
3.6. Cytotoxicity Assay
3.6.1. Cell Culture
3.6.2. Cytotixicty Assay
3.6.3. Microscopy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Johnson, L. Review of the systematics of scrophulariaceae s.L. And their current disposition. Aust. Syst. Bot. 2006, 19, 289–307. [Google Scholar]
- Al-Abbasi, T.M.; Al-Farhan, A.H.; Al-Khulaidi, W.; Hall, M.; Lewellyn, O.A.; Miller, A.G.; Patzelt, A. Important plant areas in the Arabian peninsula. Edinb. J. Bot. 2010, 67, 25–35. [Google Scholar] [CrossRef]
- Migahid, A.M. Flora of Saudi Arabia, 3rd ed.; University Libraries, King Saud University: Riyadh, Saudi Arabia, 1989; Volume II. [Google Scholar]
- Jonville, M.C.; Kodja, H.; Strasberg, D.; Pichette, A.; Ollivier, E.; Frederich, M.; Angenot, L.; Legault, J. Antiplasmodial, anti-inflammatory and cytotoxic activities of various plant extracts from the Mascarene archipelago. J. Ethnopharmacol. 2011, 136, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Mambu, L.; Grellier, P.; Florent, L.; Joyeau, R.; Ramanitrahasimbola, D.; Rasoanaivo, P.; Frappier, F. Clerodane and labdane diterpenoids from Nuxia phaerocephala. Phytochemistry 2006, 67, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.R. Chemotaxonomy of the genus Nuxia (buddlejaceae). Stud. Plant Sci. 1999, 6, 379–382. [Google Scholar]
- Laszczyk, M. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med. 2009, 15, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Jakupovic, J.; Tsichritzis, F.; Tamayo-Castillo, G.; Castro, V.; Bohlmann, F.; Boldt, E. Diterpenes from Fleischmannia hymenophylla and Brickellia laciniata. Phytochemistry 1989, 28, 2741–2744. [Google Scholar] [CrossRef]
- Caldwell, C.G.; Franzblau, S.G.; Suarez, E.; Timmermann, B.N. Oleanane triterpenes from Junellia tridens. J. Nat. Prod. 2000, 63, 1611–1614. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, K.; Arai, Y.; Masuda, K.; Takase, Y.; Ageta, T.; Ageta, H. Mass spectra of pentacyclic triterpenoids. Chem. Pharm. Bull. 1992, 40, 1683–1690. [Google Scholar] [CrossRef]
- Ngounou, F.N.; Lontsi, D.; Sondengam, B.L. Myrianthinic acid: A new triterpenoid from Myrianthus arboreus. Phytochemistry 1988, 27, 301–303. [Google Scholar] [CrossRef]
- De Souza e Silva, S.R.; de Fátima Silva, G.D.; de Almeida Barbosa, L.C.; Duarte, L.P.; Filho, S.A.V. Lupane pentacyclic triterpenes isolated from stems and branches of Maytenus imbricata (celastraceae). Helv. Chim. Acta 2005, 88, 1102–1109. [Google Scholar] [CrossRef]
- Wijeratne, D.B.T.; Kumar, V.; Uvais, M.; Sultanbawa, S.; Balasubramaniam, S. Triterpenes from Gymnosporia emarginata. Phytochemistry 1982, 21, 2422–2423. [Google Scholar] [CrossRef]
