Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media


Abstract
:
1. Introduction
2. Water-Soluble Porphyrins Bearing Cationic Substituents
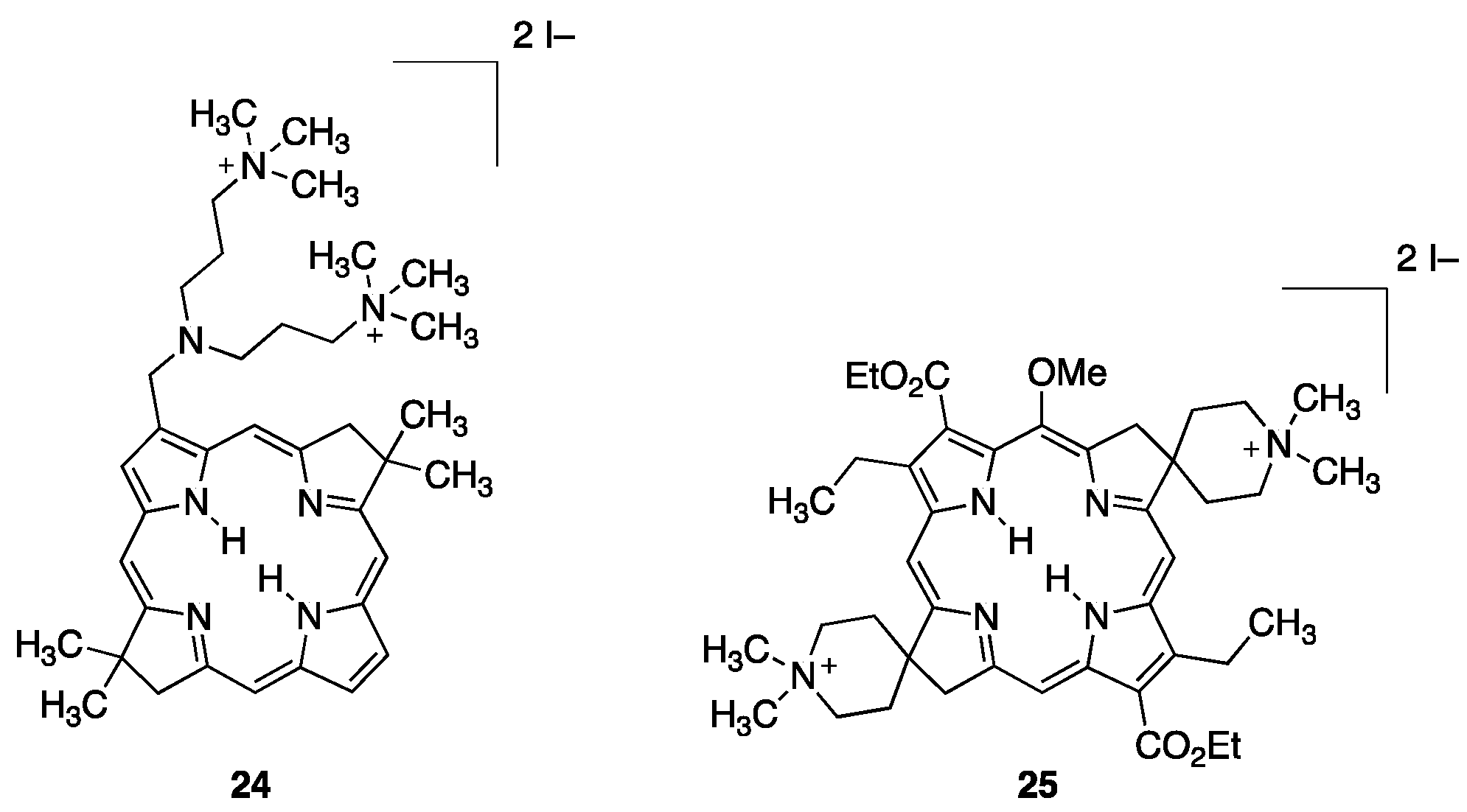
2.1. Amine/Ammonium Functionalized Porphyrins
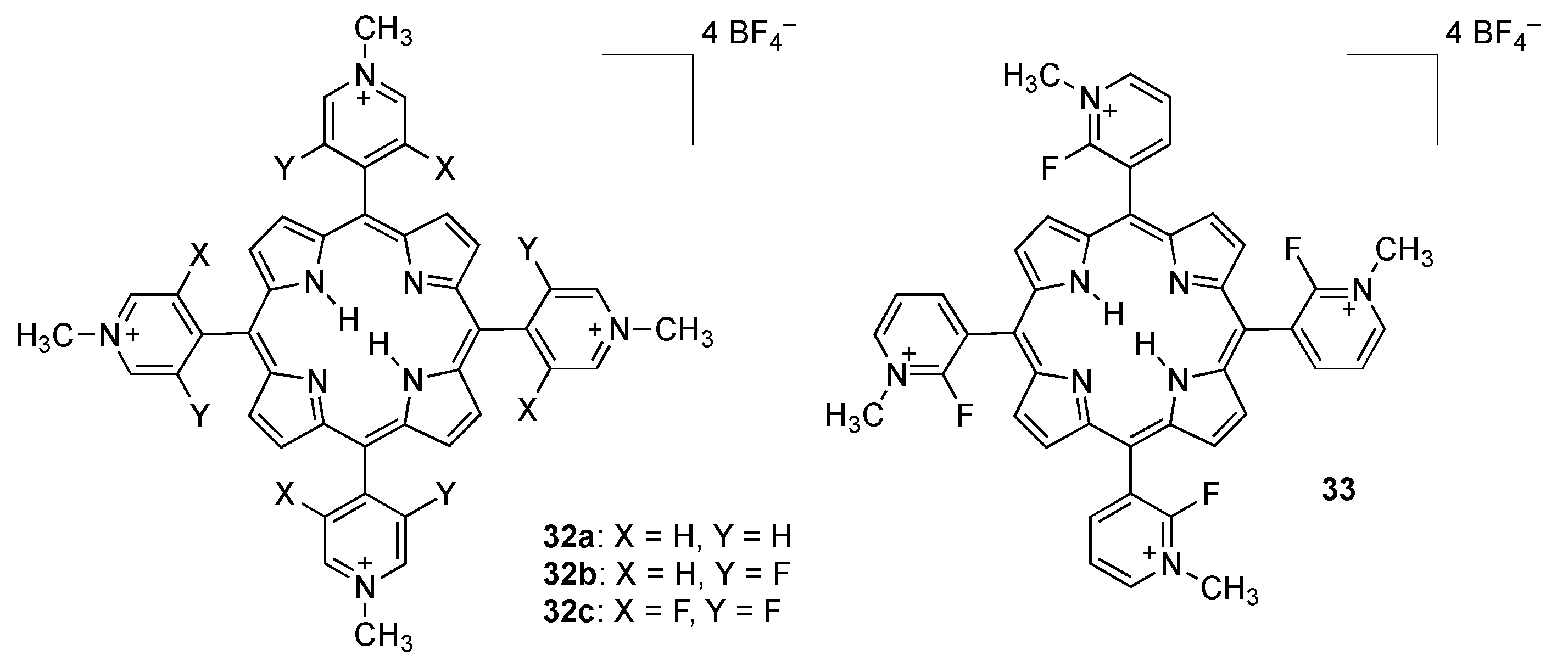
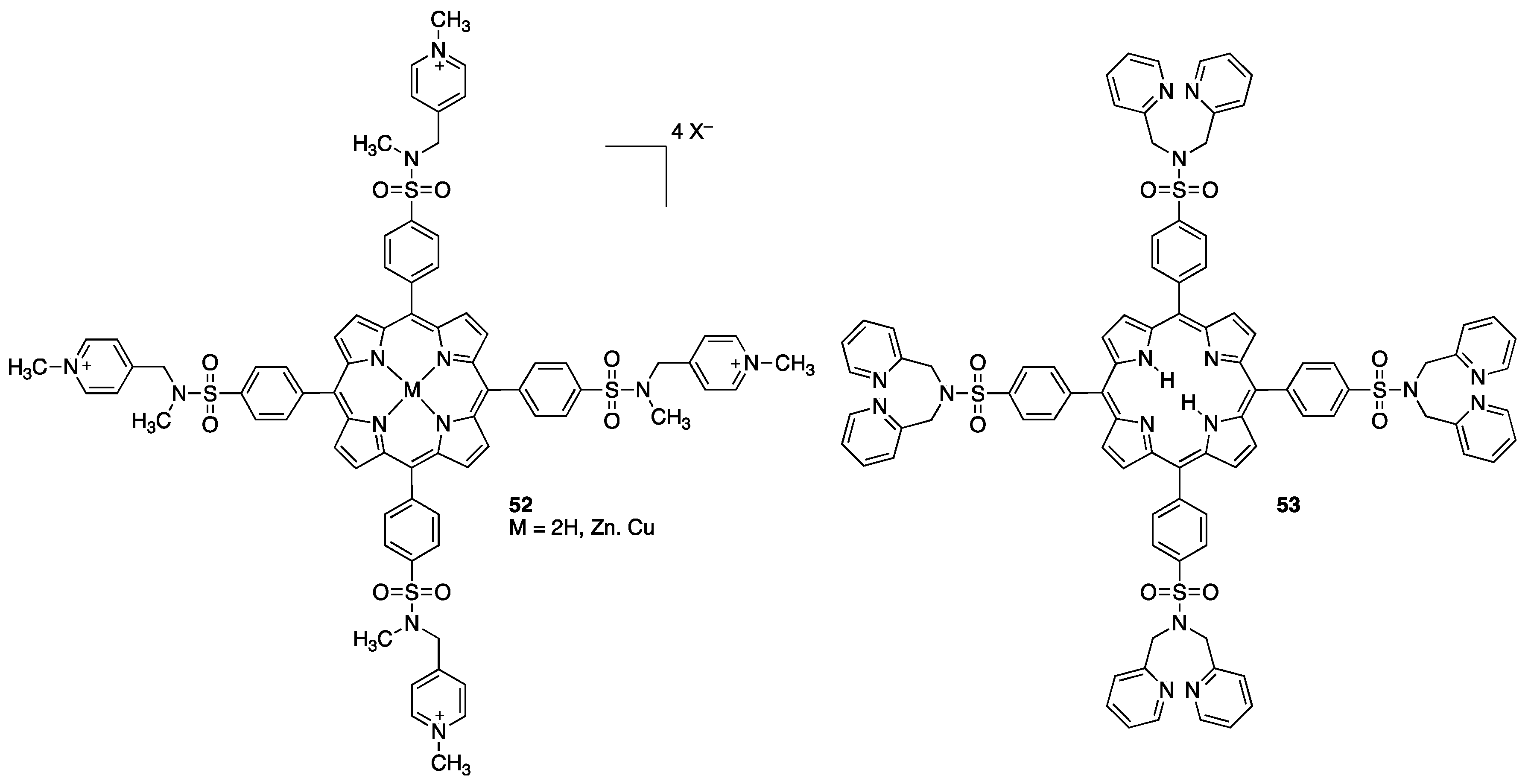
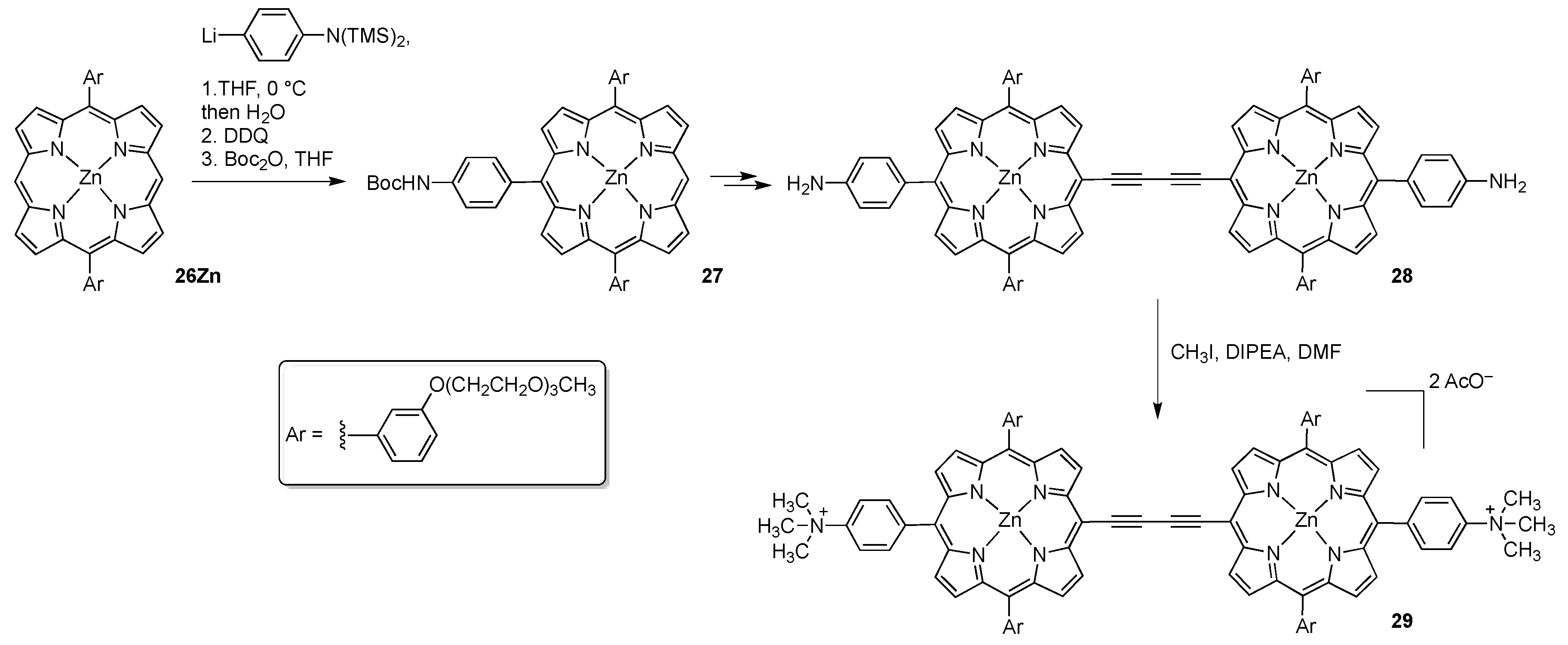
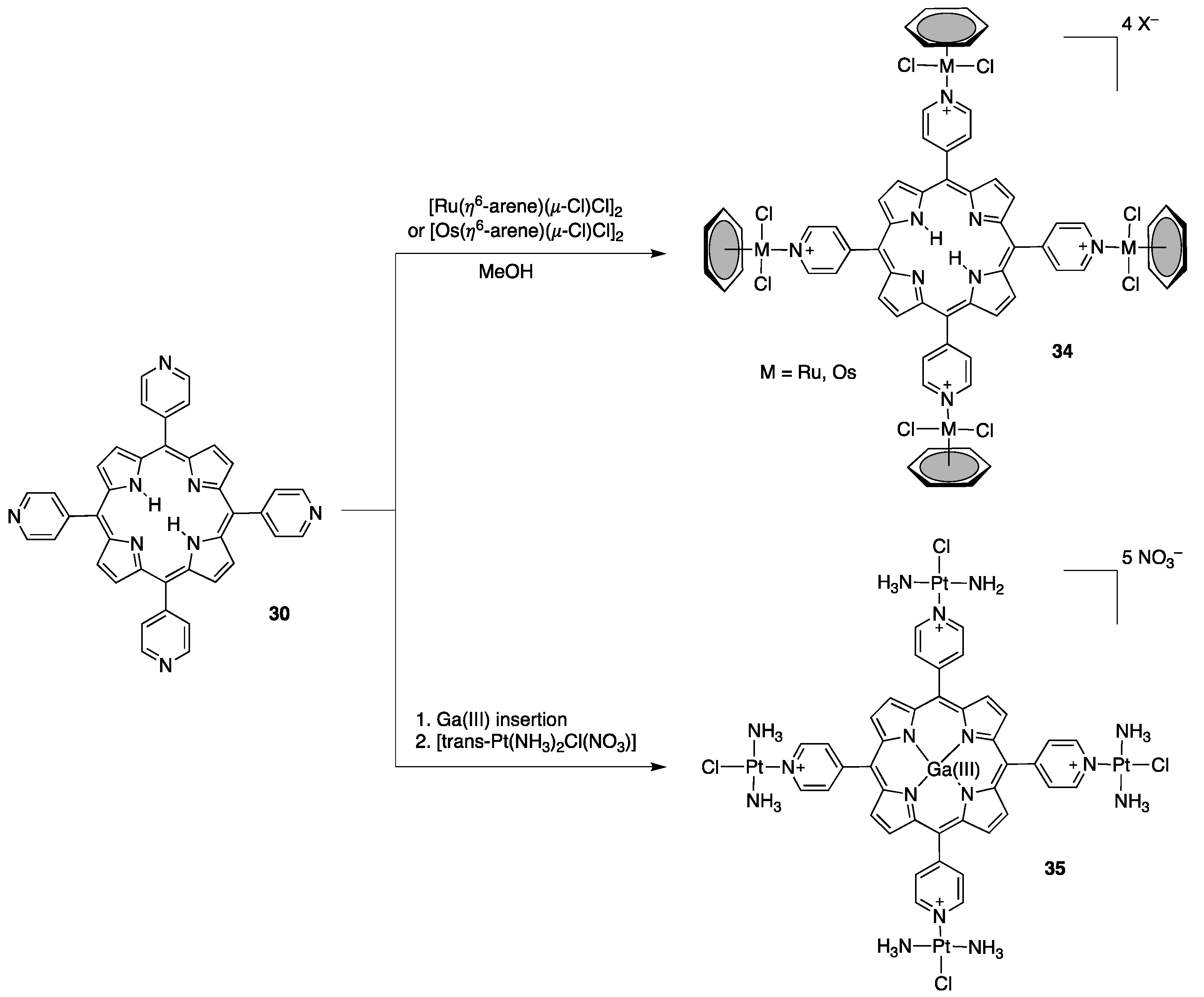
2.2. Porphyrins Carrying Pyridyl/Pyridinium Groups
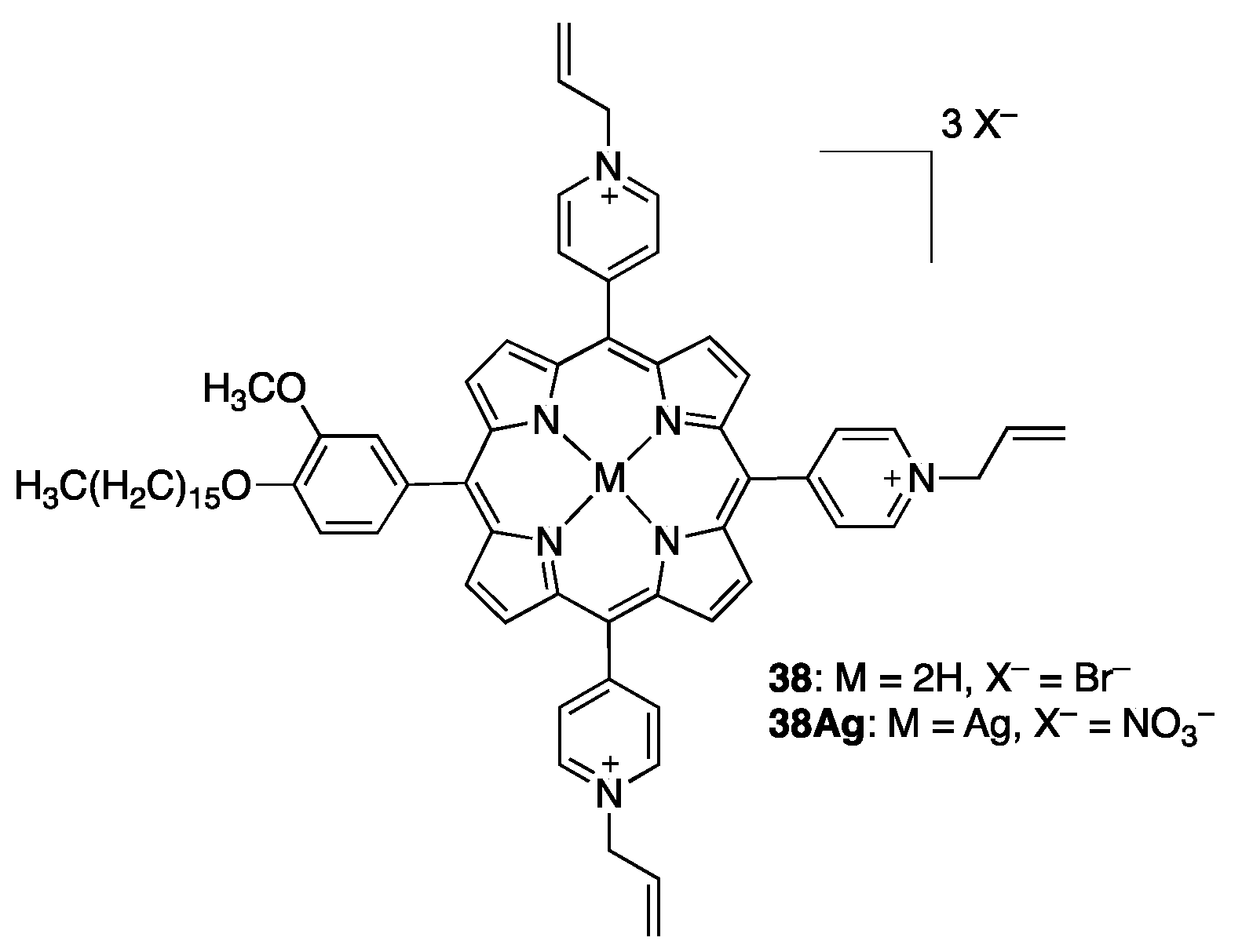
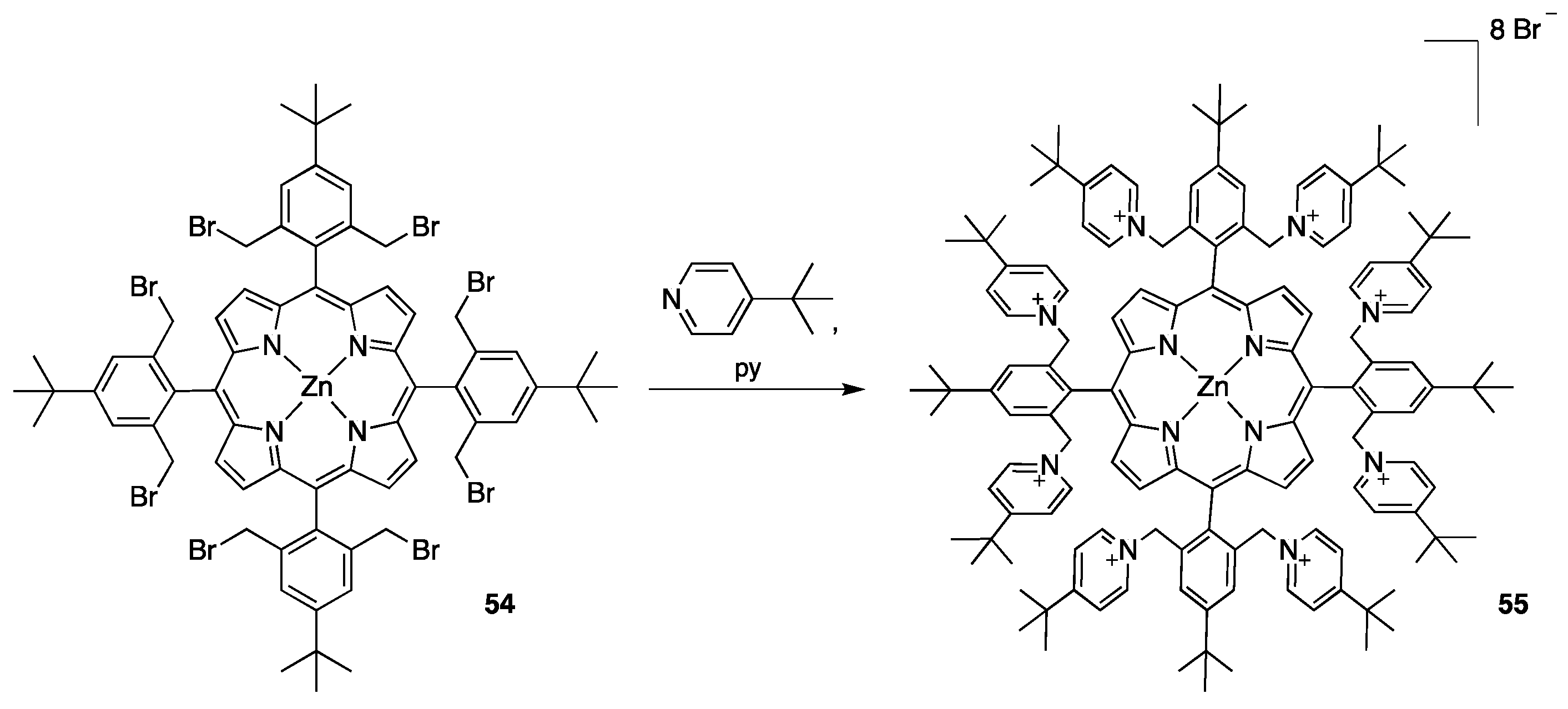
2.2.1. meso-Tetrakispyridiniumporphyrins
2.2.2. A3B and Other meso-Pyridiniumporphyrins
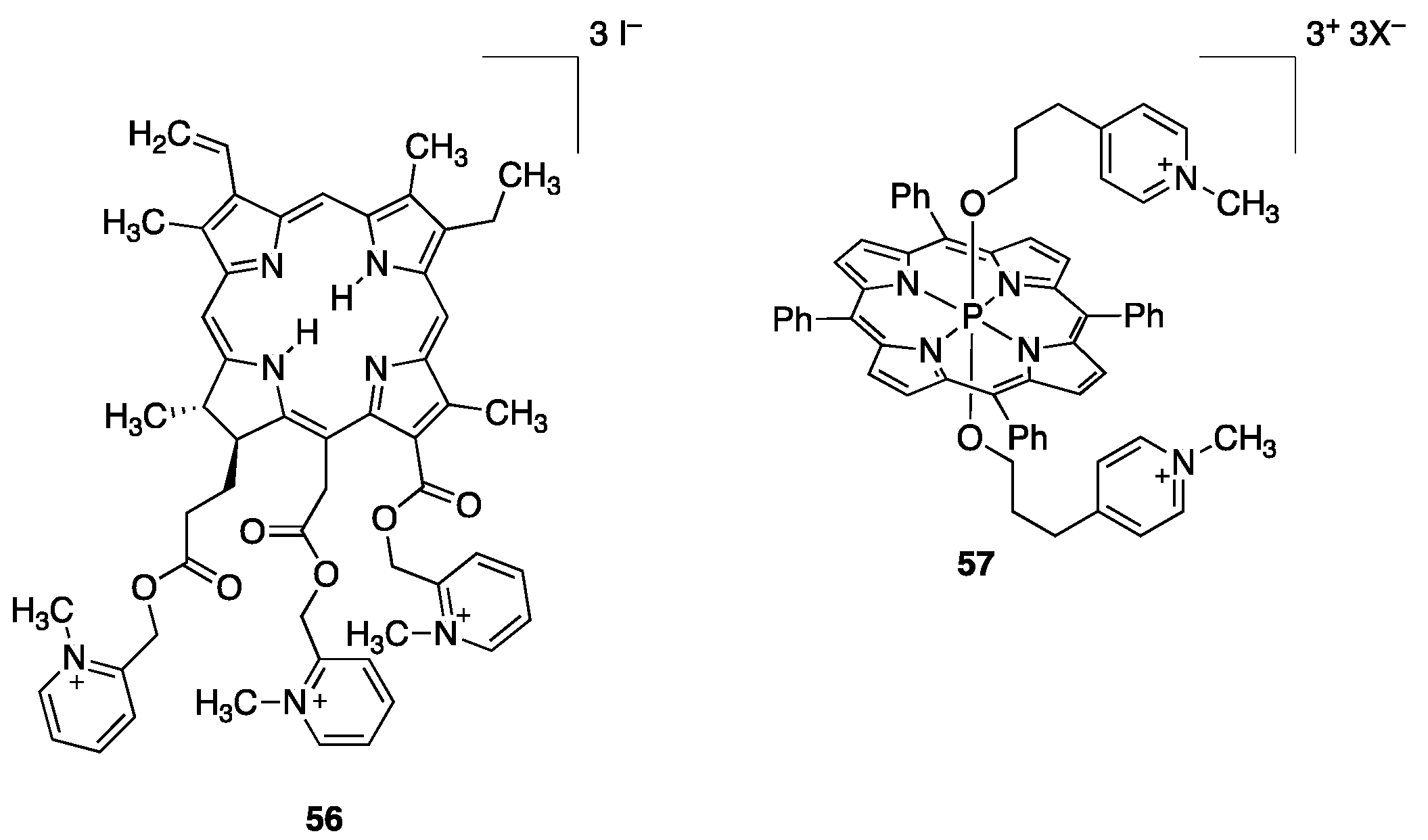
2.2.3. Other Pyridyl-Substituted Porphyrins
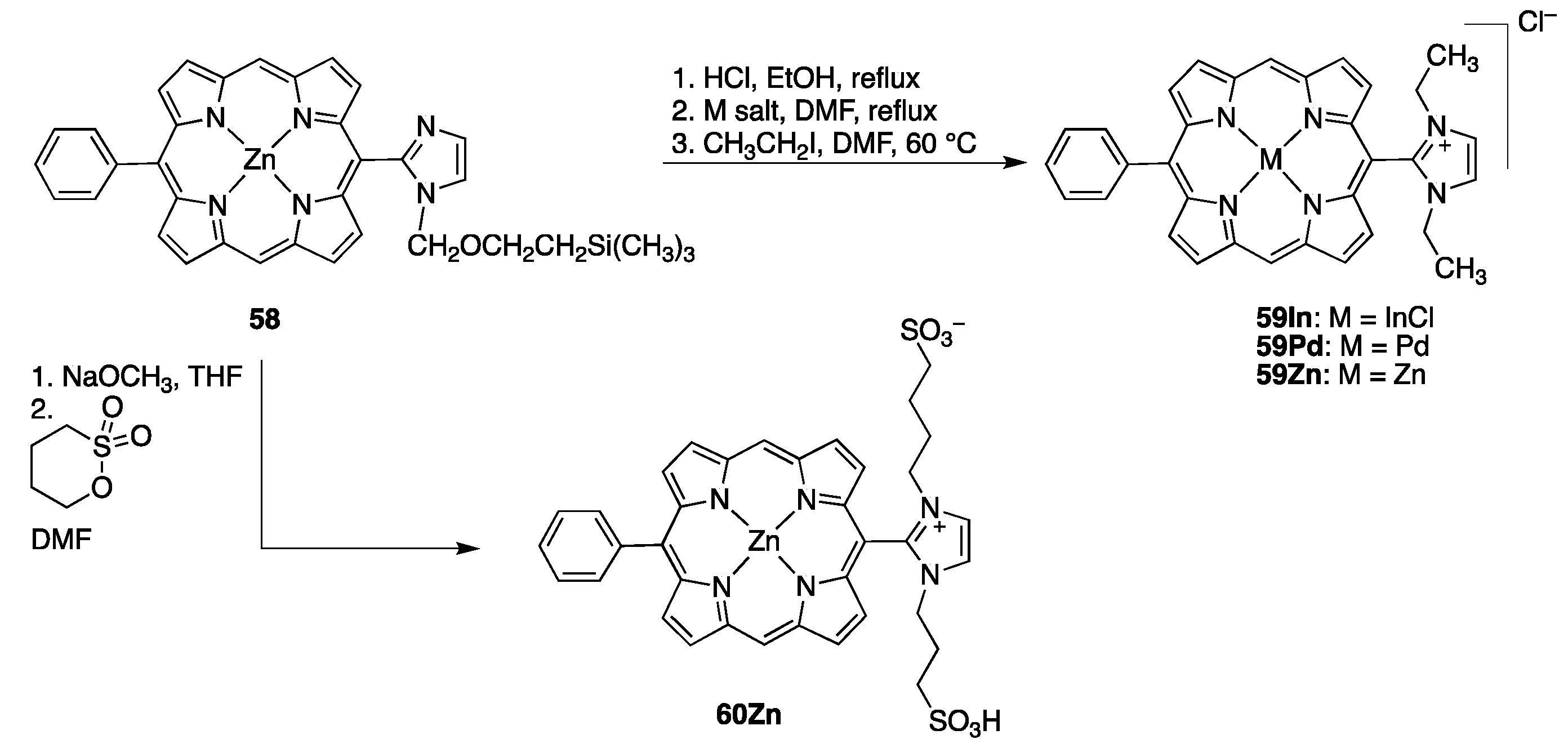
2.2.4. meso-Imidazolium Porphyrins
3. Water-Soluble Porphyrins Bearing Anionic Substituents
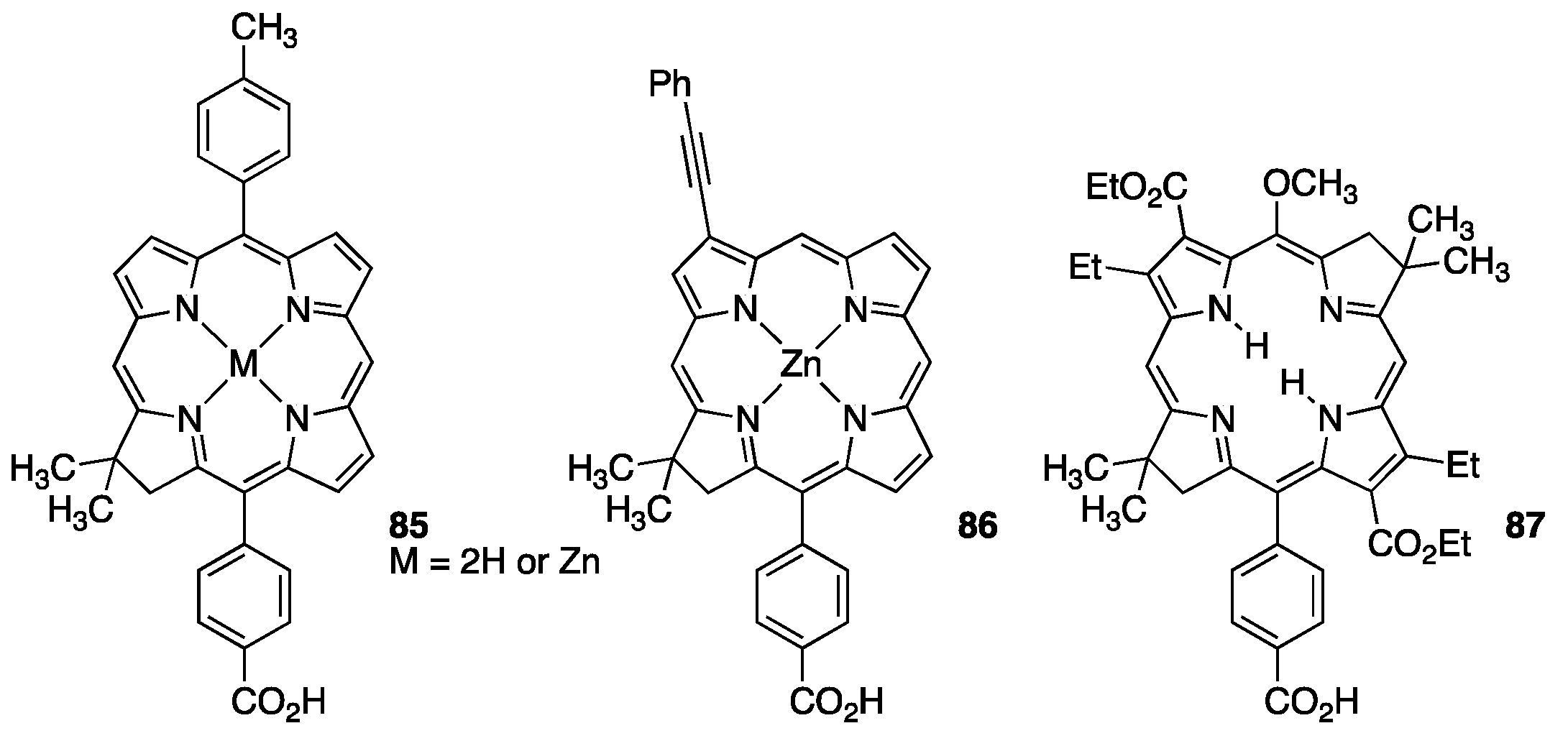
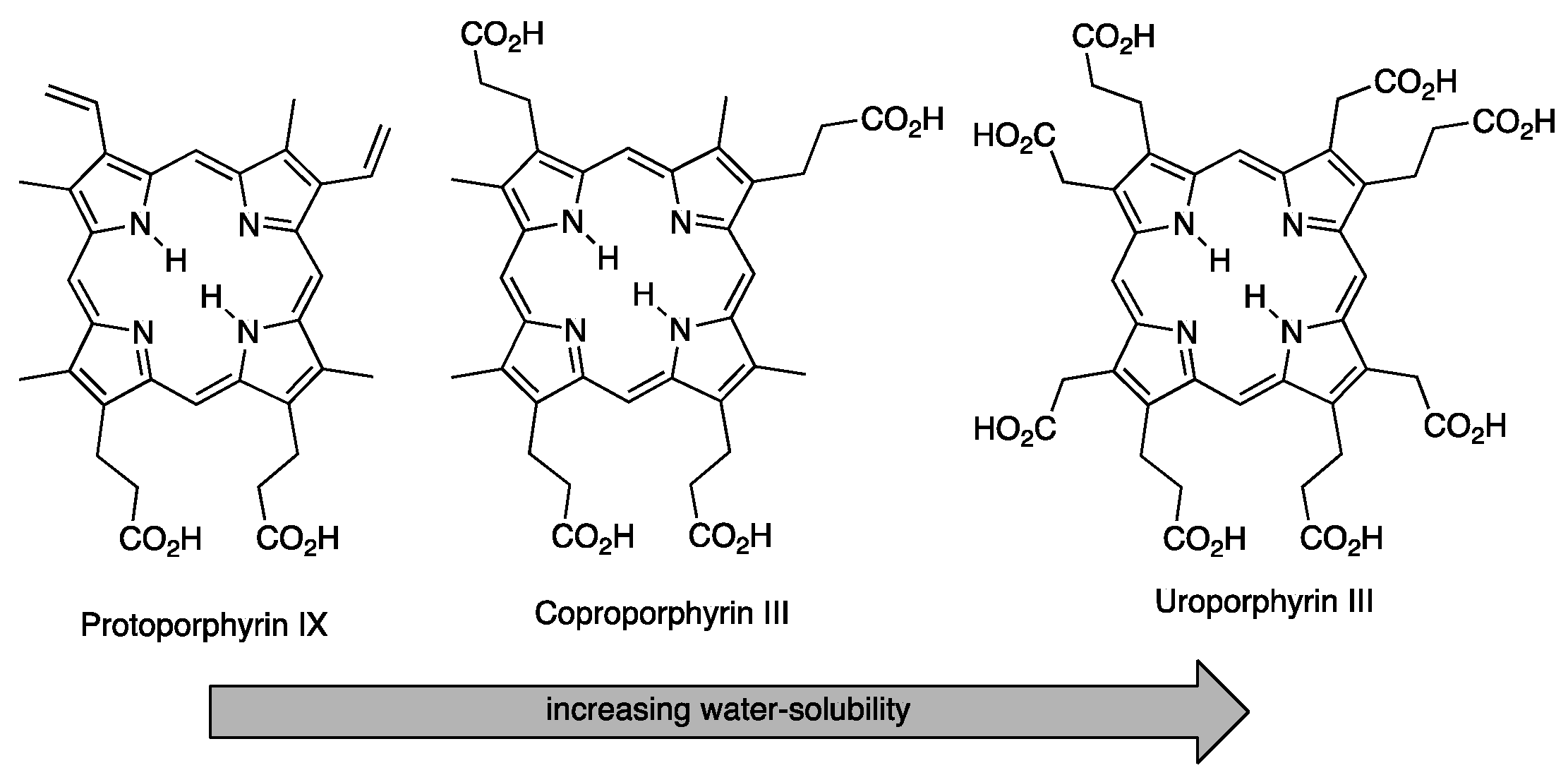
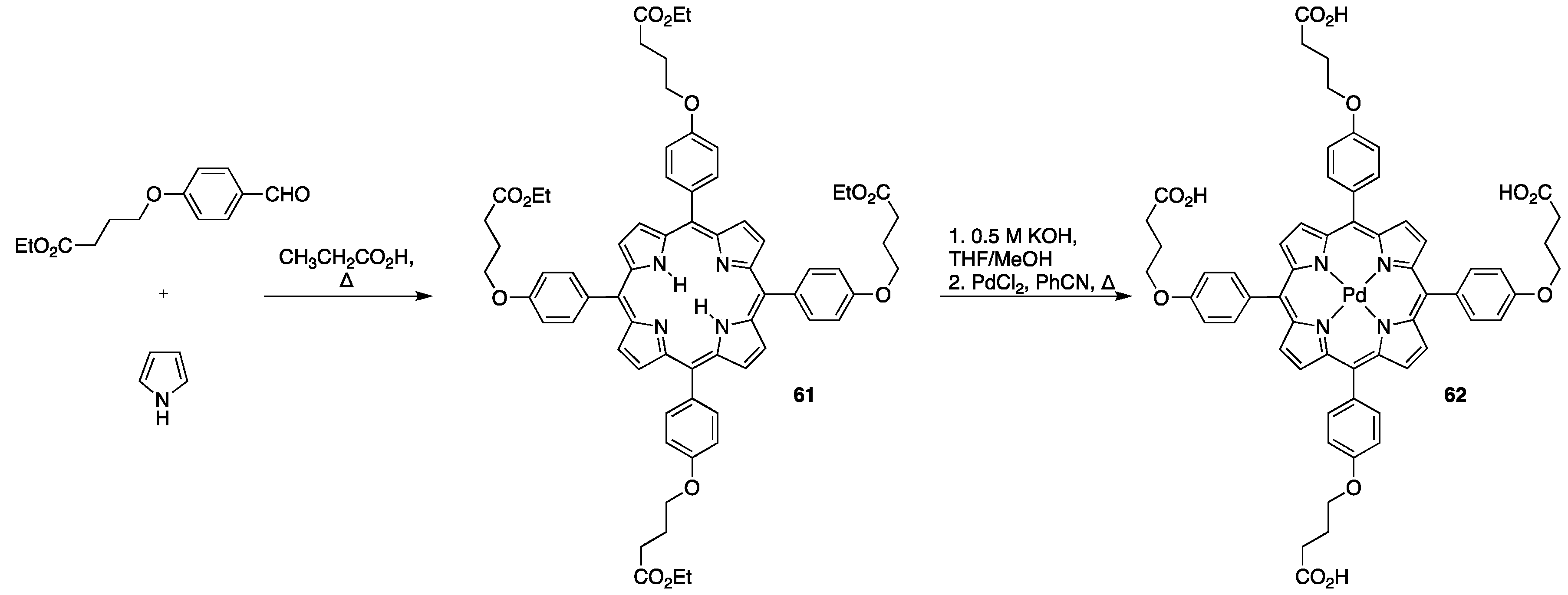
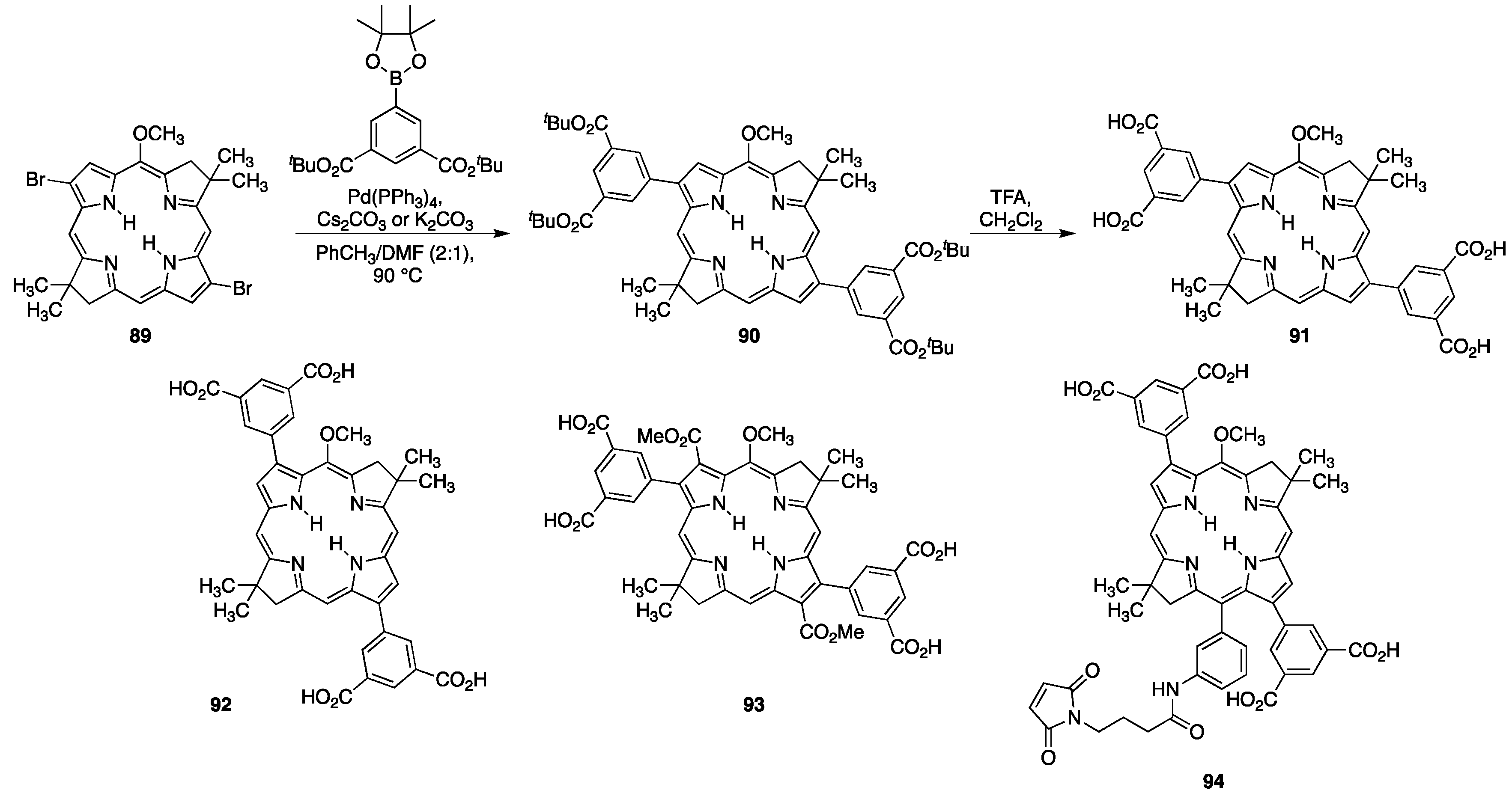
3.1. Porphyrins with Carboxylate Functional Groups
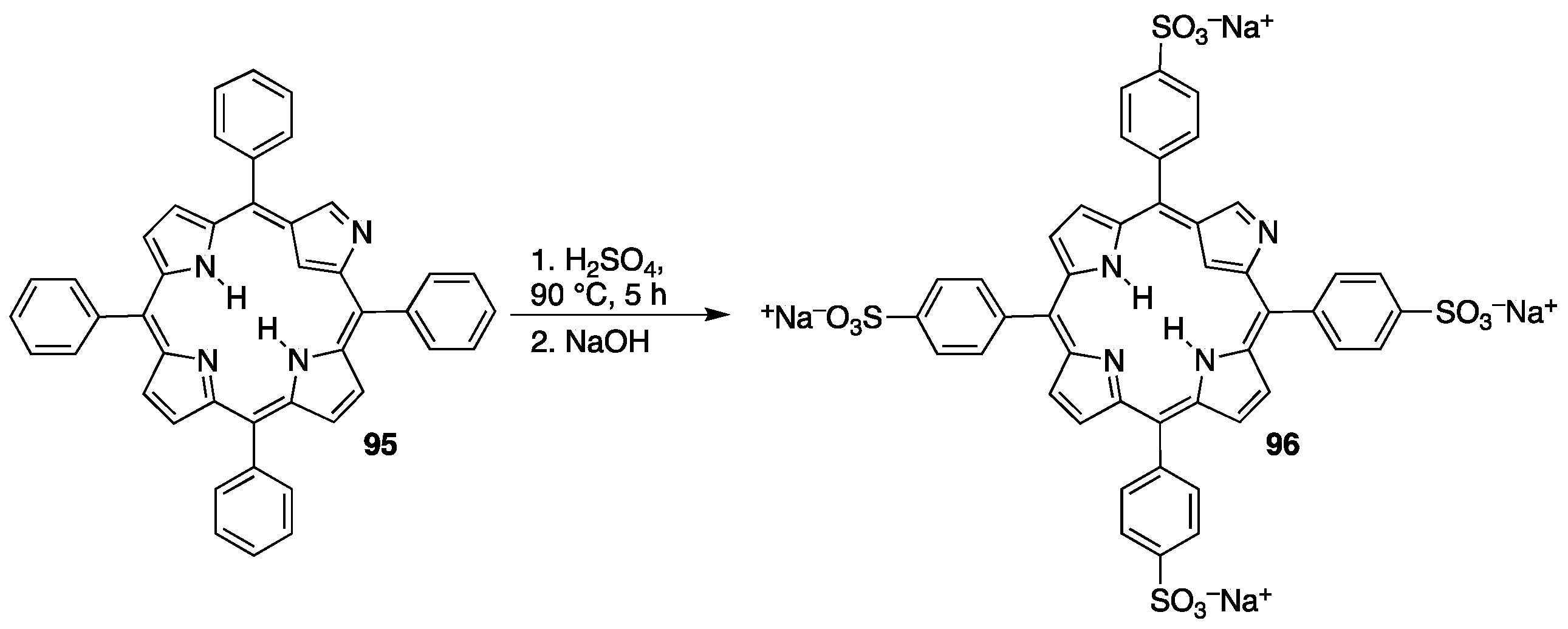
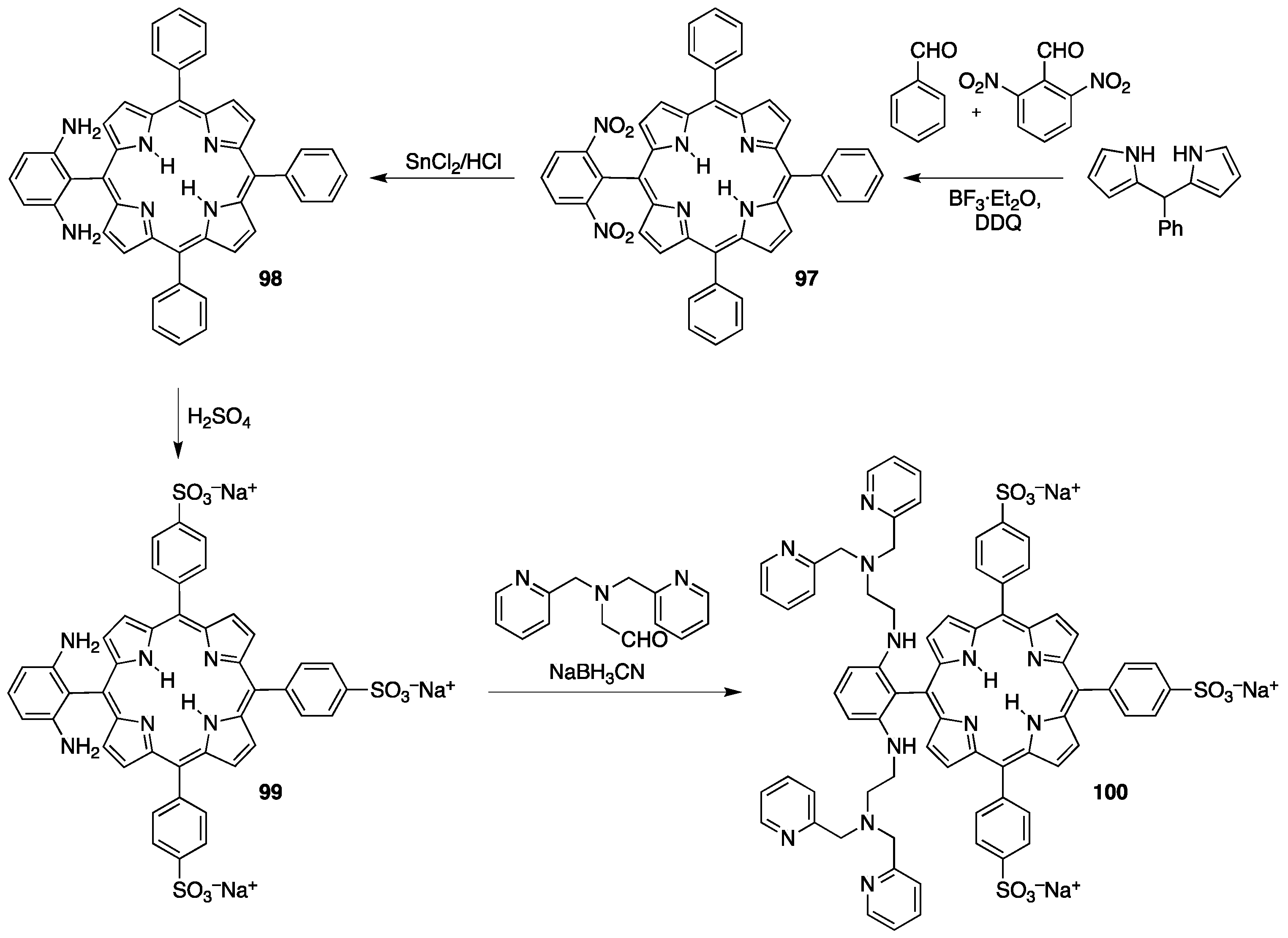
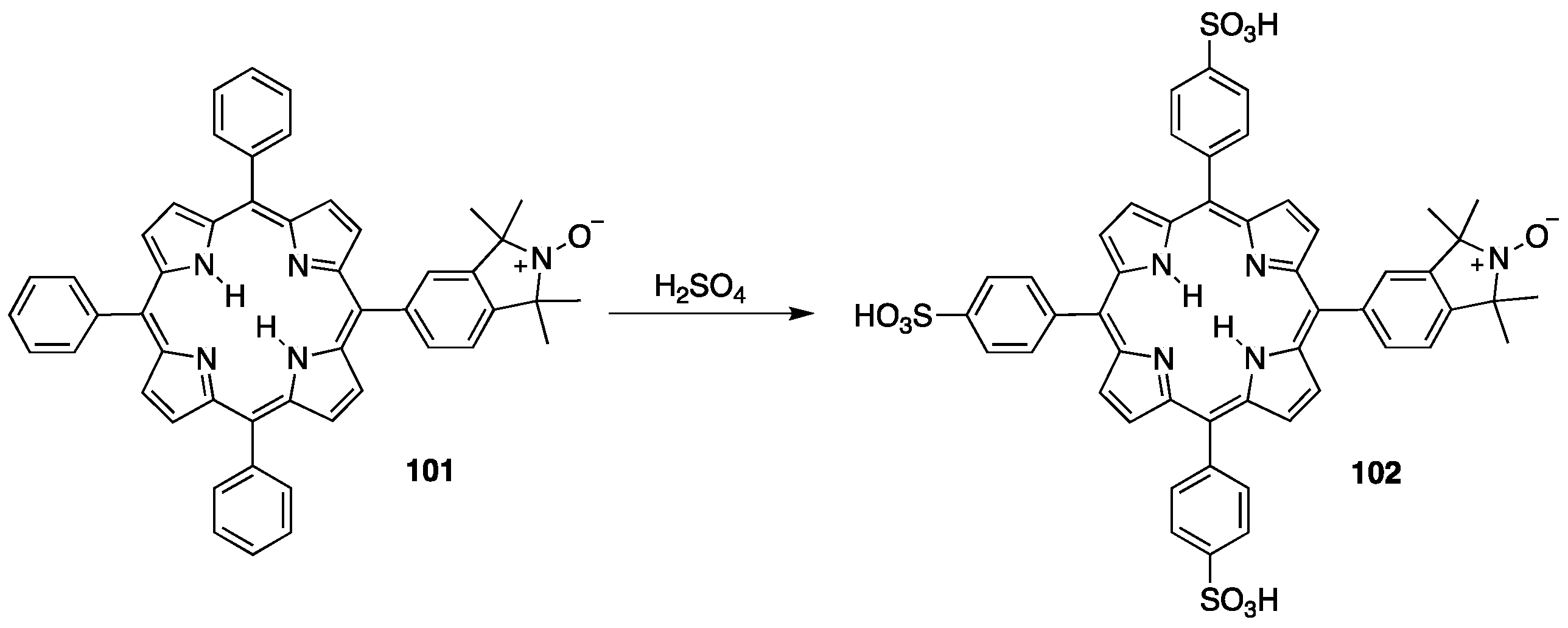
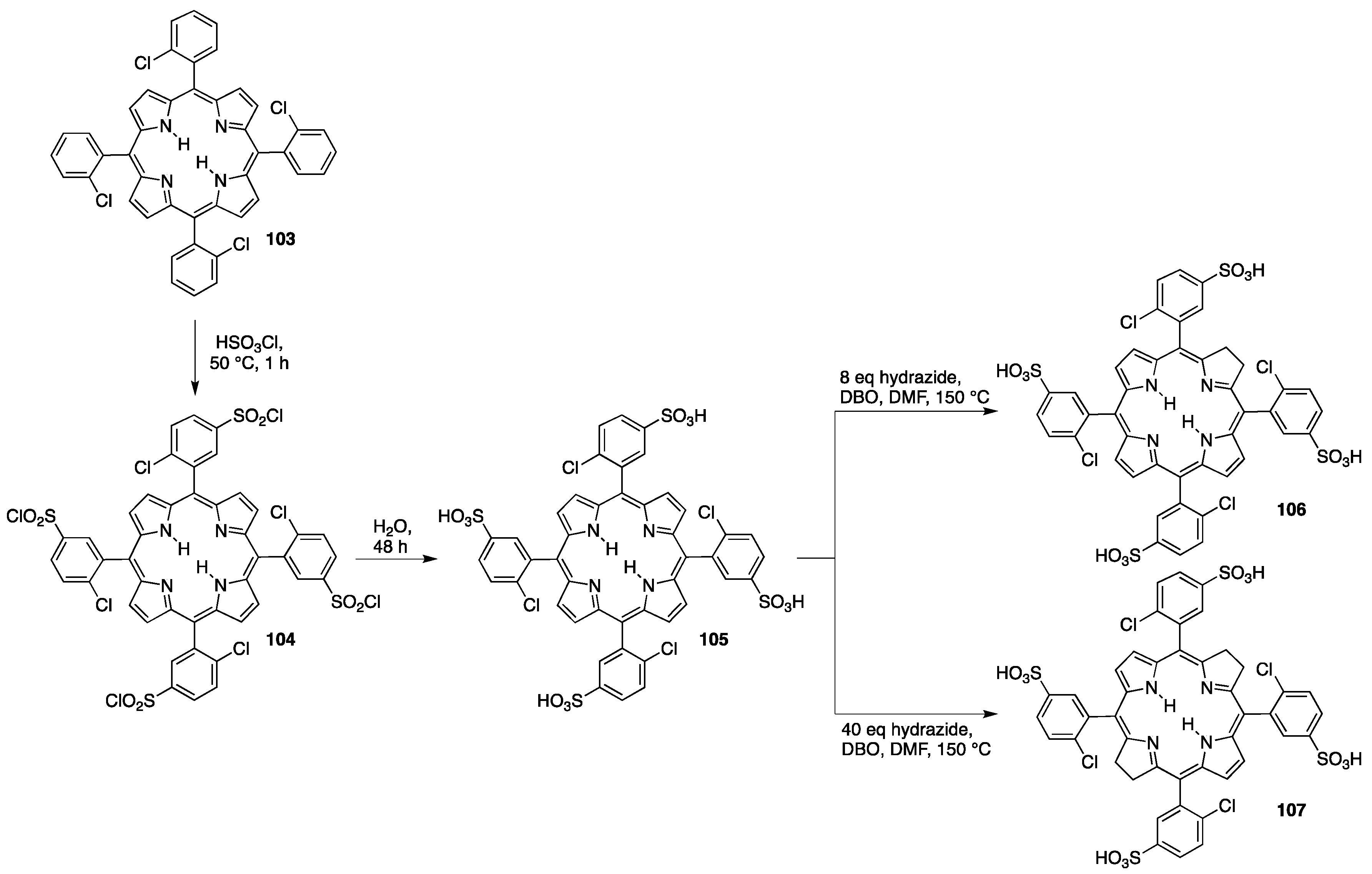
3.2. Sulfonated Porphyrins
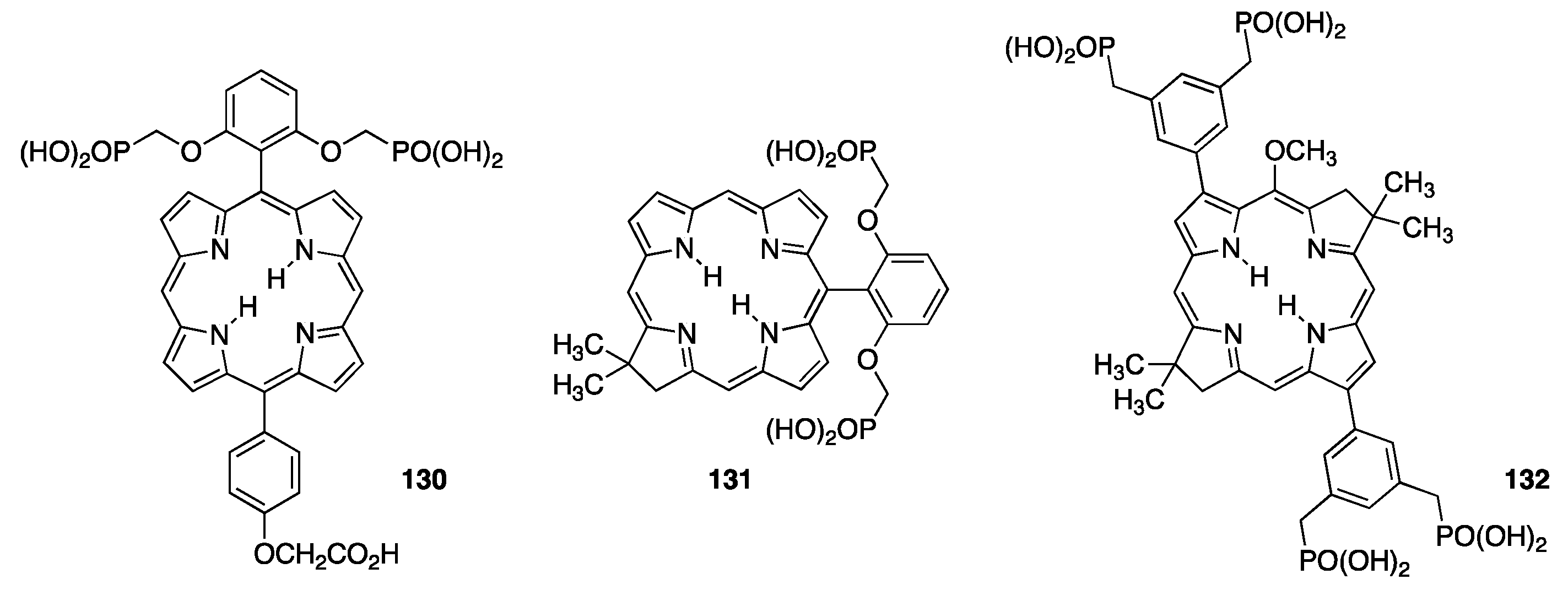
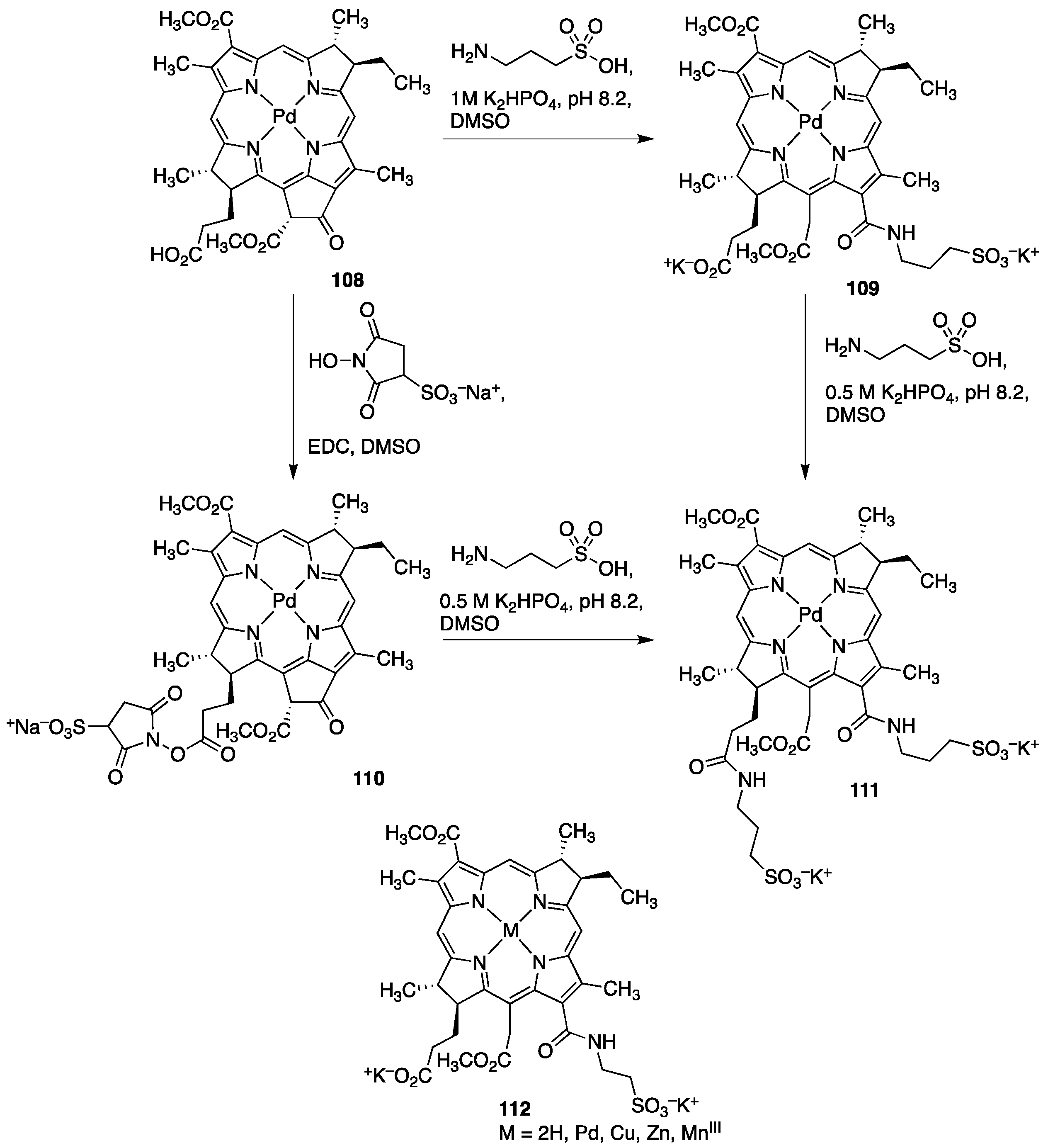
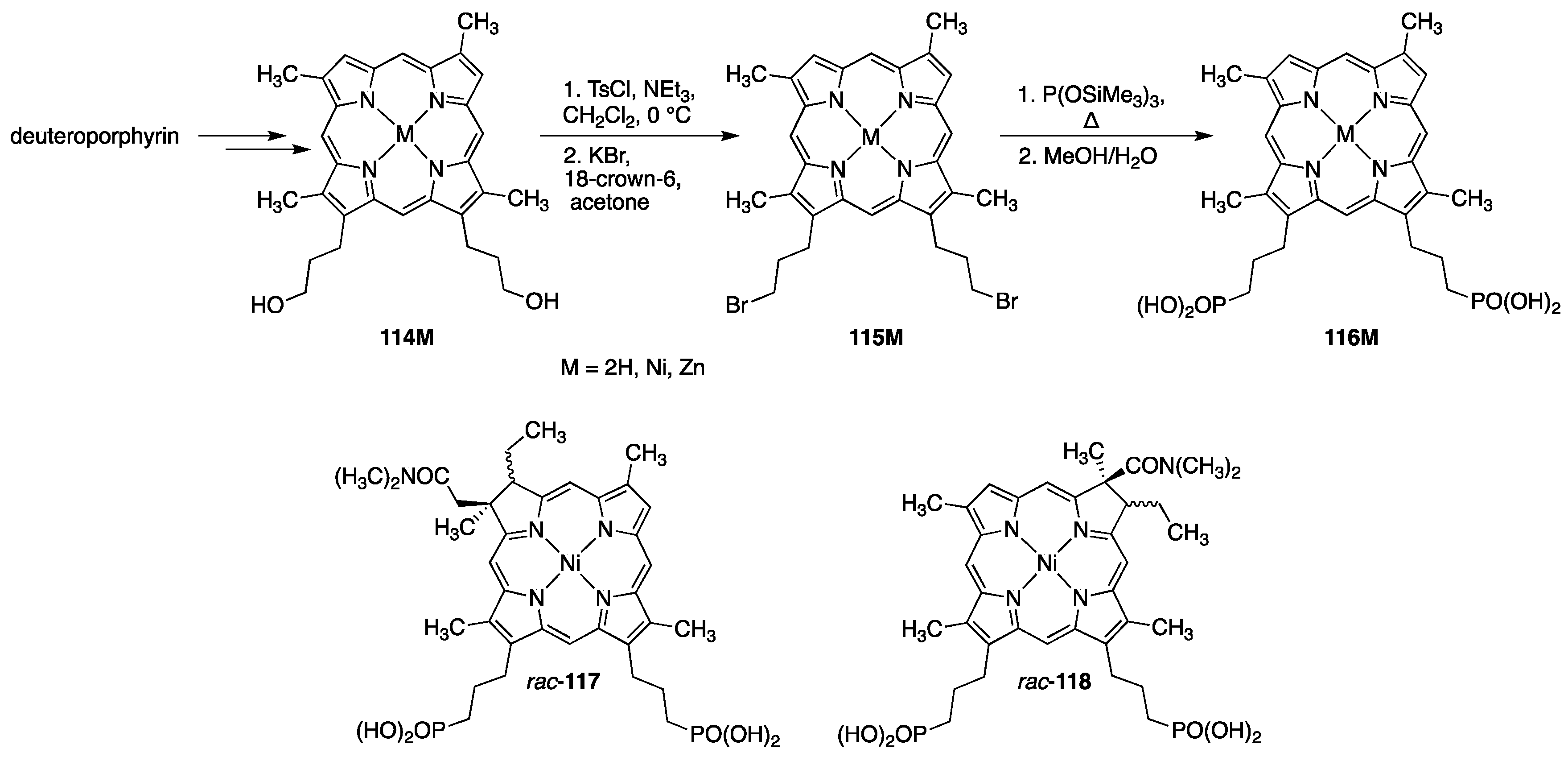
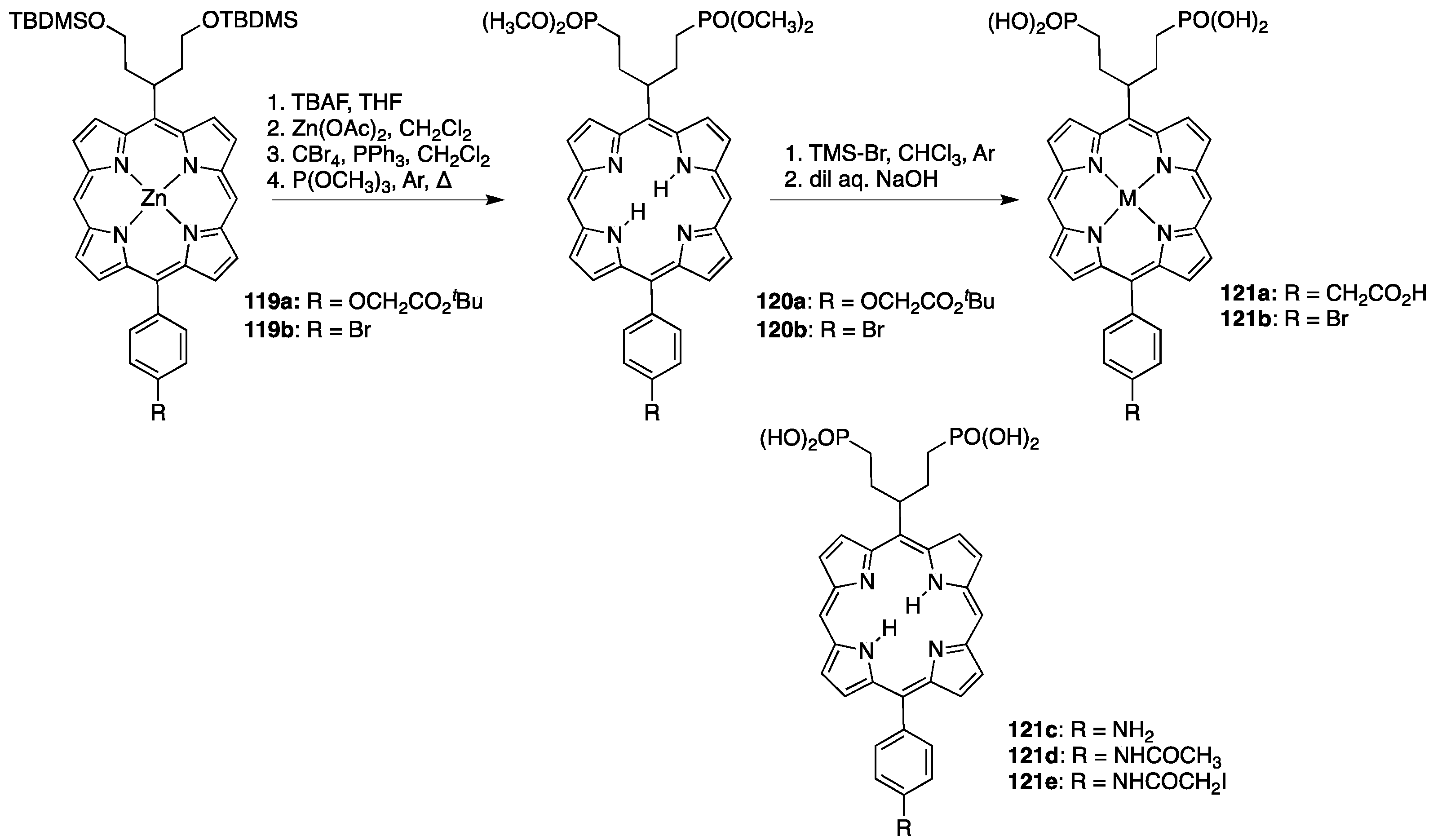
3.3. Porphyrins with Phosphate/Phosphonate Functional Groups
4. Water-Soluble Porphyrins with Neutral Groups
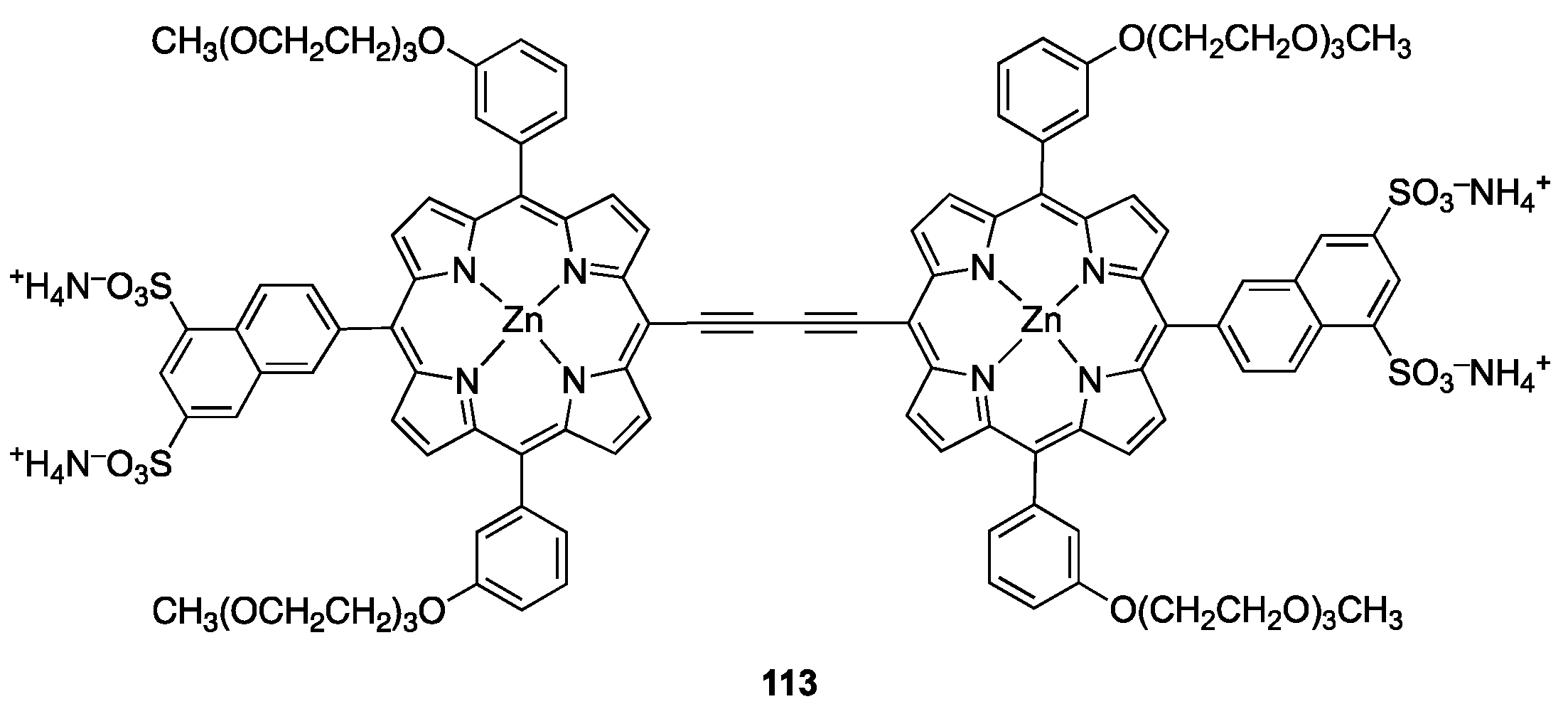
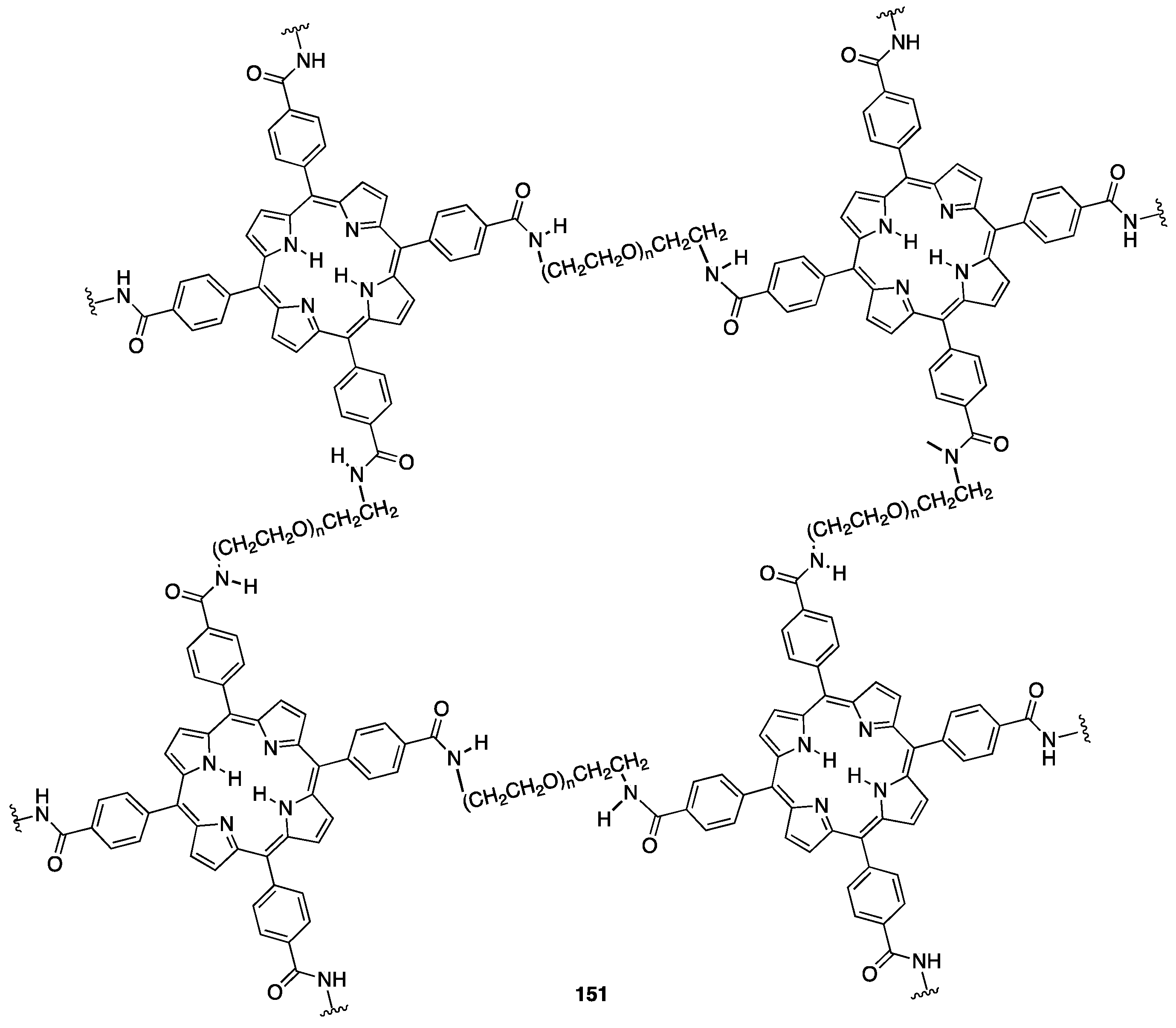
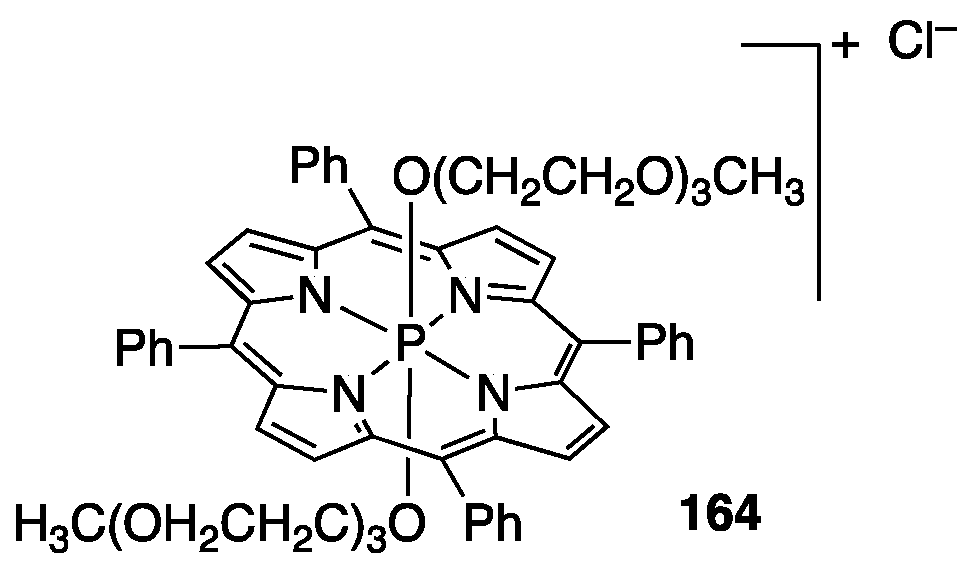
4.1. PEGylated Porphyrins
4.1.1. Porphyrinoid Macrocycle Total Synthesis Using PEGylated Building Blocks
4.1.2. PEGylation of meso-aryl Porphyrins
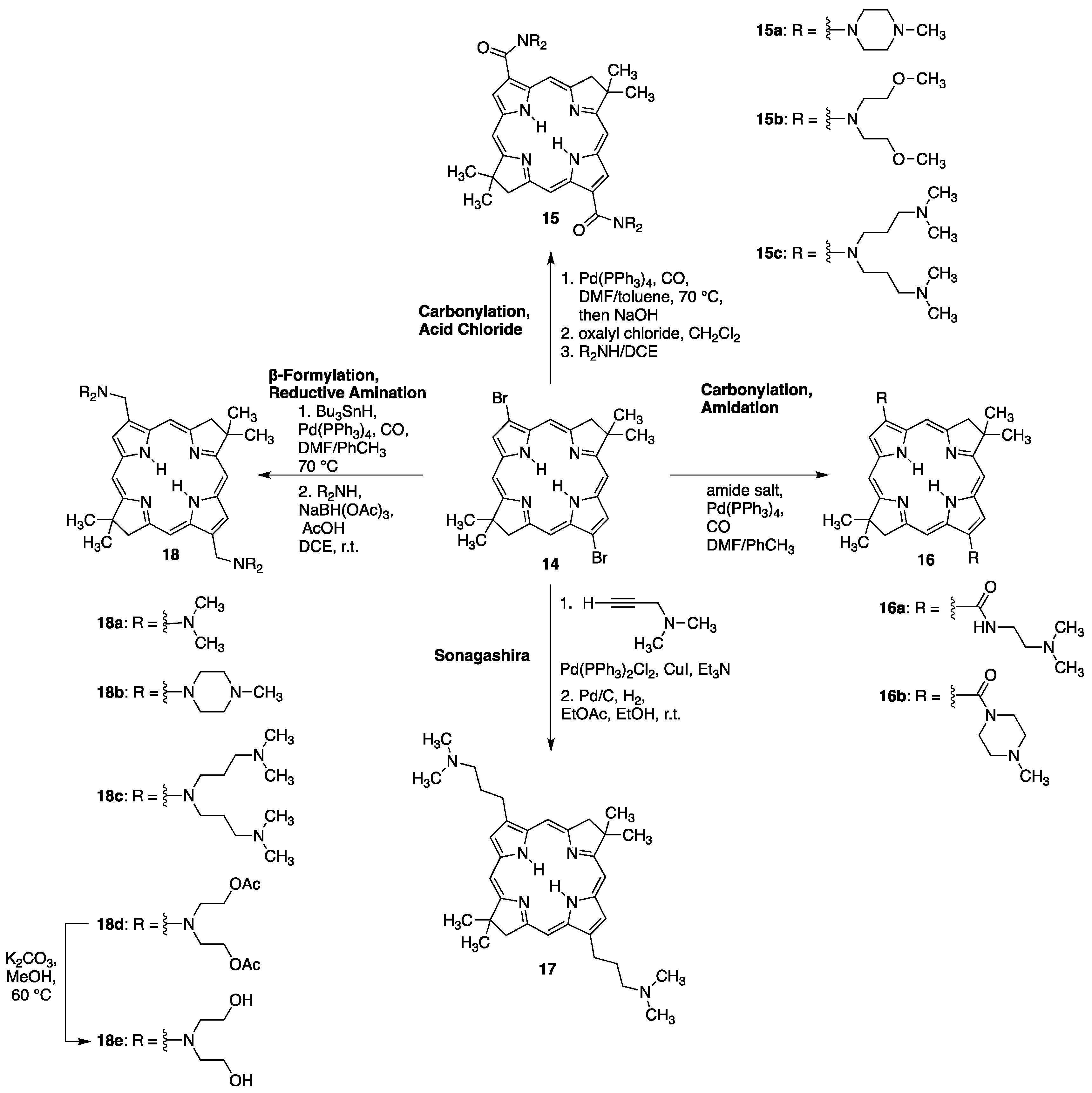
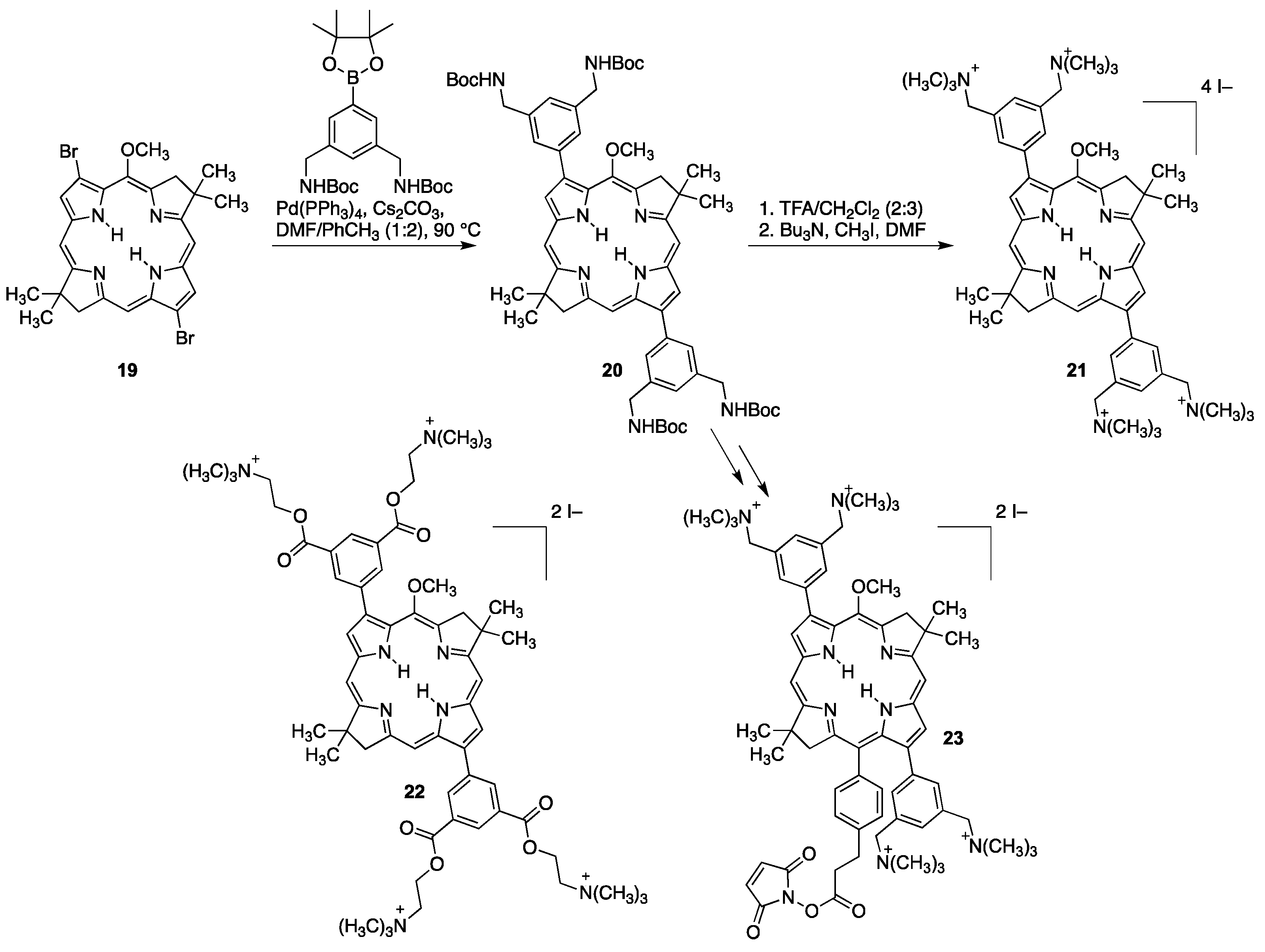
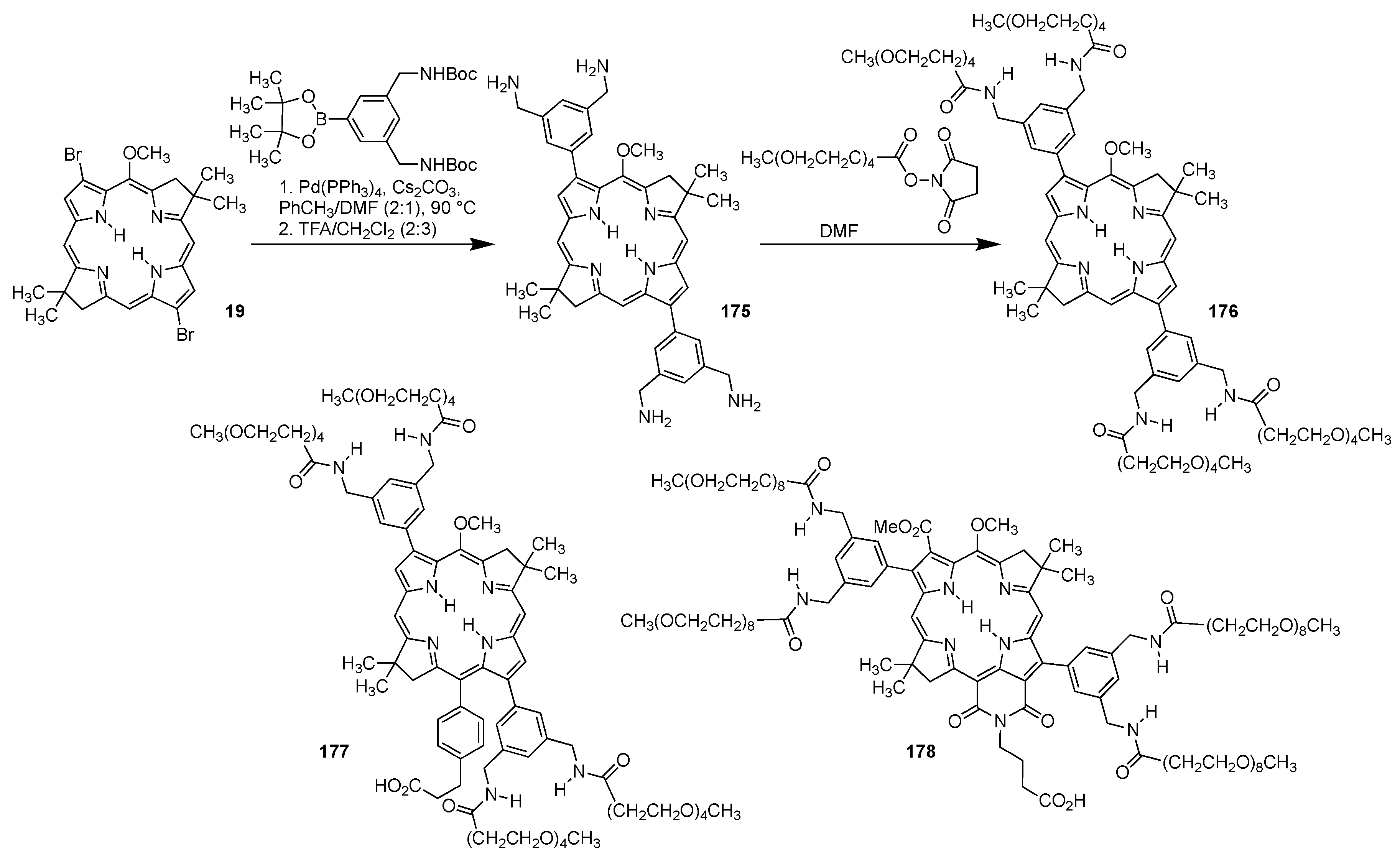
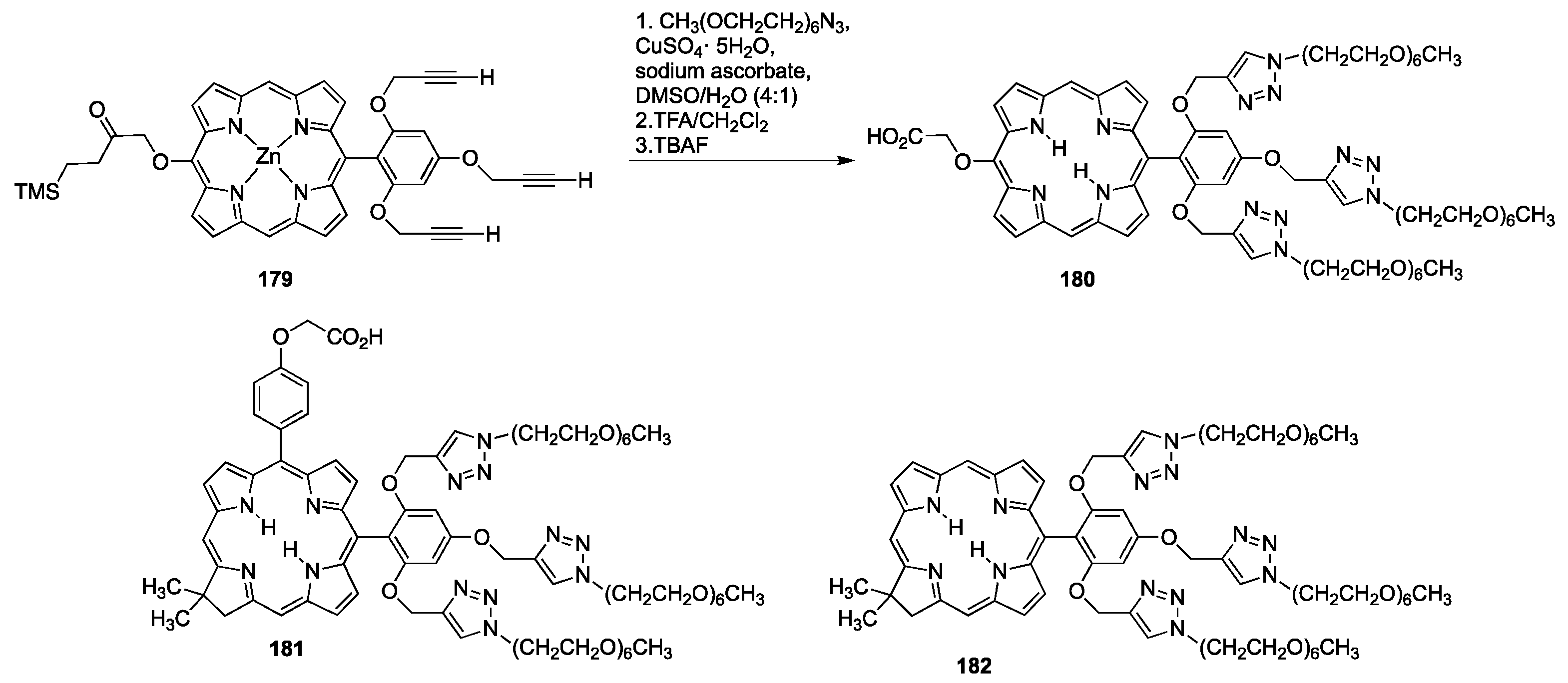
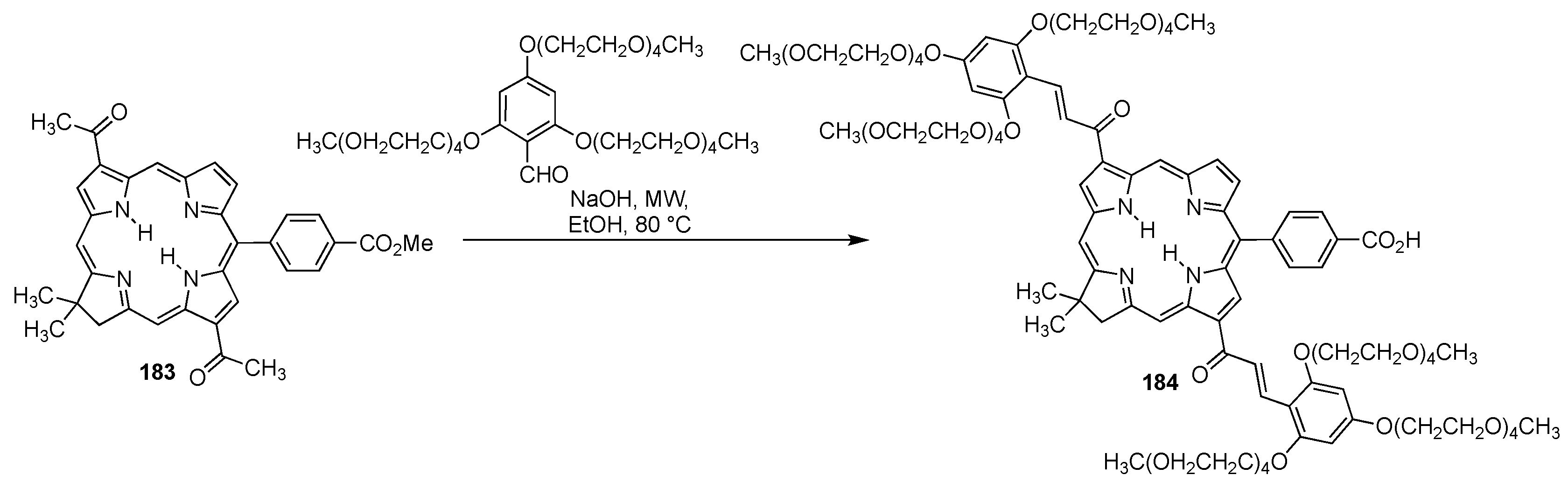
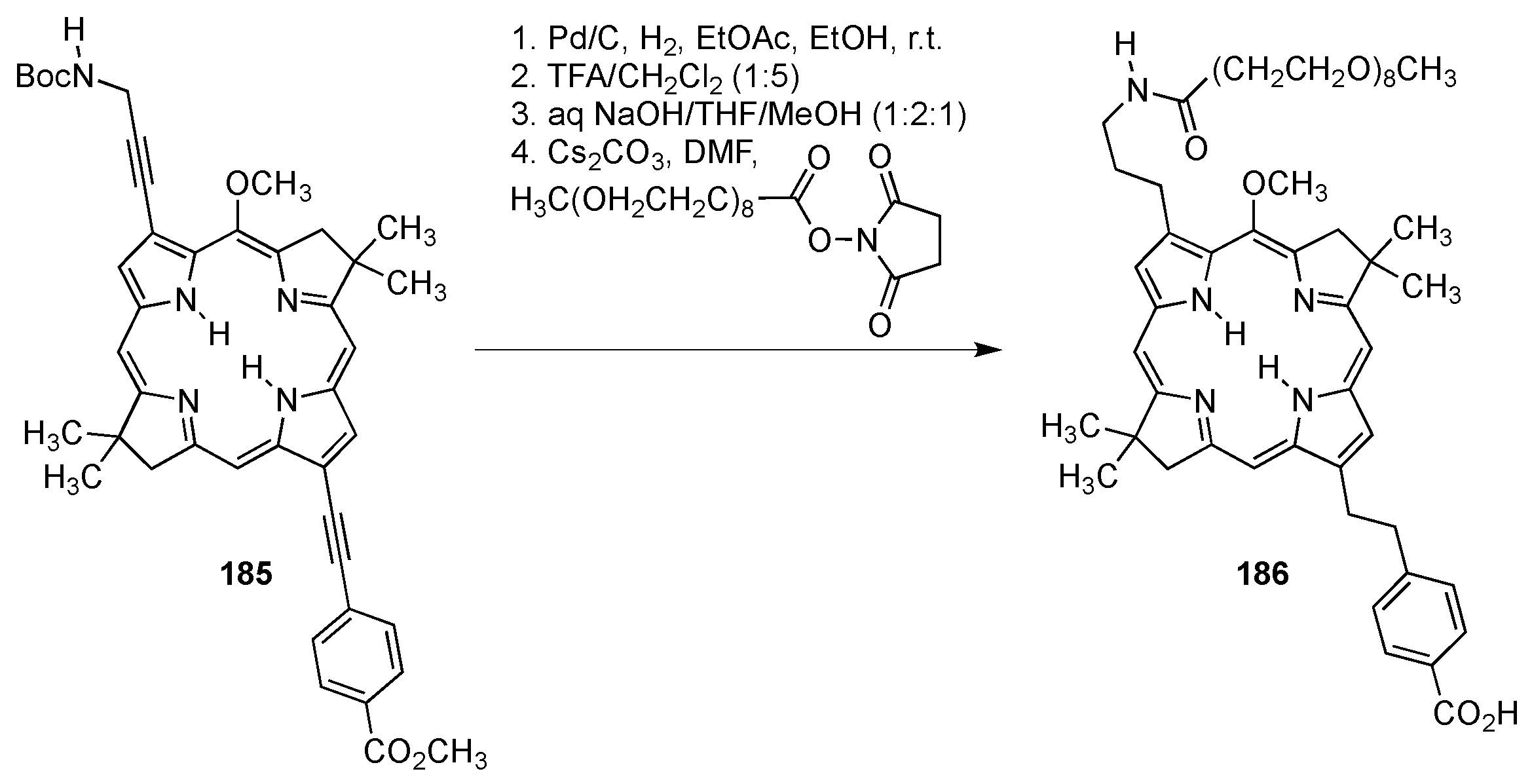
4.1.3. Porphyrins PEGylated at Their β-Positions
5. Summary and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Huang, H.; Song, W.; Rieffel, J.; Lovell, J.F. Emerging applications of porphyrins in photomedicine. Front. Phys. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, E.D.; Dolphin, D.; Brückner, C. Porphyrin-based photosensitizers for use in photodynamic therapy. Tetrahedron 1998, 54, 4151–4202. [Google Scholar] [CrossRef]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Brandis, A.S.; Salomon, Y.; Scherz, A. Bacteriochlorophyll sensitizers in photodynamic therapy. In Chlorophylls and Bacteriochlorophylls; Grimm, B., Porra, R.J., Rüdinger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 485–494. [Google Scholar]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.F.; McCarron, P.A.; Tunney, M.M. Antifungal photodynamic therapy. Microbiol. Res. 2008, 163, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Photoantimicrobials—So what’s stopping us? Photodiagn. Photodyn. Ther. 2009, 6, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; James, N.; Chen, Y.; Dobhal, M.P. Cyanine dye-based compounds for tumor imaging with and without photodynamic therapy. Top. Heterocycl. Chem. 2008, 14, 41–74. [Google Scholar]
- Weissleder, R.; Pittet, M.J. Imaging in the era of molecular oncology. Nature 2008, 452, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Favazza, C.; Wang, L.V. In vivo photoacoustic tomography of chemicals: High-resolution functional and molecular optical imaging at new depths. Chem. Rev. 2010, 110, 2756–2782. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhang, E.; Su, Y.; Cheng, T.; Shi, C. A review of NIR dyes in cancer targeting and imaging. Biomaterials 2011, 32, 7127–7138. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.V.; Hu, S. Photoacoustic tomography: In vivo imaging from organelles to organs. Science 2012, 335, 1458–1462. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Beard, P.C.; Bohndiek, S.E. Contrast agents for molecular photoacoustic imaging. Nat. Methods 2016, 13, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Luciano, M.; Erfanzadeh, M.; Zhou, F.; Zhu, H.; Bornhütter, T.; Röder, B.; Zhu, Q.; Brückner, C. In vivo photoacoustic tumor tomography using a quinoline-annulated porphyrin as NIR molecular contrast agent. Org. Biomol. Chem. 2017, 15, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Stender, A.S.; Marchuk, K.; Liu, C.; Sander, S.; Meyer, M.W.; Smith, E.A.; Neupane, B.; Wang, G.; Li, J.; Cheng, J.-X.; et al. Single cell optical imaging and spectroscopy. Chem. Rev. 2013, 113, 2469–2527. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Taniguchi, M.; Lindsey, J.S. Near-infrared tunable bacteriochlorins equipped for bioorthogonal labeling. New J. Chem. 2015, 39, 4534–4550. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Lemon, C.M.; Karnas, E.; Han, X.; Bruns, O.T.; Kempa, T.J.; Fukumura, D.; Bawendi, M.G.; Jain, R.K.; Duda, D.G.; Nocera, D.G. Micelle-encapsulated quantum dot-porphyrin assemblies as in vivo two-photon oxygen sensors. J. Am. Chem. Soc. 2015, 137, 9832–9842. [Google Scholar] [CrossRef] [PubMed]
