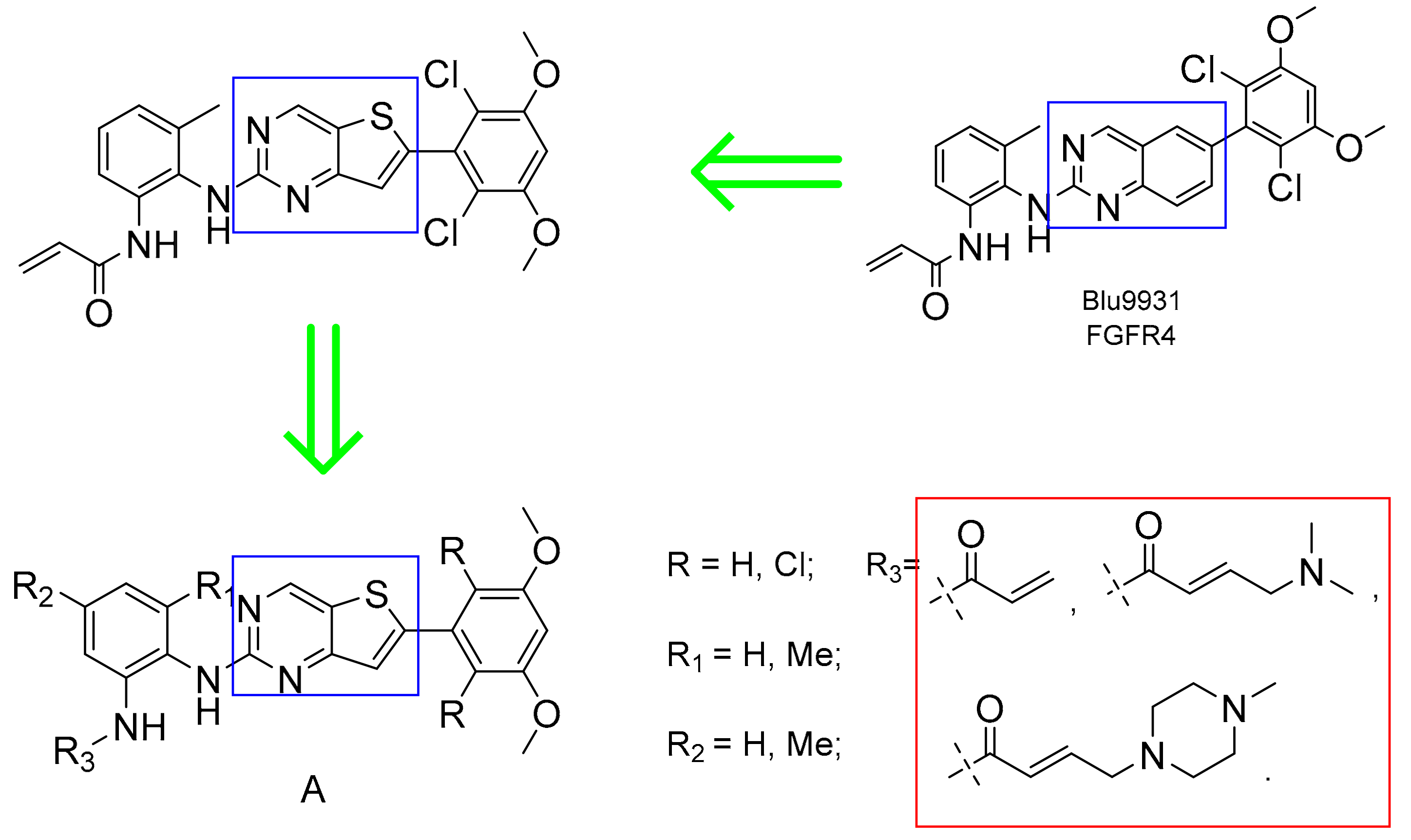
3.2. General Synthesis
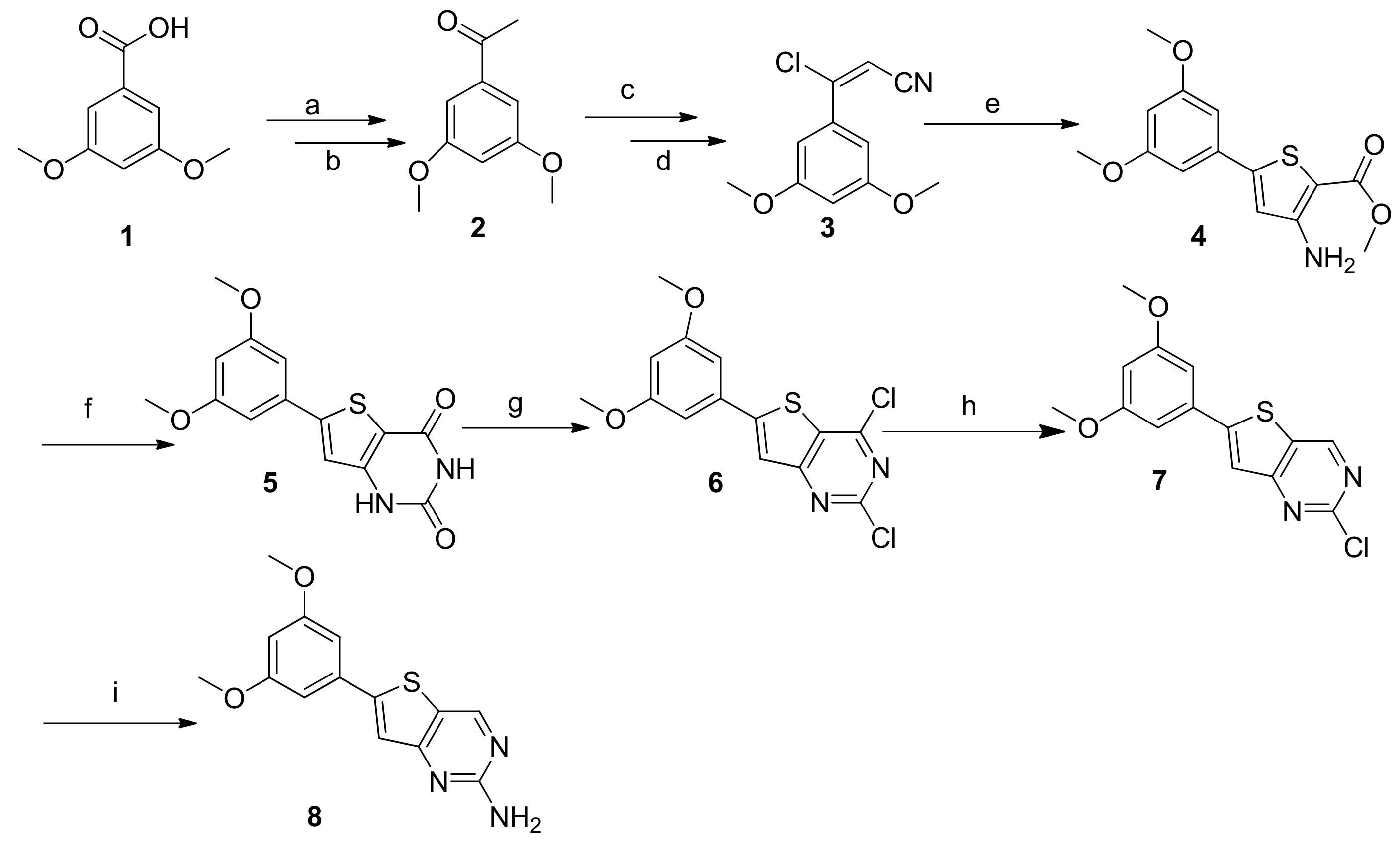
3.2.1. 1-(3,5-Dimethoxyphenyl)ethanone (2)
Step 1. 3,5-dimethoxybenzoyl chloride
3,5-dimethoxybenzoic acid (27.3 g, 150 mmol, compound 1) was suspended in 120 mL thionyl chloride, and the mixture was heated and held at reflux until 1 dissolved. Then, thionyl chloride was evaporated under reduced pressure conditions and the residue was diluted with 150 mL anhydrous acetonitrile.
Step 2. Ethyl 3,5-dimethoxybenzoate
Ethyl potassium malonate (51 g, 300 mmol) and magnesium chloride (36 g, 375 mmol) were suspended in 600 mL anhydrous acetonitrile, and 63 mL Et3N was added slowly. The mixture stirred at room temperature for 2 h. Then 3,5-dimethoxybenzoyl chloride in 50 mL anhydrous acetonitrile was added dropwise. The reaction mixture stirred at room temperature overnight. Dilute hydrochloric acid added to the mixture until pH < 5. The aqueous layer was extracted three times with ethyl acetate (EA) and the combined organic layers were washed with brine and then dried over anhydrous sodium sulfate, filtered and the solution was concentrated under reduced pressure to give the corresponding crude product.
Step 3. 1-(3,5-dimethoxyphenyl)ethanone (2)
The crude product from step 2 dissolved in 150 mL 3 M HCl and 50 mL ethanol. The reaction mixture was heated at reflux for 2 h. After cooling, the solvent was reduced under reduced pressure. The residue was separated on a silica gel column, eluting with a petroleum ether:ethyl acetate (PE/EA) (6:1 v/v) solvent system to give 2 (21.3 g, 79% over three steps); colorless oil; 1H-NMR (400 MHz, CDCl3) δ 7.08 (s, 2H), 6.64 (s, 1H), 3.83 (s, 6H), and 2.56 (s, 3H).
3.2.2. (E)-3-Chloro-3-(3,5-dimethoxyphenyl)acrylonitrile (3)
In addition, 15 mL POCl3 was added dropwise into 25 mL anhydrous DMF, the mixture stirred at room temperature for 15 min. Compound 2 (13.8 g, 78 mmol) in 30 mL anhydrous DMF was added to the mixture carefully and stirred for 20 min, and then the reaction was heated to 40 °C for 1 h (monitored by Thin Layer Chromatography, TLC). Then, the