- Tanaka, T.; Koyano, T.; Kowithayakorn, T.; Fujimoto, H.; Okuyama, E.; Hayashi, M.; Komiyama, K.; Ishibashi, M. New multiflorane-type triterpenoid acids from Sandoricum indicum. J. Nat. Prod. 2001, 64, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, J.R.; Silva, G.D.F.; Pedersoli, J.L.; Alves, R.J. Friedelane and oleanane triterpenoids from bark wood of Austroplenckia populnea. Phytochemistry 1990, 29, 3259–3261. [Google Scholar] [CrossRef]
- Chiu-Ming, C.; Ming-Tyan, C. 6-methoxybenzoxazolinone and triterpenoids from roots of Scoparia dulcis. Phytochemistry 1976, 15, 1997–1999. [Google Scholar] [CrossRef]
- Bosson, J.; Galbraith, M.; Ritchie, E.; Taylor, W. The chemical constituents of Australian Flindersia species. Xviii. The structure of ifflaionic acid. Aust. J. Chem. 1963, 16, 491–498. [Google Scholar] [CrossRef]
- Bohlmann, F.; Jakupovic, J. Neue sesquiterpene, triterpene, flavanone und andere aromatische verbindungen aus Flourensia heterolepis. Phytochemistry 1979, 18, 1189–1194. [Google Scholar] [CrossRef]
- Lan, X.; Wu, H.; Wang, W. Chemical constituents from Sinacalia davidii. Zhongguo Zhong Yao Za Zhi 2010, 35, 1001–1003. [Google Scholar] [PubMed]
- Razdan, T.K.; Kachroo, V.; Harkar, S.; Koul, G.L. Plectranthoic acid a & b, two new triterpenoids from Plectranthus rugosus. Tetrahedron 1982, 38, 991–992. [Google Scholar]
- Mahato, S.B.; Kundu, A.B. 13C-NMR Spectra of pentacyclic triterpenoids—A compilation and some salient features. Phytochemistry. 1994, 37, 1517–1575. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, L.; Liu, J.Y.; Cai, P.L.; Yang, S.L. Chemical constituents of Patrinia scabiosaefolia. Zhong Cao Yao 2011, 42, 1477–1480. [Google Scholar]
- Sample Availability: Not available.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 b |
|---|---|---|---|---|
| 1 | 1.00 m 2.13 m | 1.27 m 2.10 m | 2.03 m | 1.54 m |
| 2 | 4.94 t | 4.98 t (10.7) | 4.90 t (11.0) | 2.04 m |
| 3 | 1.17 1.71 | 1.18 br d (12.0) 1.80 br d (10.8) | 1.14 m 1.61 m | |
| 4 | 1.71 m | |||
| 5 | 1.15 br d (4.4) | 1.59 br s | 1.97 s | 0.96 br d (11.7) |
| 6 | 1.82 1.90 | 2.40 br d (16.5) 2.26 m | 1.32 m 1.33 m | |
| 7 | 5.34 br. s | 5.69 br s | 1.37 m | |
| 8 | ||||
| 9 | 1.62 | 2.03 m | 1.71 m | |
| 10 | ||||
| 11 | 1.18 1.35 | 2.03 m 2.19 m | 1.35 m | 1.81 m |
| 12 | 1.20 1.37 | 1.27 m 1.37 m | 1.37 m | 5.19 br s |
| 13 | 1.80 m | 1.95 m | 1.93 m | |
| 14 | 2.07 2.28 | 2.10 m 2.26 m | 2.03 m 2.23 m | |
| 15 | 1.93 dd 0.83 m | |||
| 16 | 0.93 d (6.7) | 0.95 d (6.2) | 0.97 d (6.7) | 1.63 m 0.97 m |
| 17 | 1.62 | 1.64 s | 1.82 s | |
| 18 | 0.87 s | 0.85 s | 1.08 s | 1.83 dd (13.4, 4.7) |
| 19 | 0.92 s | 0.90 s | 1.14 s | 1.59 br d (13.4) 1.71 m |
| 20 | 0.79 s | 1.06 s | 0.82 s | |
| 21-OAc | 2.00 | 1.97 s | 1.96 s | 1.77 m 1.28 m |
| 22 | 1.71 m | |||
| 23 | 0.78 d (6.8) | |||
| 24 | 0.78 d (6.8) | |||
| 25 | 0.86 s | |||
| 26 | 0.94 s | |||