- Drain, C.M.; Varotto, A.; Radivojevic, I. Self-organized porphyrinic materials. Chem. Rev. 2009, 109, 1630–1658. [Google Scholar] [CrossRef] [PubMed]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as molecular electronic components of functional devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Osuka, A. Conjugated porphyrin arrays: Synthesis, properties and applications for functional materials. Chem. Soc. Rev. 2015, 44, 943–969. [Google Scholar] [CrossRef] [PubMed]
- Barona-Castano, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; de Oliveira, K.T. Porphyrins as catalysts in scalable organic reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Rybicka-Jasińska, K.; Ciszewski, Ł.W.; Gryko, D. Photocatalytic reaction of diazo compounds with aldehydes. Adv. Synth. Catal. 2016, 358, 1671–1678. [Google Scholar] [CrossRef]
- Rybicka-Jasińska, K.; Shan, W.; Zawada, K.; Kadish, K.M.; Gryko, D. Porphyrins as photoredox catalysts: Experimental and theoretical studies. J. Am. Chem. Soc. 2016, 138, 15451–15458. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.K.; Ladomenou, K.; Coutsolelos, A.G. Porphyrins in bio-inspired transformations: Light-harvesting to solar cell. Coord. Chem. Rev. 2012, 256, 2601–2627. [Google Scholar] [CrossRef]
- Urbani, M.; Grätzel, M.; Nazeeruddin, M.K.; Torres, T. Meso-Substituted porphyrins for dye-sensitized solar cells. Chem. Rev. 2014, 114, 12330–12396. [Google Scholar] [CrossRef] [PubMed]
- Kärkäs, M.D.; Verho, O.; Johnston, E.V.; Åkermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef] [PubMed]
- Hedley, G.J.; Ruseckas, A.; Samuel, I.D.W. Light harvesting for organic photovoltaics. Chem. Rev. 2017, 117, 796–837. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M. Development of porphyrin syntheses. New J. Chem. 2016, 40, 5644–5649. [Google Scholar] [CrossRef]
- Hiroto, S.; Miyake, Y.; Shinokubo, H. Synthesis and functionalization of porphyrins through organometallic methodologies. Chem. Rev. 2017, 117, 2910–3043. [Google Scholar] [CrossRef] [PubMed]
- Montforts, F.-P.; Gerlach, B.; Höper, F. Discovery and synthesis of less common natural hydroporphyrins. Chem. Rev. 1994, 94, 327–347. [Google Scholar] [CrossRef]
- Brückner, C.; Samankumara, L.; Ogikubo, J. Syntheses of bacteriochlorins and isobacteriochlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NJ, USA, 2012; Volume 17, pp. 1–112. [Google Scholar]
- Lindsey, J.S. De novo synthesis of gem-dialkyl chlorophyll analogues for probing and emulating our green world. Chem. Rev. 2015, 115, 6534–6620. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Synthetic chlorins, possible surrogates for chlorophylls, prepared by derivatization of porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Müllen, K. Modifying the porphyrin core—A chemist’s jigsaw. J. Porphyr. Phthalocyanines 2011, 15, 757–779. [Google Scholar] [CrossRef]
- Brückner, C.; Akhigbe, J.; Samankumara, L. Syntheses and structures of porphyrin analogues containing non-pyrrolic heterocycles. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NJ, USA, 2014; Volume 31, pp. 1–276. [Google Scholar]
- Szyszko, B.; Latos-Grazynski, L. Core chemistry and skeletal rearrangements of porphyrinoids and metalloporphyrinoids. Chem. Soc. Rev. 2015, 44, 3588–3616. [Google Scholar] [CrossRef] [PubMed]
- Brückner, C. The breaking and mending of meso-tetraarylporphyrins: Transmuting the pyrrolic building blocks. Acc. Chem. Res. 2016, 49, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.D.; Costa, J.I.; Tome, A.C. Porphyrin macrocycle modification: Pyrrole ring-contracted or -expanded porphyrinoids. Molecules 2016, 21, 320. [Google Scholar] [CrossRef] [PubMed]
- Hambright, P. Chemistry of water-soluble porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 3, pp. 129–210. [Google Scholar]
- Pisarek, S.; Maximova, K.; Gryko, D. Strategies toward the synthesis of amphiphilic porphyrins. Tetrahedron 2014, 70, 6685–6715. [Google Scholar] [CrossRef]
- Singh, S.; Aggarwal, A.; Bhupathiraju, N.V.; Arianna, G.; Tiwari, K.; Drain, C.M. Glycosylated porphyrins, phthalocyanines, and other porphyrinoids for diagnostics and therapeutics. Chem. Rev. 2015, 115, 10261–10306. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.; Scanlan, E.M.; Senge, M.O. Chemical synthesis and medicinal applications of glycoporphyrins. Curr. Med. Chem. 2015, 22, 2238–2348. [Google Scholar] [CrossRef] [PubMed]
- Králová, J.; Kejík, Z.; Bříza, T.; Poučková, P.; Král, A.; Martásek, P.; Král, V. Porphyrin-cyclodextrin conjugates as a nanosystem for versatile drug delivery and multimodal cancer therapy. J. Med. Chem. 2010, 53, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Gotardo, M.C.A.F.; Sacco, H.C.; Filho, J.C.S.; Ferreira, A.G.; Tedesco, A.C.; Assis, M.D. A novel crowned porphyrin derived from meso-tetrakis(pentafluorophenyl)porphyrin. J. Porphyr. Phthalocyanines 2003, 7, 399–404. [Google Scholar] [CrossRef]
- Welch, C.; Archibald, S.J.; Boyle, R.W. Reductive amination—A convenient method for generating diverse, mono-functionalised 5,10,15,20-tetraphenylporphyrins. Synthesis 2009, 551–556. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Perez, M.J.; Brückner, C.; Weissleder, R. A polymeric nanoparticle preparation that eradicates tumors. Nano Lett. 2005, 5, 2552–2556. [Google Scholar] [CrossRef] [PubMed]