mixture was allowed to cool down to 25 °C, and 21.3 g hydroxylamine hydrochloride was added in batches slowly because of the intense heat released. Another 150 mL of anhydrous acetonitrile was added and the mixture stirred at room temperature for overnight. Then, the mixture poured into ice water, stirred for 5 h, filtered, and then the solid was washed by water and ether. The crude product was purified by eluting through a silica gel column with a 1:3 CH2Cl2/PE solvent system to give 3 (6.96 g, 40% over two steps); colorless oil; 1H-NMR (400 MHz, CDCl3) δ 6.76 (s, 2H), 6.58 (s, 1H), 6.00 (s, 1H), and 3.83 (s, 6H).
3.2.3. Methyl 3-Amino-5-(3,5-dimethoxyphenyl)thiophene-2-carboxylate (4)
Methyl 2-mercaptoacetate (3 mL, 33 mmol) was dissolved in 45 mL MeOH, MeONa (2.4 g, 45 mmol) was added slowly, and the mixture was stirred at 25 °C for half an hour. Compound 3 (6.72 g, 10 mmol) was added and then heated to reflux for 1 h (monitored by TLC), and then the mixture was allowed to cool down to 25 °C and poured into ice water, filtered, and then the filtrate was extracted with EA twice, and the combined organic layers were washed with brine and then dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by eluting through a silica gel column with a 1:8 EA/PE solvent system to give 4 (8.4 g, 96%); yellow oil; 1H-NMR (400 MHz, CDCl3) δ 6.74 (s, 1H), 6.72 (s, 2H), 6.46 (s, 1H), 5.46 (s, 2H), 3.84 (s, 3H), 3.82 (s, 6H).
3.2.4. 6-(3,5-Dimethoxyphenyl)thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione (5)
Compound 4 (8.2 g, 28 mmol) was reacted with 40 g urea at 180 °C for 8 h (monitored by TLC). The mixture was cooled to 120 °C, then added into 500 mL NaOH (1 M) solution, and stirred for 1 h. After filtration, the solid was washed with water and dried by vacuum to obtain 5 (7.6 g, 89%); yellow solid; M.p.: >250 °C; 1H-NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 11.26 (s, 1H), 7.24 (s, 1H), 6.84 (s, 2H), 6.61 (s, 1H), 3.82 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ 161.0, 158.8, 157.7, 151.5, 146.6, 134.0, 113.5, 110.0, 104.2, 101.5, 55.5; ESI-MS: m/z 305.1 [M + H]+.