| 27 | 1.13 s | |||
| 28 | 0.74 s | |||
| 29 | 1.04 s | |||
| 30 |
| No. | 1 | 2 | 3 | 4 b |
|---|---|---|---|---|
| 1 | 44.2 CH2 | 41.0 CH2 | 43.6 CH2 | 32.8 CH2 |
| 2 | 68.9 CH | 68.1 CH | 67.9 CH | 28.1 CH2 |
| 3 | 46.9 CH2 | 46.1 CH2 | 47.8 CH2 | 174.8 C |
| 4 | 34.6 C | 34.4 C | 33.2 C | 24.6 C |
| 5 | 49.5 CH | 49.5 CH | 62.5 CH | 46.9 CH |
| 6 | 23.5 CH2 | 34.7CH2 | 199.1 CH2 | 17.7 CH2 |
| 7 | 122.1 CH | 199.9 C | 128.5 CH | 31.1 CH2 |
| 8 | 134.8 C | 130.0 C | 158.9 C | 39.0 C |
| 9 | 54.3 CH | 167.1 C | 56.4 CH | 37.2 CH |
| 10 | 39.1 C | 42.0 C | 44.4 C | 39.5 C |
| 11 | 24.6 CH2 | 27.0 CH2 | 24.6 CH2 | 23.2 CH2 |
| 12 | 38.5 CH2 | 35.2CH2 | 38.9 CH2 | 121.9 CH |
| 13 | 30.9 CH | 31.0 CH | 30.6 CH | 144.2 C |
| 14 | 41.8 CH2 | 40.9 CH2 | 41.5 CH2 | 41.6 C |
| 15 | 179.0 C | 177.9 C | 177.9 C | 26.3 CH2 |
| 16 | 19.5 CH3 | 19.3 CH3 | 19.4 CH3 | 25.7 CH2 |
| 17 | 22.0 CH3 | 11.3 CH3 | 22.0 CH3 | 31.6 C |
| 18 | 33.0 CH3 | 32.4 CH3 | 34.0 CH3 | 47.8 CH |
| 19 | 22.5 CH3 | 22.0 CH3 | 22.6 CH3 | 42.3 CH2 |
| 20 | 14.2 CH3 | 19.2 CH3 | 15.4 CH3 | 43.1 C |
| 21 | 21.5 CH3 | 21.4 CH3 | 21.4 CH3 | 30.6 CH2 |
| 22-OAc | 170.8 C | 170.8 C | 170.7 C | 38.0 CH2 |
| 23 | 23.2 CH3 | |||
| 24 | 18.7 CH3 | |||
| 25 | 19.0 CH3 | |||
| 26 | 16.5 CH3 | |||
| 27 | 25.3 CH3 | |||
| 28 | 28.1 CH3 | |||
| 29 | 28.2 CH3 | |||
| 30 | 177.9 C |
| Compound No. | Hela (Cervical) | A549 (Lungs) | MDA (Breast) |
|---|---|---|---|
| 1 | 87.34 | 73.37 | 74.56 |
| 2 | 71.34 | 87.24 | 68.16 |
| 3 | 57.56 | 72.15 | 53.34 |
| 4 | 37.6 | 53.78 | 42.73 |
| 5 | 48.96 | 88.31 | 54.6 |
| 8 | 29.35 | 79.23 | 18.25 |
| 9 | 42.0 | 67.37 | 39.78 |
| 10 | 50.2 | 65.2 | 47.76 |
| 11 | 32.47 | 83.24 | 27.16 |
| 12 | 57.89 | 78.69 | 69.87 |
| 13 | 54.58 | 72.48 | 63.41 |
| 14 | 36.0 | 57.63 | 30.0 |
| 15 | 85.72 | 59.17 | 65.27 |
| Doxorubicin | 70.01 | 164.46 | 15.41 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Massarani, S.M.; El-Gamal, A.A.; Parvez, M.K.; Al-Dosari, M.S.; Al-Said, M.S.; Abdel-Kader, M.S.; Basudan, O.A. New Cytotoxic Seco-Type Triterpene and Labdane-Type Diterpenes from Nuxia oppositifolia. Molecules 2017, 22, 389. https://doi.org/10.3390/molecules22030389
Al-Massarani SM, El-Gamal AA, Parvez MK, Al-Dosari MS, Al-Said MS, Abdel-Kader MS, Basudan OA. New Cytotoxic Seco-Type Triterpene and Labdane-Type Diterpenes from Nuxia oppositifolia. Molecules. 2017; 22(3):389. https://doi.org/10.3390/molecules22030389
Chicago/Turabian StyleAl-Massarani, Shaza M., Ali A. El-Gamal, Mohammad K. Parvez, Mohammed S. Al-Dosari, Mansour S. Al-Said, Maged S. Abdel-Kader, and Omer A. Basudan. 2017. "New Cytotoxic Seco-Type Triterpene and Labdane-Type Diterpenes from Nuxia oppositifolia" Molecules 22, no. 3: 389. https://doi.org/10.3390/molecules22030389