- Paproski, R.J.; Forbrich, A.; Huynh, E.; Chen, J.; Lewis, J.D.; Zheng, G.; Zemp, R.J. Porphyrin nanodroplets: Sub-micrometer ultrasound and photoacoustic contrast imaging agents. Small 2016, 12, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira, J.M.; da Silva, A.R.; Senge, M.O.; Jorge, R.A. Effects of preparation conditions of poly(lactide-co-glycolide) nanoparticles loaded with amphiphilic porphyrins and their photoactivities. J. Nanosci. Nanotechnol. 2014, 14, 6274–6286. [Google Scholar] [CrossRef] [PubMed]
- Chouikrat, R.; Seve, A.; Vanderesse, R.; Benachour, H.; Barberi-Heyob, M.; Richeter, S.; Raehm, L.; Durand, J.O.; Verelst, M.; Frochot, C. Non-polymeric nanoparticles for photodynamic therapy applications: Recent developments. Curr. Med. Chem. 2012, 19, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Paszko, E.; Senge, M.O. Immunoliposomes. Curr. Med. Chem. 2012, 19, 5239–5277. [Google Scholar] [CrossRef] [PubMed]
- Huynh, E.; Lovell, J.F.; Helfield, B.L.; Jeon, M.; Kim, C.; Goertz, D.E.; Wilson, B.C.; Zheng, G. Porphyrin shell microbubbles with intrinsic ultrasound and photoacoustic properties. J. Am. Chem. Soc. 2012, 134, 16464–16467. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.F.; Jin, C.S.; Huynh, E.; Jin, H.; Kim, C.; Rubinstein, J.L.; Chan, W.C.W.; Cao, W.; Wang, L.V.; Zheng, G. Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat. Mater. 2011, 10, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Hambright, P.; Fleischer, E.B. The acid-base equilibria, kinetics of copper ion incorporation, and acid-catalyzed zinc ion displacement from the water-soluble porphyrin α, β, γ, δ-tetra(4-N-methylpyridyl)-porphine. Inorg. Chem. 1970, 9, 1757–1761. [Google Scholar] [CrossRef]
- Wheelhouse, R.T.; Sun, D.; Han, H.; Han, F.X.; Hurley, L.H. Cationic porphyrins as telomerase inhibitors: The interaction of tetra-(N-methyl-4-pyridyl)porphine with quadruplex DNA. J. Am. Chem. Soc. 1998, 120, 3261–3262. [Google Scholar] [CrossRef]
- McMillin, D.R.; Shelton, A.H.; Bejune, S.A.; Fanwick, P.E.; Wall, R.K. Understanding binding interactions of cationic porphyrins with β-form DNA. Coord. Chem. Rev. 2005, 249, 1451–1459. [Google Scholar] [CrossRef]
- Merchat, M.; Bertolini, G.; Giacomini, P.; Villaneuva, A.; Jori, G. Meso-Substituted cationic porphyrins as efficient photosensitizers of gram-positive and gram-negative bacteria. J. Photochem. Photobiol. B 1996, 32, 153–157. [Google Scholar] [CrossRef]
- Commercial Suppliers of Porphyrins, for example: Frontier Scientific: Logan, UT, USA; Sigma-Aldrich: St. Louis, MO, USA; PorphyChem SAS: Dijon, France; Porphyrin Systems GbR: Appen, Germany.
- Manono, J.; Marzilli, P.A.; Marzilli, L.G. New porphyrins bearing positively charged peripheral groups linked by a sulfonamide group to meso-tetraphenylporphyrin: Interactions with calf thymus DNA. Inorg. Chem. 2009, 48, 5636–5647. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, F.; Sol, V.; Huang, Y.-M.; Granet, R.; Guilloton, M.; Krausz, P. Synthesis and biological evaluation of polyamine-porphyrin conjugates as potential agents in photodynamic therapy (PDT). J. Porphyr. Phthalocyanines 2002, 6, 130–134. [Google Scholar] [CrossRef]
- Drogat, N.; Gady, C.; Granet, R.; Sol, V. Design and synthesis of water-soluble polyaminated chlorins and bacteriochlorins—With near-infrared absorption. Dyes Pigments 2013, 98, 609–614. [Google Scholar] [CrossRef]
- Taima, H.; Okubo, A.; Yoshioka, N.; Inoue, H. Synthesis of cationic water-soluble esters of chlorin e6. Tetrahedron Lett. 2005, 46, 4161–4164. [Google Scholar] [CrossRef]
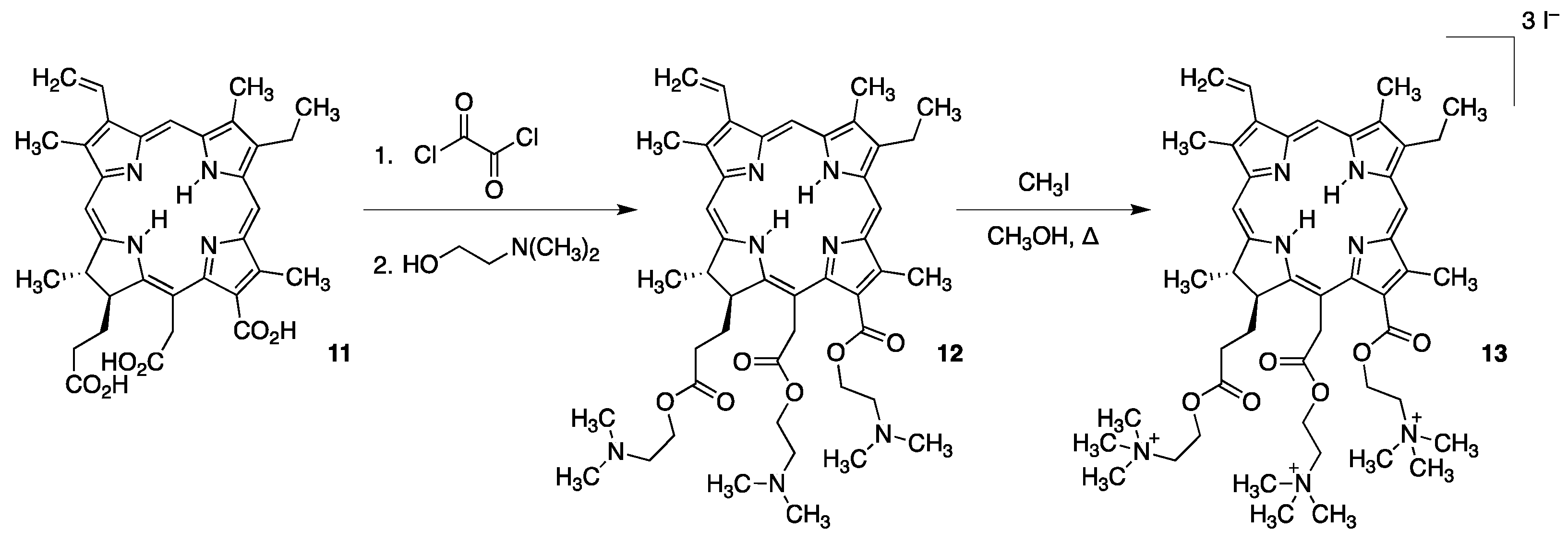
- Ruzié, C.; Krayer, M.; Balasubramanian, T.; Lindsey, J.S. Tailoring a bacteriochlorin building block with cationic, amphipathic, or lipophilic substituents. J. Org. Chem. 2008, 73, 5806–5820. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Huang, Y.Y.; Mroz, P.; Tegos, G.P.; Zhiyentayev, T.; Sharma, S.K.; Lu, Z.; Balasubramanian, T.; Krayer, M.; Ruzie, C.; et al. Stable synthetic cationic bacteriochlorins as selective antimicrobial photosensitizers. Antimicrob. Agents Chemother. 2010, 54, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yang, E.; Reddy, K.R.; Niedzwiedzki, D.M.; Kirmaier, C.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthetic bacteriochlorins bearing polar motifs (carboxylate, phosphonate, ammonium and a short PEG). Water-solubilization, bioconjugation, and photophysical properties. New J. Chem. 2015, 39, 5694–5714. [Google Scholar] [CrossRef]
- Sharma, S.K.; Krayer, M.; Sperandio, F.F.; Huang, L.; Huang, Y.Y.; Holten, D.; Lindsey, J.S.; Hamblin, M.R. Synthesis and evaluation of cationic bacteriochlorin amphiphiles with effective in vitro photodynamic activity against cancer cells at low nanomolar concentration. J. Porphyr. Phthalocyanines 2013, 17, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Thamyongkit, P.; Speckbacher, M.; Diers, J.R.; Kee, H.L.; Kirmaier, C.; Holten, D.; Bocian, D.F.; Lindsey, J.S. Swallowtail porphyrins: Synthesis, characterization and incorporation into porphyrin dyads. J. Org. Chem. 2004, 69, 3700–3710. [Google Scholar] [CrossRef] [PubMed]