3.2.5. 2,4-Dichloro-6-(3,5-dimethoxyphenyl)thieno[3,2-d]pyrimidine (6)
Compound 5 (7.5 g, 22 mmol) was dissolved in 30 mL POCl3. The solution was heated to reflux for overnight (monitored by TLC). The mixture was allowed to cool down to 40 °C, and then added to 300 mL ice water slowly, white solid precipitated slowly. The precipitate was collected, washed with water and dried to provide the desired product 6 (4.9 g, 65%); white solid; M.p.: >250 °C; 1H-NMR (400 MHz, CDCl3) δ 7.62 (s, 1H), 6.84 (d, 2H), 6.58 (s, 1H), 3.87 (s, 6H); 13C-NMR (101 MHz, CDCl3) δ 163.9, 161.5, 157.5, 156.4, 154.7, 133.7, 128.8, 119.4, 105.3, 102.5, 55.6; ESI-MS: m/z 341.0 [M + H]+.
3.2.6. 2-Chloro-6-(3,5-dimethoxyphenyl)thieno[3,2-d]pyrimidine (7)
To a solution of 6 (4.8 g, 14 mmol) and NaHCO3 (2.35 g, 28 mmol) in 20 mL EtOH and 20 mL EA, 10% Pd/C was added (960 mg, 20% by wt). The suspension was stirred at room temperature under an atmosphere of H2 for 23 h. A second portion of 10% Pd/C (980 mg, 20% by wt) was added after 12 h. The reaction mixture was filtered through Celite® with EtOAc washings. The filtrate was washed with H2O/brine (4:1), dried by MgSO4, filtered and concentrated under reduced pressure to provide 7 (3.4 g, 79%); light yellow solid; M.p.: >250 °C; 1H-NMR (400 MHz, CDCl3) δ 9.04 (s, 1H), 7.62 (s, 1H), 6.87 (s, 2H), 6.58 (s, 1H), 3.88 (s, 6H); 13C-NMR (101 MHz, CDCl3) δ 163.6, 161.4, 157.7, 157.2, 152.7, 134.1, 129.7, 119.0, 105.4, 102.2, 77.3, 77.0, 76.7, 55.6; ESI-MS: m/z 307.0 [M + H]+.
3.2.7. 6-(3,5-Dimethoxyphenyl)thieno[3,2-d]pyrimidin-2-amine (8)
Compound 7 (3.1 g, 10 mmol) was dissolved in 80 mL 4 M NH3/EtOH. The solution was heated to 150 °C for 48 h in sealed tube. The reaction mixture was cooled to room temperature and then poured into ice water, filtered through Celite® with EtOAc washings. The solid dried by vacuum to obtain compound 8 (1.9 g, 67%); Off-white solid; M.p.: 249.4–250.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.65 (s, 1H), 6.96 (s, 2H), 6.62 (s, 1H), 6.57 (s, 2H), 3.83 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ 162.7, 161.8, 160.9 152.8, 152.3, 134.7, 119.5, 119.2, 104.5, 101.5, 55.5; ESI-MS: m/z 288.1 [M + H]+.
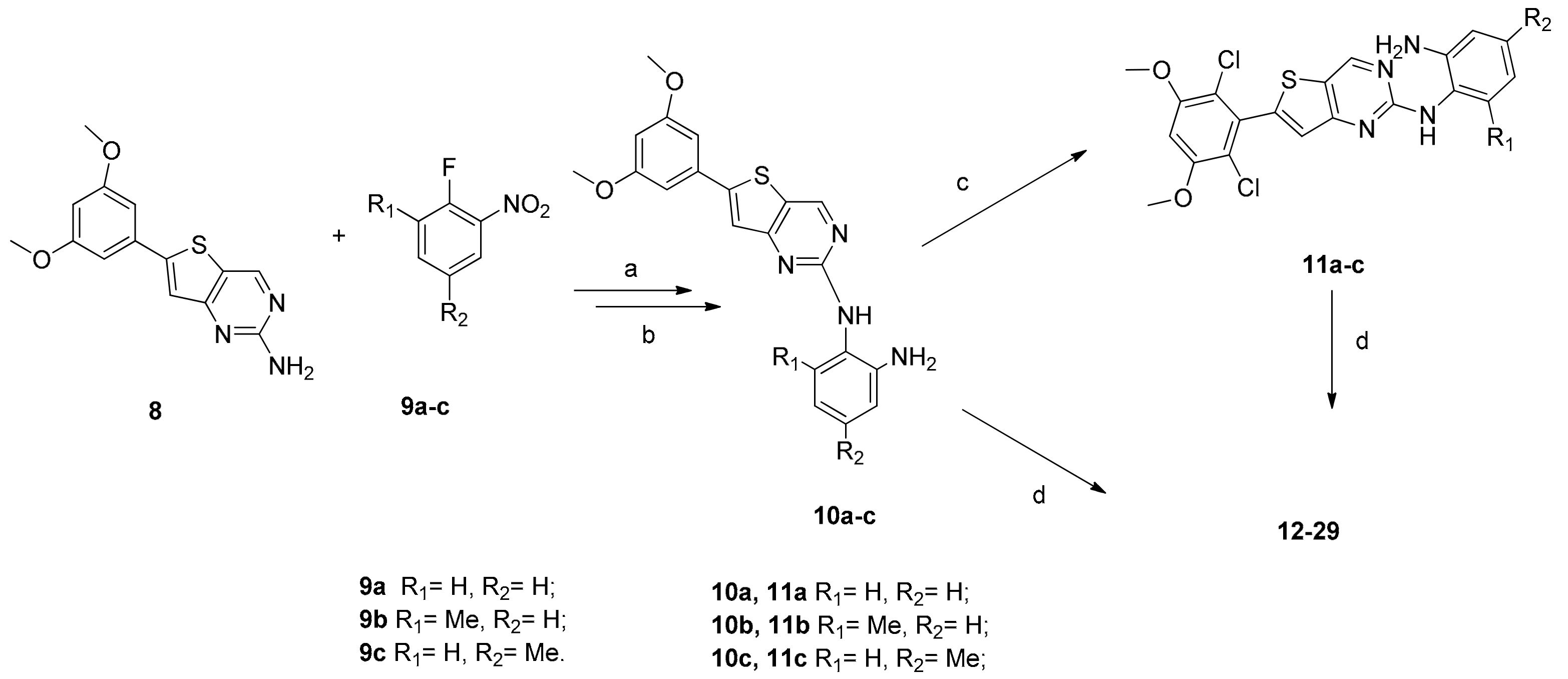
3.2.8. General Procedure 1 of the Synthesis of Compounds 10a–c
The solution of compound 8 (287 mg, 1 mmol) and NaH (60%, 80 mg, 2 mmol) in 10 mL anhydrous DMF was stirred for 30 min, corresponding o-fluoronitrobenzene (9a–c, 1.05 mmol) in 5 mL anhydrous DMF was added to the mixture dropwise. The reaction mixture was stirred for overnight and then poured into 100 mL water, filtered through Celite with water washings. The solid dried by vacuum to give corresponding crude products. To a solution of corresponding crude products (0.8 mmol) in 15 mL EtOH, Fe(OH)3 (43mg, 0.4 mmol) and Hydrazine hydrate (80%, 0.5 mL, 8 mmol) were added. The mixture was heated to 70 °C for about 4 h (monitored by TLC). The reaction mixture was cooled to room temperature and then filtered through Celite® with EtOAc washings. The filtrate was concentrated under reduced pressure to provide crude products and then the residue was purified through a silica gel column to give the corresponding 10a–c.