- Thamyongkit, P.; Lindsey, J.S. Synthesis of swallowtail-substituted multiporphyrin rods. J. Org. Chem. 2004, 69, 5796–5799. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Lubian, E.; Pavan, M.P.; Kim, H.-J.; Yang, E.; Holten, D.; Lindsey, J.S. Synthetic bacteriochlorins with integral spiro-piperidine motifs. New J. Chem. 2013, 37, 1157–1173. [Google Scholar] [CrossRef]
- Balaz, M.; Collins, H.A.; Dahlstedt, E.; Anderson, H.L. Synthesis of hydrophilic conjugated porphyrin dimers for one-photon and two-photon photodynamic therapy at NIR wavelengths. Org. Biomol. Chem. 2009, 7, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Banfi, S.; Caruso, E.; Buccafurni, L.; Battini, V.; Zazzaron, S.; Barbieri, P.; Orlandi, V. Antibacterial activity of tetraaryl-porphyrin photosensitizers: An in vitro study on gram negative and gram positive bacteria. J. Photochem. Photobiol. B 2006, 85, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.G.; Babayan, N.S.; Sahakyan, L.A.; Shahkhatuni, A.G.; Gasparyan, G.H.; Aroutiounian, R.M.; Ghazaryan, R.K. Synthesis and in vitro anticancer activity of water-soluble cationic pyridylporphyrins and their metallocomplexes. J. Porphyr. Phthalocyanines 2008, 12, 1100–1110. [Google Scholar] [CrossRef]
- Diabate, P.D.; Laguerre, A.; Pirrotta, M.; Desbois, N.; Boudon, J.; Gros, C.P.; Monchaud, D. DNA structure-specific sensitization of a metalloporphyrin leads to an efficient in vitro quadruplex detection molecular tool. New J. Chem. 2016, 40, 5683–5689. [Google Scholar] [CrossRef]
- Laguerre, A.; Chang, Y.; Pirrotta, M.; Desbois, N.; Gros, C.P.; Lesniewska, E.; Monchaud, D. Surface-promoted aggregation of amphiphilic quadruplex ligands drives their selectivity for alternative DNA structures. Org. Biomol. Chem. 2015, 13, 7034–7039. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.J.; Yun, K.J.; Kang, M.S.; Park, J.; Lee, K.T.; Park, S.B.; Shin, J.H. Synthesis and in vitro photodynamic activities of water-soluble fluorinated tetrapyridylporphyrins as tumor photosensitizers. Bioorg. Med. Chem. Lett. 2007, 17, 2789–2794. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.; Govindaswamy, P.; Süss-Fink, G.; Ang, W.H.; Dyson, P.J.; Juillerat-Jeanneret, L.; Therrien, B. Ruthenium porphyrin compounds for photodynamic therapy of cancer. J. Med. Chem. 2008, 51, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.; Govindaswamy, P.; Zava, O.; Süss-Fink, G.; Juillerat-Jeanneret, L.; Therrien, B. Combined arene ruthenium porphyrins as chemotherapeutics and photosensitizers for cancer therapy. J. Biol. Inorg. Chem. 2009, 14, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ogawa, K.; Kiwada, T.; Odani, A. Water-soluble metalloporphyrinates with excellent photo-induced anticancer activity resulting from high tumor accumulation. J. Inorg. Biochem. 2017, 170, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gianferrara, T.; Serli, B.; Zangrando, E.; Iengo, E.; Alessio, E. Pyridylporphyrins peripherally coordinated to ruthenium-nitrosyls, including the water-soluble Na4[Zn·4′TPyP{RuCl4(NO)}4]: Synthesis and structural characterization. New J. Chem. 2005, 29, 895–903. [Google Scholar] [CrossRef]
- Haeubl, M.; Reith, L.M.; Gruber, B.; Karner, U.; Müller, N.; Knör, G.; Schoefberger, W. DNA interactions and photocatalytic strand cleavage by artificial nucleases based on water-soluble gold(III) porphyrins. J. Biol. Inorg. Chem. 2009, 14, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.M.; Clarke, O.J.; Fernandez, N.; Boyle, R.W. Porphyrin, chlorin, and bacteriochlorin isothiocyanates: Useful reagents for the synthesis of photoactive bioconjugates. Bioconjug. Chem. 2002, 13, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Kim, Y.-S.; Lee, C.O.; Sohn, Y.S. Synthesis and antitumor activity of DNA binding cationic porphyrin–platinum(II) complexes. Tetrahedron Lett. 2003, 44, 1537–1540. [Google Scholar] [CrossRef]
- Spagnul, C.; Alberto, R.; Gasser, G.; Ferrari, S.; Pierroz, V.; Bergamo, A.; Gianferrara, T.; Alessio, E. Novel water-soluble 99mTc(I)/Re(I)-porphyrin conjugates as potential multimodal agents for molecular imaging. J. Inorg. Biochem. 2013, 122, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Gianferrara, T.; Spagnul, C.; Alberto, R.; Gasser, G.; Ferrari, S.; Pierroz, V.; Bergamo, A.; Alessio, E. Towards matched pairs of porphyrin–ReI/99mTcI conjugates that combine photodynamic activity with fluorescence and radio imaging. ChemMedChem 2014, 9, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Mion, G.; Mari, C.; Da Ros, T.; Rubbiani, R.; Gasser, G.; Gianferrara, T. Towards the synthesis of new tumor targeting photosensitizers for photodynamic therapy and imaging applications. ChemistrySelect 2017, 2, 190–200. [Google Scholar] [CrossRef]
- Feng, X.-X.; Zhang, J.-X.; Wu, Y.; Zhang, Q.; Liu, J.-C. Detailed profiling on DNA binding affinity, cytotoxicity and pathway of induced cell death of novel water-soluble Cu(II)-based acylhydrazone porphyrin derivatives. Dyes Pigm. 2017, 136, 773–781. [Google Scholar] [CrossRef]
- Entract, G.M.; Bryden, F.; Domarkas, J.; Savoie, H.; Allott, L.; Archibald, S.J.; Cawthorne, C.; Boyle, R.W. Development of PDT/PET theranostics: Synthesis and biological evaluation of an 18F-radiolabeled water-soluble porphyrin. Mol. Pharm. 2015, 12, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.; Babayan, N.; Poghosyan, D.; Margaryan, K.; Harutyunyan, B.; Grigoryan, R.; Sarkisyan, N.; Spasojevic, I.; Mamyan, S.; Sahakyan, L.; et al. Novel amphiphilic cationic porphyrin and its Ag(II) complex as potential anticancer agents. J. Inorg. Biochem. 2014, 140, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Suemoto, Y.; Kanemaru, H.; Takemori, K.; Shigehara, M.; Miyamoto, A.; Yokoi, H.; Yasuda, M. Alkyl substituent effect on photosensitized inactivation of Escherichia coli by pyridinium-bonded P-porphyrins. J. Photochem. Photobiol. B 2017, 168, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Shinbara, T.; Tanimura, S.-I.; Matsumoto, T.; Shiragami, T.; Yokoi, H.; Nosaka, Y.; Okazaki, S.; Hirakawa, K.; Yasuda, M. Water-soluble phosphorus porphyrins with high activity for visible light-assisted inactivation of Saccharomyces cerevisiae. Photochem. Photobiol. A 2011, 218, 178–184. [Google Scholar] [CrossRef]
- Aravindu, K.; Mass, O.; Vairaprakash, P.; Springer, J.W.; Yang, E.; Niedzwiedzki, D.M.; Kirmaier, C.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Amphiphilic chlorins and bacteriochlorins in micellar environments. Molecular design, de novo synthesis, and photophysical properties. Chem. Sci. 2013, 4, 3459–3477. [Google Scholar] [CrossRef]
- Reeve, J.E.; Collins, H.A.; Mey, K.D.; Kohl, M.M.; Thorley, K.J.; Paulsen, O.; Clays, K.; Anderson, H.L. Amphiphilic porphyrins for second harmonic generation imaging. J. Am. Chem. Soc. 2009, 131, 2758–2759. [Google Scholar] [CrossRef] [PubMed]
- Reeve, J.E.; Corbett, A.D.; Boczarow, I.; Kaluza, W.; Barford, W.; Bayley, H.; Wilson, T.; Anderson, H.L. Porphyrins for probing electrical potential across lipid bilayer membranes by second harmonic generation. Angew. Chem. Int. Ed. Engl. 2013, 52, 9044–9048. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.P.; Michelin, C.; Depotter, G.; Desbois, N.; Clays, K.; Cui, Y.; Zeng, L.; Fang, Y.; Ngo, H.M.; Lopez, C. Non-linear optical, electrochemical and spectroelectrochemical properties of amphiphilic inner salt porphyrinic systems. J. Porphyr. Phthalocyanines 2016, 20, 1002–1015. [Google Scholar] [CrossRef]
- Manono, J.; Marzilli, P.A.; Fronczek, F.R.; Marzilli, L.G. New porphyrins bearing pyridyl peripheral groups linked by secondary or tertiary sulfonamide groups: Synthesis and structural characterization. Inorg. Chem. 2009, 48, 5626–5635. [Google Scholar] [CrossRef] [PubMed]
- Perera, T.; Abhayawardhana, P.; Marzilli, P.A.; Fronczek, F.R.; Marzilli, L.G. Formation of a metal-to-nitrogen bond of normal length by a neutral sufonamide group within a tridentate ligand. A new approach to radiopharmaceutical bioconjugation. Inorg. Chem. 2013, 52, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Guldi, D.M.; Zilbermann, I.; Anderson, G.; Li, A.; Balbinot, D.; Jux, N.; Hatzimarinaki, M.; Hirsch, A.; Prato, M. Multicomponent redox gradients on photoactive electrode surfaces. Chem. Commun. 2004, 726–727. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Kubo, T.; Shinbara, T.; Matsuda, N.; Shiragami, T.; Fujitsuka, M.; Majima, T.; Yasuda, M. Spectroscopic analysis of the interaction of human serum albumin with tricationic phosphorus porphyrins bearing axial pyridinio groups. Bull. Chem. Soc. Jpn. 2013, 86, 1240–1247. [Google Scholar] [CrossRef]
- Milgrom, L.R.; Bone, S.; Bruce, D.W.; Macdonald, M.P. Evidence for solid-state proton conductivity and low-dimensional ordering in meso-tetrakis (2-imidazolyl)porphyrin. J. Mol. Electron. 1991, 7, 95–100. [Google Scholar]
- Bhaumik, J.; Yao, Z.; Borbas, K.E.; Taniguchi, M.; Lindsey, J.S. Masked imidazolyl-dipyrromethanes in the synthesis of imidazole-substituted porphyrins. J. Org. Chem. 2006, 71, 8807–8817. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.; Bhaumik, J.; Dogutan, D.K.; Aly, Z.; Kamal, Z.; Khalid, L.; Kee, H.L.; Bocian, D.F.; Holten, D.; Lindsey, J.S.; et al. Imidazole metalloporphyrins as photosensitizers for photodynamic therapy: Role of molecular charge, central metal and hydroxyl radical production. Cancer Lett. 2009, 282, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Fuhrhop, J.-H.; Smith, K.M. Laboratory Methods in Porphyrin and Metalloporphyrin Research; Elsevier: Amsterdam, The Netherlands, 1975. [Google Scholar]
- Longo, F.R.; Finarelli, M.G.; Kim, J.B. The synthesis and some physical properties of ms-tetra (pentafluorophenyl)-porphin and ms-tetra (pentachlorophenyl) porphin. J. Heterocycl. Chem. 1969, 6, 927–931. [Google Scholar] [CrossRef]
- Datta-Gupta, N.; Bardos, T. Synthetic porphyrins. I. Synthesis and spectra of some para-substituted meso-tetraphenylporphines. J. Heterocycl. Chem. 1966, 3, 495–502. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler-Longo reactions revisited: Synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Kostas, I.D.; Coutsolelos, A.G.; Charalambidis, G.; Skondra, A. The first use of porphyrins as catalysts in cross-coupling reactions: A water-soluble palladium complex with a porphyrin ligand as an efficient catalyst precursor for the Suzuki–Miyaura reaction in aqueous media under aerobic conditions. Tetrahedron Lett. 2007, 48, 6688–6691. [Google Scholar] [CrossRef]