Preparation of 10a. From compound 9c (131 mg, 1.05 mmol), as that described in procedure 1, gave pure 10a (298 mg, 96%); off-white solid; M.p.: 209.2–210.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 8.51 (s, 1H), 7.80 (s, 1H), 7.42 (d, J = 7.1 Hz, 1H), 6.99 (d, J = 2.1 Hz, 2H), 6.93–6.86 (m, 1H), 6.75 (d, J = 6.9 Hz, 1H), 6.61 (dd, J = 7.0, 5.0 Hz, 2H), 4.88 (s, 2H), 3.83 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ 162.3, 161.0, 159.4, 152.8, 142.3, 134.6, 125.3, 124.8, 120.7, 119.6, 116.2, 115.6, 104.4, 101.7, 55.5; ESI-MS: m/z 379.1 [M + H]+.
Preparation of 10b. From compound 9b (131 mg, 1.05 mmol), as that described in procedure 1, gave pure 10b (275 mg, 88%); yellow solid; M.p.: 180.0–181.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.28 (s, 1H), 7.76 (s, 1H), 6.95 (s, 2H), 6.88 (t, J = 7.6 Hz, 1H), 6.60 (s, 2H), 6.46 (d, J = 7.3 Hz, 1H), 4.71 (s, 2H), 3.81 (s, 6H), 2.06 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 162.6, 160.9, 160.1, 152.9, 152.5, 145.6, 136.5, 134.7, 126.4, 123.4, 120.3, 119.7, 117.8, 112.8, 104.4, 101.6, 55.5, 18.3; ESI-MS: m/z 393.2 [M + H]+.
Preparation of 10c. From compound 9b (131 mg, 1.05 mmol), as that described in procedure 1, gave pure 10c (282 mg, 90%); yellow solid; M.p.: 192.0–193.2 °C; 1H-NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 7.40 (s, 1H), 7.28 (s, 1H), 6.84 (d, J = 1.8 Hz, 2H), 6.82 (s, 1H), 6.68 (s, 1H), 6.65 (d, J = 8.0 Hz, 1H), 6.53 (s, 1H), 3.85 (s, 7H), 2.30 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 163.0, 161.2, 159.8, 154.5, 152.4, 141.9, 136.8, 135.1, 126.3, 123.2, 122.2, 120.0, 119.2, 117.7, 105.0, 101.7, 55.5, 21.2; ESI-MS: m/z 393.2 [M + H]+.
3.2.9. General Procedure 2 of the Synthesis of Compounds 11a–c
Corresponding 10a–c (0.35 mmol) was dissolved in anhydrous THF or anhydrous DCM (5 mL), and sulfonyl chloride (290 mg, 2.1 mmol) was added to the solution in portions at −10 °C. The resulting mixture stirred at 0 °C for 1 h (monitored by TLC). EA (20 mL) and Na2CO3 solution (20 mL) were added, and the layers were partitioned and separated. The organic layers were washed with water and brine, dried over anhydrous sodium sulfate, filtered and then the filtration was concentrated in the vacuum to give corresponding 11a–c.
Preparation of 11a. From compound 10a (133 mg, 0.35 mmol), as that described in procedure 2, gave pure 11a (128 mg, 82%); yellow solid; M.p.: >250 °C; 1H-NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 7.55–7.39 (m, 1H), 7.13 (s, 1H), 7.07 (m, 1H), 6.92–6.81 (m, 2H), 6.74 (s, 1H), 6.68 (s, 1H), 3.97 (s, 6H), 3.93–3.78 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ 162.2, 159.3, 154.7, 152.7, 148.4, 141.4, 133.4, 126.5, 126.1, 125.6, 124.7, 123.7, 119.4, 117.2, 115.3, 98.2, 56.7; ESI-MS: m/z 447.0 [M + H]+.
Preparation of 11b. From compound 10b (138 mg, 0.35 mmol), as that described in procedure 2, gave pure 11b (127 mg, 79%); yellow solid; M.p.: >250 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 8.32 (s, 1H), 7.14 (s, 1H), 7.07 (s, 1H), 6.87 (t, J = 7.7 Hz, 1H), 6.59 (d, J = 7.8 Hz, 1H), 6.46 (d, J = 7.3 Hz, 1H), 4.72 (s, 2H), 3.97 (s, 6H), 2.07 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 161.8, 160.1, 154.5, 153.2, 145.7, 136.5, 132.6, 126.5, 124.2, 123.3, 121.5, 117.7, 113.4, 112.8, 99.4, 56.9, 18.3; ESI-MS: m/z 461.0 [M + H]+.
Preparation of 11c. From compound 10c (138 mg, 0.35 mmol), as that described in procedure 2, gave pure 11c (131 mg, 81%); yellow solid; M.p.: 229.6–231.4 °C; 1H-NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 7.27 (m, 1H), 7.11 (s, 1H), 6.80 (s, 1H), 6.67 (s, 2H), 3.97 (s, 6H), 2.29 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 162.3, 159.7, 154.7, 152.7, 148.3, 141.9, 136.8, 133.4, 126.3, 124.7, 123.4, 123.2, 120.0, 117.7, 115.3, 98.2, 56.1, 21.2; ESI-MS: m/z 461.0 [M + H]+.
3.2.10. General Procedure 3 of the Synthesis of Compounds 12–17
One of corresponding 10a–c and 11a–c (0.1 mmol) was dissolved in anhydrous DCM. DIPEA (0.04 mL) and 0.1 mL 10% Acryloyl chloride (anhydrous THF) were added to the solution dropwise at −10 °C. The resulting mixture was stirred for 1 h (monitored by TLC), DCM (20 mL) and NaHCO3 solution (20 mL) were then added, and the layers were partitioned and separated. The organic layers were washed with NH4Cl solution, washed with water and brine, dried over anhydrous sodium sulfate, filtered and the solution was then concentrated under reduced pressure to give corresponding crude product 12–17, which was furthermore purified through a silica gel column.
Preparation of 12. From compound 10a (38 mg, 0.1 mmol), as that described in procedure 3, gave pure 12 (21 mg, 50%); yellow solid; M.p.: 217.1–218.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 9.08 (s, 1H), 8.64 (s, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.84 (s, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.00 (s, 2H), 6.63 (s, 1H), 6.51 (dd, J = 16.6, 10.2 Hz, 1H), 6.28 (d, J = 17.0 Hz, 1H), 5.77 (d, J = 10.5 Hz, 1H), 3.83 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ 163.8, 162.1, 161.0, 158.4, 153.4, 153.0, 134.5, 132.8, 131.5, 129.7, 127.2, 125.3, 124.6, 124.0, 123.4, 121.8, 119.5, 104.5, 101.8, 55.5; HPLC: method 1, room temperature, tg = 11.72 min, UV254 = 98%; HRMS (ESI) m/z calcd for C23H20N4O3S [M + H]+: 433.1329, found: 433.1350.
Preparation of 13. From compound 10b (39 mg, 0.1 mmol), as that described in procedure 3, gave pure 13 (18 mg, 40%); yellow solid; M.p.:184.5–185.6 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.99 (s, 1H), 8.33 (s, 1H), 7.77 (s, 1H), 7.71 (d, J = 7.0 Hz, 1H), 7.18 (t, J = 7.9 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 1.8 Hz, 2H), 6.60 (s, 1H), 6.51 (dd, J = 16.8, 10.1 Hz, 1H), 6.21 (d, J = 16.7 Hz, 1H), 5.69 (d, J = 10.3 Hz, 1H), 3.81 (s, 6H), 2.16 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 168.8, 163. 5, 162.4, 160.9, 159.5, 152.9, 150.8, 136.9, 134.9, 134.6, 131.8, 126.8, 126.5, 125.9, 121.0, 120.0, 104.4, 101.7, 55.5, 18.5; HPLC: method 2, room temperature, tg = 11.67 min, UV254 = 97%; HRMS (ESI) m/z calcd for C24H22N4O3S [M + H]+: 447.1485, found: 447.1495.
Preparation of 14. From compound 10c (39 mg, 0.1 mmol), as that described in procedure 3, gave pure 14 (16 mg, 36%); light yellow solid; M.p.: 239.0–240.1 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 9.05 (s, 1H), 8.55 (s, 1H), 7.82 (s, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.40 (s, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 2.1 Hz, 2H), 6.62 (s, 1H), 6.50 (dd, J = 17.0, 10.2 Hz, 1H), 6.28 (d, J = 17.0 Hz, 1H), 5.76 (t, J = 5.0 Hz, 1H), 3.83 (s, 6H), 3.34 (s, 4H), 2.50 (s, 4H), 2.31 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 163.6, 162.2, 161.0, 158.6, 153.2, 153.0, 134.5, 132.7, 131.5, 130.1, 129.8, 127.1, 125.8, 124.7, 124.3, 121.5, 119.6, 104.5, 101.8, 55.5, 20.5; HPLC: method 2, room temperature, tg = 12.05 min, UV254 = 98%; HRMS (ESI) m/z calcd for C24H22N4O3S [M+Na]+: 469.1305, found: 469.1299.
Preparation of 15. From compound 11a (45 mg, 0.1 mmol), as that described in procedure 3, gave pure 15 (22 mg, 44%); white solid; M.p.: 236.6–236.9 °C; 1H-NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 8.63 (s, 1H), 7.93 (s, 1H), 7.52 (s, 1H), 7.38 (s, 1H), 7.22 (d, J = 8.2 Hz, 1H), 7.15 (s, 1H), 6.68 (s, 1H), 6.35 (d, J = 16.9 Hz, 1H), 6.20 (dd, J = 16.8, 10.3 Hz, 1H), 5.69 (d, J = 10.2 Hz, 1H), 3.98 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ 163.7, 161.2, 158.3, 154.5, 153.3, 148.0, 132.6, 132.4, 131.5, 130.1, 127.1, 125.3, 124.5, 124.4, 124.2, 123.7, 123.0, 113.3, 99.5, 56.9; HPLC: method 2, room temperature, tg = 11.37 min, UV254 = 97%; HRMS (ESI) m/z calcd for C23H18Cl2N4O3S [M+Na]+: 523.0369, found: 523.0371.
Preparation of 16. From compound 11b (46 mg, 0.1 mmol), as that described in procedure 3, gave pure 16 (24 mg, 47%); light yellow solid; M.p.: 142.5–143.8 °C; 1H-NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.49 (s, 1H), 8.05 (s, 1H), 7.24 (m, 1H), 7.13 (s, 1H), 7.09 (d, J = 7.5 Hz, 1H), 6.68 (s, 1H), 6.60 (s, 1H), 6.30 (d, J = 17.0 Hz, 1H), 6.14 (dd, J = 16.8, 10.2 Hz, 1H), 5.65 (d, J = 10.2 Hz, 1H), 3.98 (s, 6H), 2.31 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 163.5, 161.6, 159.5, 154.5, 153.2, 147.4, 136.9, 135.0, 132.5, 131.9, 126.8, 126.4, 125.9, 124.2, 122.2, 120.8, 113.4, 99.4, 56.9, 18.5; HPLC: method 2, room temperature, tg = 11.12 min, UV254 = 98%; HRMS (ESI) m/z calcd for C24H20Cl2N4O3S [M + H]+: 515.0706, found: 515.0715.