- Murashima, T.; Tsujimoto, S.; Yamada, T.; Miyazawa, T.; Uno, H.; Ono, N.; Sugimoto, N. Synthesis of water-soluble porphyrin and the corresponding highly planar benzoporphyrin without meso-substituents. Tetrahedron Lett. 2005, 46, 113–116. [Google Scholar] [CrossRef]
- Ogawa, K.; Hasegawa, H.; Inaba, Y.; Kobuke, Y.; Inouye, H.; Kanemitsu, Y.; Kohno, E.; Hirano, T.; Ogura, S.; Okura, I. Water-soluble bis(imidazolylporphyrin) self-assemblies with large two-photon absorption cross sections as potential agents for photodynamic therapy. J. Med. Chem. 2006, 49, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Ogawa, K.; Kobuke, Y. Synthesis and properties of acetylene-linked bis- and tris- porphyrins toward two-photon photodynamic therapy. J. Porphyr. Phthalocyanines 2007, 11, 406–417. [Google Scholar] [CrossRef]
- Muresan, A.Z.; Lindsey, J.S. Design and synthesis of water-soluble bioconjugatable trans-AB-porphyrins. Tetrahedron 2008, 64, 11440–11448. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, M.; Shetty, S.J.; Srivastava, T.S.; Noronha, O.P.D.; Samuel, A.M.; Mukhtar, H. Water-soluble 99mTc-labeled dendritic novel porphyrins tumor imaging and diagnosis. Biochem. Biophys. Res. Commun. 2001, 281, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Eggenspiller, A.; Michelin, C.; Desbois, N.; Richard, P.; Barbe, J.-M.; Denat, F.; Licona, C.; Gaiddon, C.; Sayeh, A.; Choquet, P.; et al. Design of porphyrin-DOTA-like scaffolds as all-in-one multimodal heterometallic complexes for medical imaging. Eur. J. Org. Chem. 2013, 2013, 6629–6643. [Google Scholar] [CrossRef]
- Gros, C.P.; Eggenspiller, A.; Nonat, A.; Barbe, J.-M.; Denat, F. New potential bimodal imaging contrast agents based on DOTA-like and porphyrin macrocycles. MedChemComm 2011, 2, 119–125. [Google Scholar] [CrossRef]
- Luo, J.; Chen, L.-F.; Hu, P.; Chen, Z.-N. Tetranuclear gadolinium(III) porphyrin complex as a theranostic agent for multimodal imaging and photodynamic therapy. Inorg. Chem. 2014, 53, 4184–4191. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Gibson, S.L.; Hilf, R.; Davies, S.R.; Oseroff, A.R.; Roy, I.; Ohulchanskyy, T.Y.; Bergey, E.J.; Detty, M.R. Water-soluble, core-modified porphyrins. 3. Synthesis, photophysical properties, and in vitro studies of photosensitization, uptake, and localization with carboxylic acid-substituted derivatives. J. Med. Chem. 2003, 46, 3734–3747. [Google Scholar] [CrossRef] [PubMed]
- Sour, A.; Jenni, S.; Ortí-Suárez, A.; Schmitt, J.; Heitz, V.; Bolze, F.; Loureiro de Sousa, P.; Po, C.; Bonnet, C.S.; Pallier, A.; et al. Four gadolinium(III) complexes appended to a porphyrin: A water-soluble molecular theranostic agent with remarkable relaxivity suited for mri tracking of the photosensitizer. Inorg. Chem. 2016, 55, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Vairaprakash, P.; Reddy, K.R.; Sahin, T.; Pavan, M.P.; Lubian, E.; Lindsey, J.S. Hydrophilic tetracarboxy bacteriochlorins for photonics applications. Org. Biomol. Chem. 2014, 12, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.S.; Tsutsui, M. Preparation and purification of tetrasodium meso-tetra(p-sulfophenyl)porphine. An easy procedure. J. Org. Chem. 1973, 38, 2103. [Google Scholar] [CrossRef]
- Mahammed, A.; Goldberg, I.; Gross, Z. Highly selective chlorosulfonation of tris(pentafluorophenyl)corrole as a synthetic tool for the preparation of amphiphilic corroles and metal complexes of planar chirality. Org. Lett. 2001, 3, 3443–3446. [Google Scholar] [CrossRef] [PubMed]
- Saltsman, I.; Mahammed, A.; Goldberg, I.; Tkachenko, E.; Botoshansky, M.; Gross, Z. Selective substitution of corroles: Nitration, hydroformylation, and chlorosulfonation. J. Am. Chem. Soc. 2002, 124, 7411–7420. [Google Scholar] [CrossRef] [PubMed]
- Vestfrid, J.; Kothari, R.; Kostenko, A.; Goldberg, I.; Tumanskii, B.; Gross, Z. Intriguing physical and chemical properties of phosphorus corroles. Inorg. Chem. 2016, 55, 6061–6067. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.P.; Saneesh Babu, P.S.; Asha Nair, S.; Ramakrishnan, S.; Ramaiah, D.; Chandrashekar, T.K.; Srinivasan, A.; Radhakrishna Pillai, M. Meso-Tetrakis(p-sulfonatophenyl)-N-confused porphyrin tetrasodium salt: A potential sensitizer for photodynamic therapy. J. Med. Chem. 2012, 55, 5110–5120. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Kim, Y.-S.; Sohn, Y.S. Synthesis and selective tumor targetting properties of water soluble porphyrin-Pt(II) conjugates. J. Inorg. Biochem. 2002, 83, 83–88. [Google Scholar] [CrossRef]
- Zhang, X.A.; Lovejoy, K.S.; Jasanoff, A.; Lippard, S.J. Water-soluble porphyrins as a dual-function molecular imaging platform for MRI and fluorescence zinc sensing. Proc. Natl. Acad. Sci. USA 2007, 104, 10780–10785. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zou, T.J.; Tan, Z.L.; Chen, S.; Wu, Z.H.; Yan, G.P.; Zhang, Q.; Liang, S.C.; Yang, J. Isoindoline nitroxide-labeled porphyrins as potential fluorescence-suppressed spin probes. Org. Biomol. Chem. 2017, 15, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.J.P.; Pereira, M.M.; Pinto, S.M.A.; Simões, A.V.C.; Sá, G.F.F.; Arnaut, L.G.; Formosinho, S.J.; Simões, S.; Wyatt, M.F. Synthesis of amphiphilic sulfonamide halogenated porphyrins: MALDI-TOFMS characterization and evaluation of 1-octanol/water partition coefficients. Tetrahedron 2008, 64, 5132–5138. [Google Scholar] [CrossRef]
- Dabrowski, J.M.; Arnaut, L.G.; Pereira, M.M.; Monteiro, C.J.; Urbanska, K.; Simoes, S.; Stochel, G. New halogenated water-soluble chlorin and bacteriochlorin as photostable PDT sensitizers: Synthesis, spectroscopy, photophysics, and in vitro photosensitizing efficacy. ChemMedChem 2010, 5, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
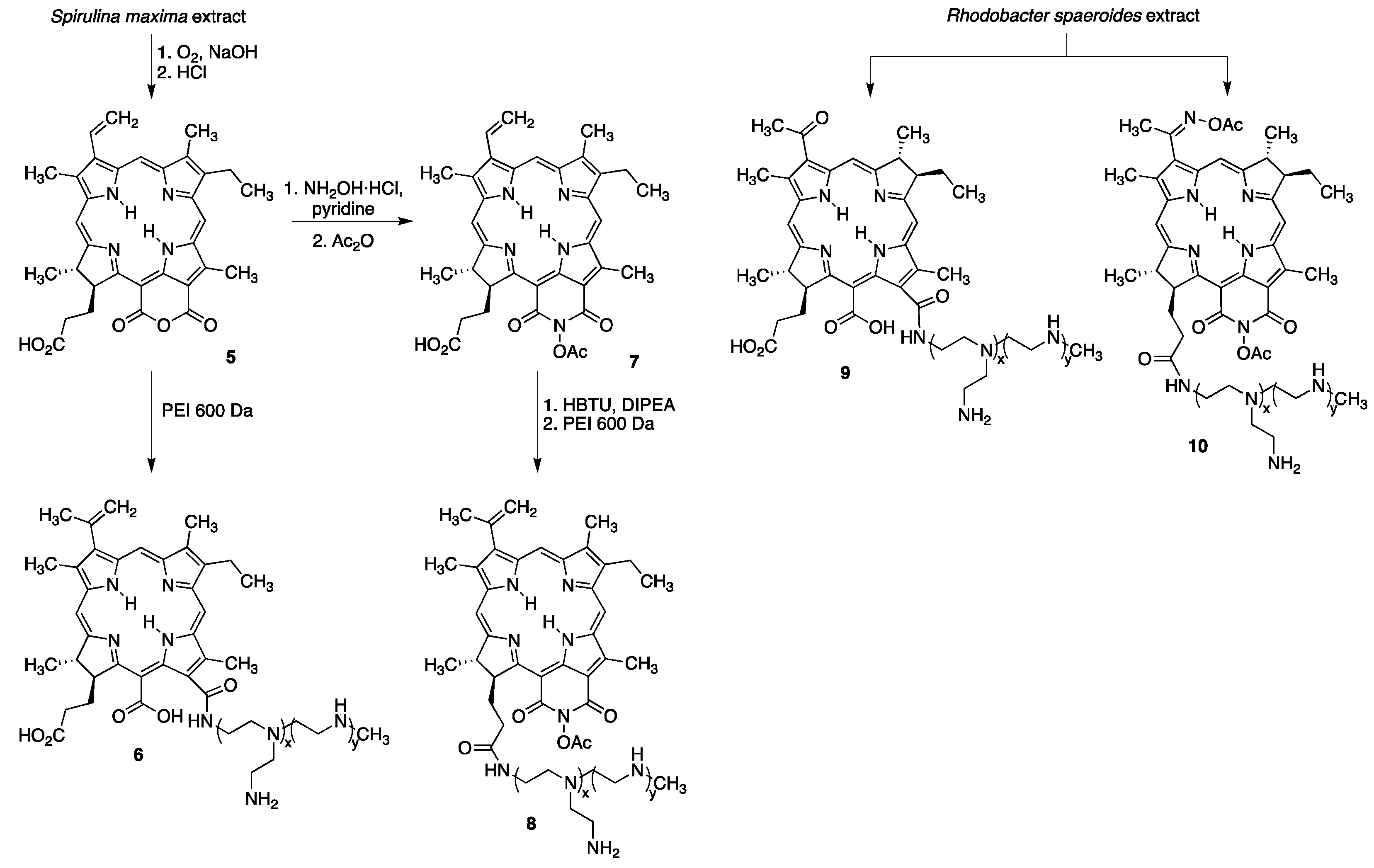
- Brandis, A.; Mazor, O.; Neumark, E.; Rosenbach-Belkin, V.; Salomon, Y.; Scherz, A. Novel water-soluble bacteriochlorophyll derivatives for vascular-targeted photodynamic therapy: Synthesis, solubility, phototoxicity and the effect of serum proteins. Photochem. Photobiol. 2005, 81, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Mazor, O.; Brandis, A.; Plaks, V.; Neumark, E.; Rosenbach-Belkin, V.; Salomon, Y.; Scherz, A. WSTI 1, a novel water-soluble bacteriochlorophyll derivative; cellular uptake, pharmacokinetics, biodistribution and vascular-targeted photodynamic activity using melanoma tumors as a model. Photochem. Photobiol. 2005, 81, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Wedel, M.; Walter, A.; Montforts, F.-P. Synthesis of metalloporphyrins and metallochlorins for immobilization on electrode surfaces. Eur. J. Org. Chem. 2001, 2001, 1681–1687. [Google Scholar] [CrossRef]
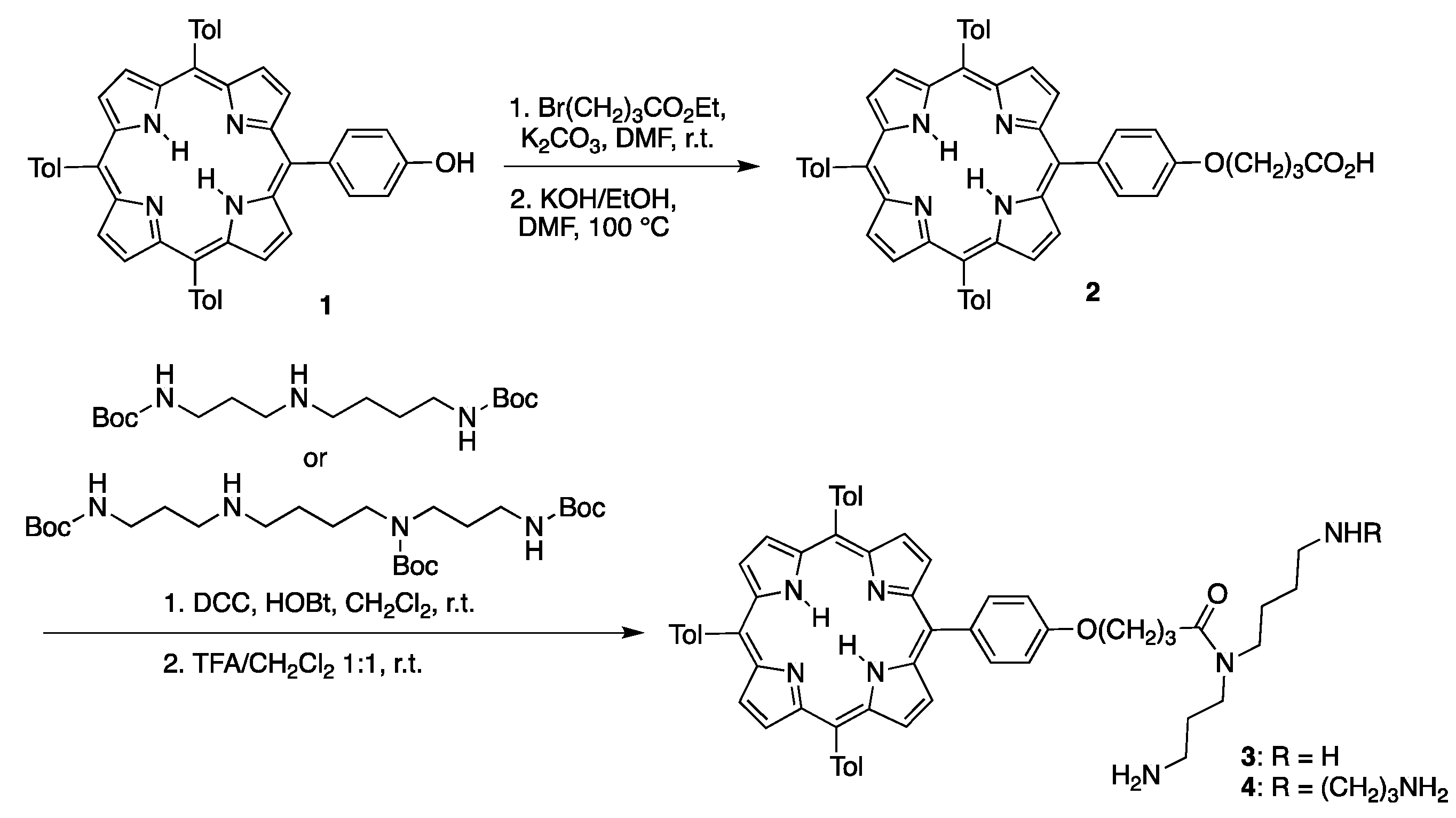
- Borbas, K.E.; Mroz, P.; Hamblin, M.R.; Lindsey, J.S. Bioconjugatable porphyrins bearing a compact swallowtail motif for water solubility. Bioconjug. Chem. 2006, 17, 638–653. [Google Scholar] [CrossRef] [PubMed]
- Borbas, K.E.; Kee, H.L.; Holten, D.; Lindsey, J.S. A compact water-soluble porphyrin bearing an iodoacetamido bioconjugatable site. Org. Biomol. Chem. 2008, 6, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Borbas, K.E.; Chandrashaker, V.; Muthiah, C.; Kee, H.L.; Holten, D.; Lindsey, J.S. Design, synthesis, and photophysical characterization of water-soluble chlorins. J. Org. Chem. 2008, 73, 3145–3158. [Google Scholar] [CrossRef] [PubMed]
- Sahin, T.; Vairaprakash, P.; Borbas, K.E.; Balasubramanian, T.; Lindsey, J.S. Hydrophilic bioconjugatable trans-AB-porphyrins and peptide conjugates. J. Porphyr. Pthalocyanines 2015, 19, 663–678. [Google Scholar] [CrossRef]
- Zhang, N.; Jiang, J.; Liu, M.; Taniguchi, M.; Mandal, A.K.; Evans-Storms, R.B.; Pitner, J.B.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Bioconjugatable, PEGylated hydroporphyrins for photochemistry and photomedicine. Narrow-band, near-infrared-emitting bacteriochlorins. New J. Chem. 2016, 40, 7750–7767. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, C.-Y.; Mandal, A.K.; Chandrashaker, V.; Evans-Storms, R.B.; Pitner, J.B.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Bioconjugatable, pegylated hydroporphyrins for photochemistry and photomedicine. Narrow-band, red-emitting chlorins. New J. Chem. 2016, 40, 7721–7740. [Google Scholar] [CrossRef] [PubMed]
- Benaglia, M.; Danelli, T.; Fabris, F.; Sperandio, D.; Pozzi, G. Poly(ethylene glycol)-supported tetrahydroxy-phenyl porphyrin: A convenient, recyclable catalyst for photooxidation reactions. Org. Lett. 2002, 4, 4229–4232. [Google Scholar] [CrossRef] [PubMed]
- Worlinsky, J.L.; Halepas, S.; Ghandehari, M.; Khalil, G.; Brückner, C. High pH sensing with water-soluble porpholactone derivatives and their incorporation into a Nafion® optode membrane. Analyst 2015, 140, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Worlinsky, J.L.; Halepas, S.; Brückner, C. PEGylated meso-arylporpholactone metal complexes as optical cyanide sensors in water. Org. Biomol. Chem. 2014, 12, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Lottner, C.; Bart, K.-C.; Bernhardt, G.; Brunner, H. Soluble tetraarylporphyrin-platinum conjugates as cytotoxic and phototoxic antitumor agents. J. Med. Chem. 2002, 45, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Reuther, T.; Kübler, A.C.; Zillmann, U.; Flechtenmacher, C.; Sinn, H. Comparison of the in vivo efficiency of photofrin II-, mTHPC-, mTHPC-PEG- and mTHPCnPEG-mediated PDT in a human xenografted head and neck carcinoma. Lasers Surg. Med. 2001, 29, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Topkaya, D.; Lafont, D.; Poyer, F.; Garcia, G.; Albrieux, F.; Maillard, P.; Bretonnière, Y.; Dumoulin, F. Design of an amphiphilic porphyrin exhibiting high in vitro photocytotoxicity. New J. Chem. 2016, 40, 2044–2050. [Google Scholar] [CrossRef]
- Zhang, J.-L.; Che, C.-M. Soluble polymer-supported ruthenium porphyrin catalysts for epoxidation, cyclopropanation, and aziridination of alkenes. Org. Lett. 2002, 4, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Nawalany, K.; Kozik, B.; Kepczynski, M.; Zapotoczny, S.; Kumorek, M.; Nowakowska, M.; Jachimska, B. Properties of polyethylene glycol supported tetraarylporphyrin in aqueous solution and its interaction with liposomal membranes. J. Phys. Chem. B 2008, 112, 12231–12239. [Google Scholar] [CrossRef] [PubMed]
- Mineo, P.; Scamporrino, E.; Vitalini, D. Synthesis and characterization of uncharged water-soluble star polymers containing a porphyrin core. Macromol. Rapid Commun. 2002, 23, 681–687. [Google Scholar] [CrossRef]