Preparation of 17. From compound 11c (46 mg, 0.1 mmol), as that described in Procedure 3, gave pure 17 (22 mg, 44%); white solid; M.p.: 258.2–259.6 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 9.11 (s, 1H), 8.59 (s, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.41 (s, 1H), 7.23 (s, 1H), 7.08 (s, 1H), 7.03 (d, J = 8.3 Hz, 1H), 6.50 (dd, J = 17.0, 10.2 Hz, 1H), 6.27 (dd, J = 17.0, 1.7 Hz, 1H), 5.76 (dd, J = 10.1, 1.7 Hz, 1H), 3.98 (s, 6H), 2.30 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 163.6, 161.3, 158.6, 154.5, 153.3, 147.7, 133.0, 132.4, 131.5, 130.2, 129.9, 127.0, 125.8, 124.7, 124.6, 124.2, 122.7, 113.3, 99.4, 56.9, 20.5; HPLC: method 2, room temperature, tg = 11.54 min, UV254 = 98%; HRMS (ESI) m/z calcd for C24H20Cl2N4O3S [M + H]+: 515.0706, found: 515.0723.
3.2.11. General Procedure 4 of the Synthesis of Compounds 18–29
Step 1. One of corresponding 10a–c and 11a–c (0.2 mmol) was dissolved in anhydrous DCM. DIPEA (0.07 mL) and (E)-4-bromobut-2-enoyl chloride (0.01 g/mL in anhydrous THF, 3 mL) were added to the solution at −5 °C. The resulting mixture was stirred for 2 h (monitored by TLC), DCM (10 mL) and water (10 mL) were added, the layers were partitioned and separated. The organic layers were washed with NaHCO3 solution, water and brine, dried over anhydrous sodium sulfate, filtered and the solution was concentrated under reduced pressure, used directly in the next step.
Step 2. One of corresponding crude product in the Step 1 (0.1 mmol) was dissolved in anhydrous DMF. NaI (46 mg, 0.3 mmol) and 0.07 mL (40%) dimethylamine solution in water were added. The resulting mixture was stirred at room temperature (monitored by TLC). EA (10 mL) and water (10 mL) were added, and the layers were partitioned and separated. The organic layers were washed with water and brine, dried over anhydrous sodium sulfate, filtered and concentrated, and then the residue was purified through a silica gel column to give corresponding 18–23.
Preparation of 18. From compound 10a (38 mg, 0.1 mmol), as that described in step 1 and step 2 of procedure 4, gave pure 18 (24 mg, 49%); yellow solid; M.p.: 175.1–175.9 °C; 1H-NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 8.55 (s, 1H), 7.88 (s, 1H), 7.58 (s, 2H), 7.40 (s, 1H), 7.22 (d, J = 4.1 Hz, 2H), 6.92 (dt, J = 12.2, 5.9 Hz, 1H), 6.84 (d, J = 2.1 Hz, 2H), 6.53 (t, J = 2.1 Hz, 1H), 6.06 (d, J = 15.4 Hz, 1H), 3.85 (s, 6H), 3.05 (d, J = 5.7 Hz, 2H), 2.22 (s, 6H); 13C-NMR (101 MHz, CDCl3) δ 172.5, 164.0, 162.6, 161.3, 159.1, 155.3, 152.3, 141.8, 134.9, 131.7, 126.2, 125.6, 124.9, 124.6, 122.9, 118.9, 105.0, 101.9, 60.2, 55.6, 45.3; HPLC: method 1, room temperature, tg = 11.94 min, UV280 = 96%; HRMS (ESI) m/z calcd for C26H27N5O3S [M + H]+: 490.1907, found: 490.1940.
Preparation of 19. From compound 10b (39 mg, 0.1 mmol), as that described in step 1 and step 2 of procedure 4, gave pure 19 (20 mg, 40%); yellow solid; M.p.: 151.2–152.9 °C; 1H-NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 8.37 (s, 1H), 8.08 (s, 1H), 7.36 (s, 1H), 7.24 (m, J = 8.0 Hz, 1H), 7.08 (d, J = 7.5 Hz, 1H), 6.91–6.84 (m, 2H), 6.83 (d, J = 2.2 Hz, 2H), 6.53 (t, J = 2.1 Hz, 1H), 5.99 (d, J = 15.3 Hz, 1H), 3.85 (s, 6H), 3.10–2.90 (m, 2H), 2.27 (s, 3H), 2.19 (s, 6H); 13C-NMR (101 MHz, CDCl3) δ 163.7, 162. 9, 161.3, 159.7, 155.4, 152.5, 141.7, 135.5, 135.2, 134.9, 127.2, 126.6, 126.3, 122.9, 120.8, 118.9, 105.0, 101.9, 60.2, 55.7, 45.4, 18.7; HPLC: method 1, room temperature, tg = 11.87 min, UV254 = 98%; HRMS (ESI) m/z calcd for C27H29N5O3S [M + H]+: 504.2064, found: 504.2082.
Preparation of 20. From compound 10c (39 mg, 0.1 mmol), as that described in step 1 and step 2 of procedure 4, gave pure 20 (17 mg, 34%); yellow solid; M.p.: 170.8–171.5 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.78 (s, 1H), 9.06 (s, 1H), 8.56 (s, 1H), 7.82 (s, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.41 (s, 1H), 7.04 (d, J = 8.5 Hz, 1H), 7.00 (s, 2H), 6.84–6.71 (m, 1H), 6.64 (s, 1H), 6.39 (d, J = 15.9 Hz, 1H), 3.84 (s, 6H), 3.27 (s, 2H), 2.32 (s, 9H); 13C-NMR (101 MHz, DMSO-d6) δ 163.5, 162.2, 161.0, 158.6, 153.2, 153.0, 134.5, 132.7, 130.0, 126.3, 125.7, 124.6, 124.4, 121.5, 119.6, 104.5, 101.8, 59.3, 55.5, 44.7, 20.5; HPLC: method 1, room temperature, tg = 12.46 min, UV254 = 99%; HRMS (ESI) m/z calcd for C27H29N5O3S [M + H]+: 504.2064, found: 504.2092.
Preparation of 21. From compound 11a (45 mg, 0.1 mmol), as that described in step 1 and step 2 of procedure 4, gave pure 21 (21 mg, 38%); yellow solid; M.p.: 231.7–232.5 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.79 (s, 1H), 9.14 (s, 1H), 8.70 (s, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.26 (s, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 7.08 (s, 1H), 6.76 (dt, J = 15.2, 5.8 Hz, 1H), 6.34 (d, J = 15.5 Hz, 1H), 3.98 (s, 6H), 3.05 (d, J = 5.5 Hz, 2H), 2.16 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ 163.8, 161.3, 158.4, 154.5, 153.4, 147. 8, 141.8, 132.4, 130.2, 125.4, 125.1, 124.4, 124.2, 123.6, 123.0, 113.3, 99.5, 59.7, 56.9, 45.2; HPLC: method 1, room temperature, tg = 14.20 min, UV254 = 98%; HRMS (ESI) m/z calcd for C26H25Cl2N5O3S [M + H]+: 558.1128, found: 558.1158.
Preparation of 22. From compound 11b (46 mg, 0.1 mmol), as that described in step 1 and step 2 of procedure 4, gave pure 22 (24 mg, 42%); yellow solid; M.p.: >250 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 9.04 (s, 1H), 8.40 (s, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.18 (t, J = 7.8 Hz, 2H), 7.08 (d, J = 8.2 Hz, 2H), 6.70 (dt, J = 15.3, 6.2 Hz, 1H), 6.41 (d, J = 15.4 Hz, 1H), 3.97 (s, 6H), 3.21 (d, J = 4.4 Hz, 2H), 2.28 (s, 6H), 2.16 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 163.2, 161.6, 159.5, 154.5, 153.2, 147.4, 136.9, 135.2, 132.5, 130.3, 126.3, 125.9, 124.2, 122.2, 120.6, 113.3, 99.4, 58.9, 56.9, 44.0, 18.5; HPLC: method 2, room temperature, tg = 11.35 min, UV254 = 99%; HRMS (ESI) m/z calcd for C27H27Cl2N5O3S [M + H]+: 572.1284, found: 572.1327.
Preparation of 23. From compound 11c (46 mg, 0.1 mmol), as that described in step 1 and step 2 of procedure 4, gave pure 23 (19 mg, 33%); yellow solid; M.p.: 214.0–214.6 °C; 1H-NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.45 (s, 1H), 7.80 (s, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.16 (s, 1H), 7.14 (s, 1H), 7.00 (d, J = 7.8 Hz, 1H), 6.91 (dt, J = 15.0, 6.0 Hz, 1H), 6.68 (s, 1H), 6.03 (d, J = 15.4 Hz, 1H), 5.30 (s, 1H), 3.98 (s, 6H), 3.07 (d, J = 5.8 Hz, 2H), 2.36 (s, 3H), 2.24 (s, 6H); 13C-NMR (101 MHz, CDCl3) δ 163.8, 161.9, 159.3, 154.7, 152.8, 149.0, 141.5, 133.2, 126.4, 126.3, 125.2, 124.7, 124.4, 124.1 115.2, 98.2, 60.2, 56.7, 45.3, 21.2; HPLC: method 1, room temperature, tg = 12.66 min, UV254 = 95%; HRMS (ESI) m/z calcd for C27H27Cl2N5O3S [M + Na]+: 594.1104, found: 594.1091.
Step 3. Corresponding crude product in the step 1 (0.1 mmol) was dissolved in anhydrous DMF. NaI (46 mg, 0.3 mmol) and 0.02 mL N-Methylpiperazine were added. The resulting mixture was stirred at room temperature (monitored by TLC). EA (10 mL) and water (10 mL) were added, and the layers were partitioned and separated. The organic layers were washed with water and brine, and dried over anhydrous sodium sulfate. Filtered and the solution concentrated under reduced pressure, and then the residue was purified through a silica gel column to give corresponding 24–29.
Preparation of 24. From compound 10a (38 mg, 0.1 mmol), as that described in step 1 and step 3 of procedure 4, gave pure 24 (23 mg, 43%); yellow solid; M.p.: 182.9–183.7 °C; 1H-NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 8.50 (s, 1H), 7.88 (s, 1H), 7.55 (s, 1H), 7.48 (s, 1H), 7.41 (s, 1H), 7.21 (s, 2H), 6.91 (d, J = 9.3 Hz, 1H), 6.84 (d, J = 2.0 Hz, 2H), 6.53 (s, 1H), 6.04 (d, J = 15.3 Hz, 1H), 3.85 (s, 6H), 3.09 (d, J = 5.5 Hz, 2H), 2.42 (m, 8H), 2.23 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 163.9, 162.5, 161.3, 159.1, 155.4, 152.3, 141.7, 134.8, 131.7, 126.2, 125.7, 124.9, 124.7, 122.8, 118.8, 105.0, 101.8, 59.2, 55.6, 54.9, 53.2, 45.9; HPLC: method 1, room temperature, tg = 11.04 min, UV280 = 96%; HRMS (ESI) m/z calcd for C29H32N6O3S [M+Na]+: 567.2149, found: 567.2169.
Preparation of 25. From compound 10b (39 mg, 0.1 mmol), as that described in step 1 and step 3 of procedure 4, gave pure 25 (20 mg, 36%); yellow solid; M.p.: 156.5–157.8 °C;1H-NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 8.58 (s, 1H), 8.00 (s, 1H), 7.39 (s, 1H), 7.23 (s, 1H), 7.10 (d, J = 7.0 Hz, 1H), 6.84 (s, 2H), 6.82–6.69 (m, 2H), 6.54 (s, 1H), 5.99 (d, J = 15.0 Hz, 1H), 3.86 (s, 6H), 3.32 (s, 2H), 3.16 (s, 2H), 2.83 (s, 6H), 2.68 (s, 3H), 2.29 (s, 4H); 13C-NMR (101 MHz, MeOD) δ 166.3, 164.0, 162.9, 160.7, 156.5, 153.9, 141.6, 138.4, 136.1, 135.9, 132.1, 128.9, 127.9, 127.7, 123.3, 119.7, 105.8, 102.7, 59.5, 56.1, 55.2, 52.4, 44.8, 18.8; HPLC: method 1, room temperature, tg = 11.26 min, UV280 = 97%; HRMS (ESI) m/z calcd for C30H34N6O3S [M + H]+: 559.2486, found: 559.2494.
Preparation of 26. From compound 10c (39 mg, 0.1 mmol), as that described in step 1 and step 3 of procedure 4, gave pure 26 (19 mg, 34%); yellow solid; M.p.: 182.0–183.2 °C; 1H-NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 7.73 (s, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.37 (s, 1H), 7.02 (d, J = 7.3 Hz, 1H), 6.83 (d, J = 2.0 Hz, 2H), 6.82–6.72 (m, 1H), 6.53 (t, J = 1.9 Hz, 1H), 6.08 (d, J = 15.0 Hz, 1H), 3.85 (s, 6H), 3.17 (s, 2H), 3.14 (d, J = 5.5 Hz, 2H), 2.80 (s, 6H), 2.63 (s, 3H), 2.36 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 163.5, 161.3, 155.3, 139.8, 135.6, 134.8, 131.4, 129.0, 126.5, 125.0, 118.8, 105.0, 101.8, 58.1, 55.6, 53.8, 49.7, 43.5, 21.1; HPLC: method 1, room temperature, tg = 11.87 min, UV254 = 99%; HRMS (ESI) m/z calcd for C30H34N6O3S [M+Na]+: 581.2305, found: 581.2278.
Preparation of 27. From compound 11a (45 mg, 0.1 mmol), as that described in step 1 and step 3 of procedure 4, gave pure 27 (21 mg, 34%); off-white solid; M.p.: 160.1–160.7 °C; 1H-NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 8.46 (s, 1H), 7.88 (s, 1H), 7.56 (s, 1H), 7.29 (s, 1H), 7.22 (s, 2H), 7.16 (s, 1H), 6.93 (dt, J = 15.1, 6.1 Hz, 1H), 6.68 (s, 1H), 6.05 (d, J = 15.4 Hz, 1H), 3.98 (s, 6H), 3.11 (d, J = 5.8 Hz, 2H), 2.45 (s, 8H), 2.27 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 163.9, 161.8, 159.0, 154.7, 152.7, 149.1, 141.9, 133.1, 131.6, 126.2, 125.7, 124.9, 124.7, 124.4, 124.2, 115.1, 98.2, 59.3, 56.7, 55.0, 53.3, 46.0; HPLC: method 1, room temperature, tg = 11.63 min, UV254 = 97%; HRMS (ESI) m/z calcd for C29H30Cl2N6O3S [M + H]+: 613.1550, found: 613.1577.
Preparation of 28. From compound 11b (46 mg, 0.1 mmol), as that described in step 1 and step 3 of procedure 4, gave pure 28 (18 mg, 29%); white solid; M.p.: 225.0–226.2 °C; 1H-NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.46 (s, 1H), 8.01 (s, 1H), 7.23 (t, J = 7.9 Hz, 1H), 7.09 (s, 1H), 7.07 (d, J = 7.4 Hz, 1H), 6.96 (s, 1H), 6.87 (dt, J = 15.3, 6.2 Hz, 1H), 6.68 (s, 1H), 6.01 (d, J = 15.3 Hz, 1H), 3.97 (s, 6H), 3.07 (d, J = 6.1 Hz, 2H), 2.43 (s, 8H), 2.28 (s, 3H), 2.26 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 172.6, 163.7, 162.1, 159.5, 154.7, 152.9, 149.1, 141.5, 135.6, 135.1, 133.1, 127.1, 126.7, 124.4, 124.0, 115.2, 98.3, 59.2, 56.7, 55.0, 53.2, 45.9, 18.8; HPLC: method 1, room temperature, tg = 11.50 min, UV254 = 99%; HRMS (ESI) m/z calcd for C30H32Cl2N6O3S [M + H]+: 627.1706, found: 627.1736.
Preparation of 29. From compound 11c (46 mg, 0.1 mmol), as that described in step 1 and step 3 of procedure 4, gave pure 29 (22 mg, 35%); white solid; M.p.: 212.4–212.9 °C; 1H-NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 8.39 (s, 1H), 7.69 (s, 1H), 7.30 (d, J = 6.6 Hz, 1H), 7.14 (d, J = 13.1 Hz, 1H), 7.07 (s, 1H), 6.93 (d, J = 7.6 Hz, 1H), 6.85 (dd, J = 13.8, 7.4 Hz, 1H), 6.61 (s, 1H), 5.95 (d, J = 15.3 Hz, 1H), 3.90 (s, 6H), 3.03 (d, J = 5.9 Hz, 2H), 2.40 (s, 8H), 2.29 (s, 3H), 2.22 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 163.7, 161.3, 158.6, 154.5, 153.3, 147.7, 141.1, 133.1, 132.4, 130.3, 125.8, 125.7, 124.7, 125.6, 124.2, 122.7, 113.3, 99.4, 58.3, 56.8, 54.4, 52.4, 45.4, 20.5; HPLC: method 1, room temperature, tg = 12.03 min, UV254 = 96%; HRMS (ESI) m/z calcd for C30H32Cl2N6O3S [M + Na]+: 649.1526, found: 649.1482.
3.3. General Procedure for In Vitro Cell-Proliferation Assays
NCI-H1975, Ramos, and SNU-16 cells were maintained in Revolutions-Per-minute Indicator 1640 (RPMI 1640) medium (Gibco, Grand Island, NY, USA), A431 was maintained in Dulbecco’s Modified Eagle Medium (DMEM) medium (Gibco), MDA-MB-231 was cultured in L-15 medium (Gibco), and NCI-H1581 was cultured in ACL-4 (Gibco), and all of them were supplemented with 10% heat-inactivated fetal calf serum (FBS; Gibco) at 37 °C in a 5% CO2 humidified environment. Cells in 96-well plates were treated with gradient concentrations of compounds at 37 °C for 72 h. Cell proliferation assays in NCI-H1975, A431, MDA-MB-231 and NCI-H1581 were determined by using sulforhodamine B (SRB; Sigma, St. Louis, MO, USA). The antiproliferative activity of compounds, examined in Ramos and SUN-16 cell lines, was determined by using CCK-8 (Dojindo Laboratories, Kamimashiki-gun, Japan). The dosages corresponding to the half-maximal inhibition (IC50) were calculated using SoftMax pro-based nonlinear 4-parameter regression analysis (n = 3).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