- Kim, W.J.; Kang, M.S.; Kim, H.K.; Kim, Y.; Chang, T.; Ohulchanskyy, T.Y.; Prasad, P.N.; Lee, K.-S. Water-soluble porphyrin-polyethylene glycol conjugates with enhanced cellular uptake for photodynamic therapy. J. Nanosci. Nanotechnol. 2009, 9, 7130–7135. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Romeo, A.; Angelini, N.; Micali, N.; Longo, A.; Mazzaglia, A.; Scolaro, L.M. Structural features of meso-tetrakis(4-carboxyphenyl)porphyrin interacting with amino-terminated poly(propylene oxide). Macromolecules 2006, 39, 5489–5496. [Google Scholar] [CrossRef]
- Lovell, J.F.; Roxin, A.; Ng, K.K.; Qi, Q.; McMullen, J.D.; DaCosta, R.S.; Zheng, G. Porphyrin-cross-linked hydrogel for fluorescence-guided monitoring and surgical resection. Biomacromolecules 2011, 12, 3115–3118. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.-L.; Shieh, M.-J.; Tsai, M.-H.; Chang, C.-C.; Lai, P.-S. Self-assembled star-shaped chlorin-core poly(ε-caprolactone)–poly(ethylene glycol) diblock copolymer micelles for dual chemo-photodynamic therapies. Biomaterials 2008, 29, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Sibrian-Vazquez, M.; Jensen, T.J.; Hammer, R.P.; Vicente, M.G.H. Peptide-mediated cell transport of water soluble porphyrin conjugates. J. Med. Chem. 2006, 49, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Gianferrara, T.; Bergamo, A.; Bratsos, I.; Milani, B.; Spagnul, C.; Sava, G.; Alessio, E. Ruthenium-porphyrin conjugates with cytotoxic and phototoxic antitumor activity. J. Med. Chem. 2010, 53, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Mion, G.; Gianferrara, T.; Bergamo, A.; Gasser, G.; Pierroz, V.; Rubbiani, R.; Vilar, R.; Leczkowska, A.; Alessio, E. Phototoxic activity and DNA interactions of water-soluble porphyrins and their rhenium(I) conjugates. ChemMedChem 2015, 10, 1901–1914. [Google Scholar] [CrossRef] [PubMed]
- Zingg, A.; Felber, B.; Gramlich, V.; Fu, L.; Collman, J.P.; Diederich, F. Dendritic iron(II) porphyrins as models for hemoglobin and myoglobin: Specific stabilization of O2 complexes in dendrimers with H-bond-donor centers. Helv. Chim. Acta 2002, 85, 333–351. [Google Scholar] [CrossRef]
- Akhtar, M.A.; Riaz, S.; Hayat, A.; Nasir, M.; Muhammad, N.; Rahim, A.; Nawaz, M.H. Poly(ethylene oxide) tethered trans-porphyrin: Synthesis, self-assembly with fullerene (C60) and DNA binding studies. J. Mol. Liq. 2017, 225, 235–239. [Google Scholar] [CrossRef]
- Ogikubo, J.; Worlinsky, J.L.; Fu, Y.-J.; Brückner, C. A two-step, one-pot route to swap the pyrroline moiety in meso-tetraaryldihydroxy-chlorins with an O/N-substituted oxazoline. Tetrahedron Lett. 2013, 54, 1707–1710. [Google Scholar] [CrossRef]
- Ahmed, S.; Davoust, E.; Savoie, H.; Boa, A.N.; Boyle, R.W. Thioglycosylated cationic porphyrins—Convenient synthesis and photodynamic activity in vitro. Tetrahedron Lett. 2004, 45, 6045–6047. [Google Scholar] [CrossRef]
- Hamblin, M.R.; Miller, J.L.; Rizvi, I.; Ortel, B.; Maytin, E.V.; Hasan, T. PEGylation of a chlorin e6 polymer conjugate increases tumor targeting of photosensitizer. Cancer Res. 2001, 61, 7155–7162. [Google Scholar] [PubMed]
- Kimani, S.; Ghosh, G.; Ghogare, A.; Rudshteyn, B.; Bartusik, D.; Hasan, T.; Greer, A. Synthesis and characterization of mono-, di-, and tri-poly(ethylene glycol) chlorin e6 conjugates for the photokilling of human ovarian cancer cells. J. Org. Chem. 2012, 77, 10638–10647. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Sawa, T.; Fang, J.; Tanaka, S.; Miyamoto, Y.; Akaike, T.; Maeda, H. Pegylated zinc protoporphyrin: A water-soluble heme oxygenase inhibitor with tumor-targeting capacity. Bioconjug. Chem. 2002, 13, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Regehly, M.; Greish, K.; Rancan, F.; Maeda, H.; Böhm, F.; Röder, B. Water-soluble polymer conjugates of ZnPP for photodynamic tumor therapy. Bioconjug. Chem. 2007, 18, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Lottner, C.; Bart, K.-C.; Bernhardt, G.; Brunner, H. Hematoporphyrin-derived soluble porphyrin-platinum conjugates with combined cytotoxic and phototoxic antitumor activity. J. Med. Chem. 2002, 45, 2064–2078. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Song, R.; Hyun Kim, D.; Jun, M.J.; Sohn, Y.S. Synthesis, biodistribution and antitumor activity of hematoporphyrin-platinum(II) conjugates. Biorg. Med. Chem. 2003, 11, 1753–1760. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Karmakova, T.A.; Kassies, R.; Pljutinskaya, A.D.; Grin, M.A.; Refregiers, M.; Yakubovskaya, R.I.; Mironov, A.F.; Maurizot, J.-C.; Vigny, P.; et al. Cycloimide bacteriochlorin p derivatives: Photodynamic properties and cellular and tissue distribution. Free Radic. Biol. Med. 2006, 40, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Taniguchi, M.; Lindsey, J.S. Regioselective 15-bromination and functionalization of a stable synthetic bacteriochlorin. J. Org. Chem. 2007, 72, 5350–5357. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Sahin, T.; Liu, M.; Lindsey, J.S.; Bocian, D.F.; Holten, D. Photophysical comparisons of PEGylated porphyrins, chlorins and bacteriochlorins in water. New J. Chem. 2016, 40, 9648–9656. [Google Scholar] [CrossRef]





































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
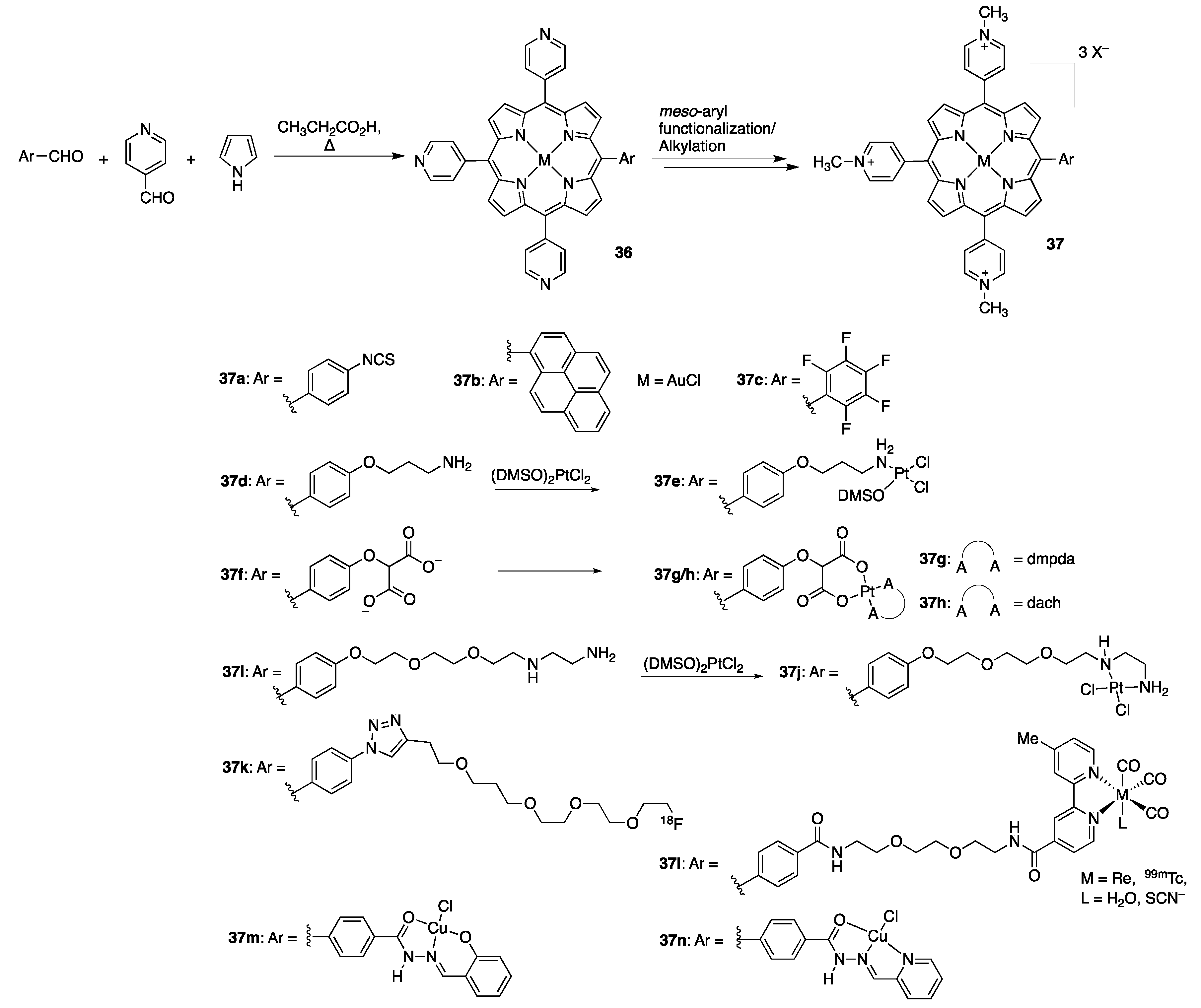{kind=link}
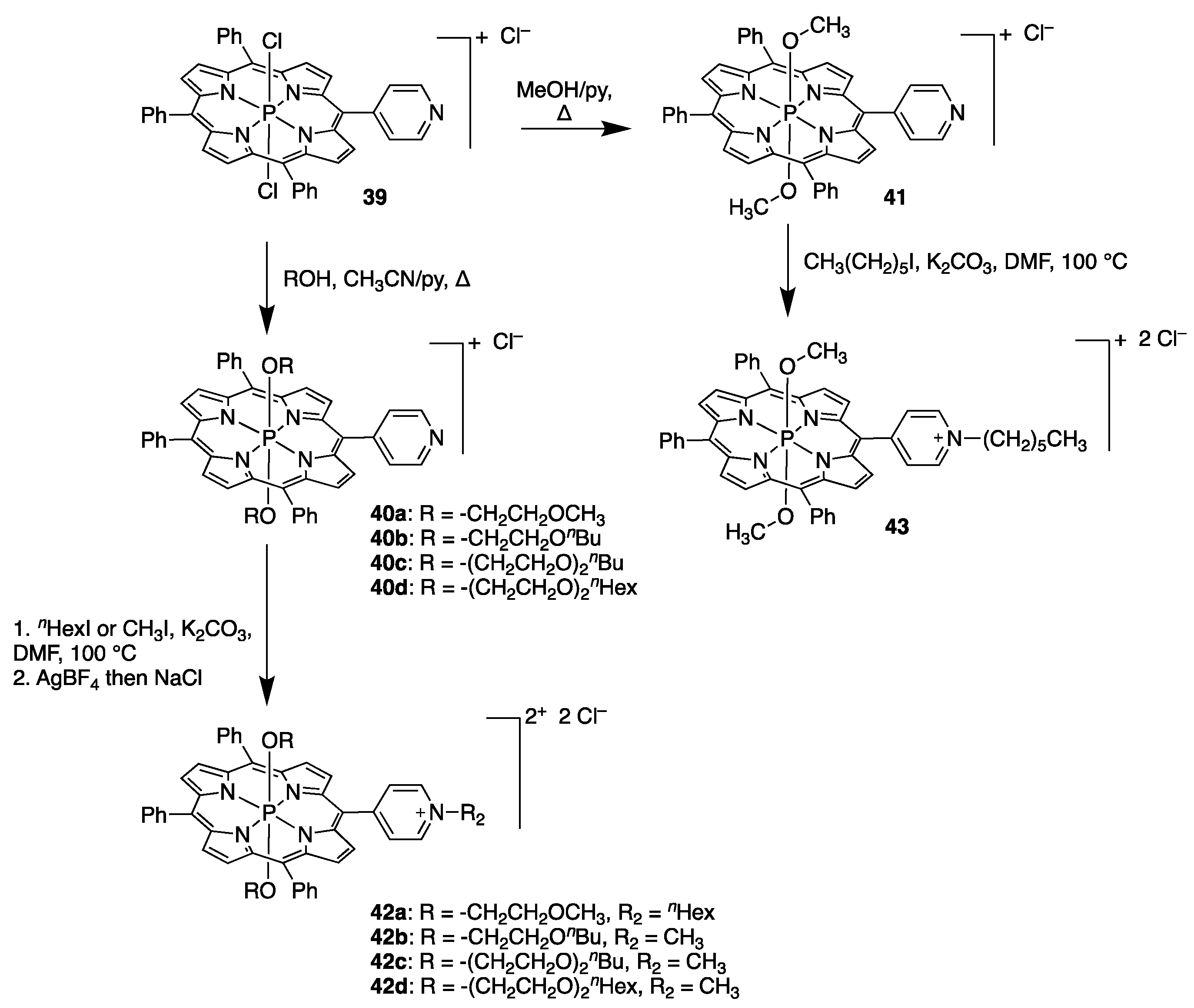{kind=link}
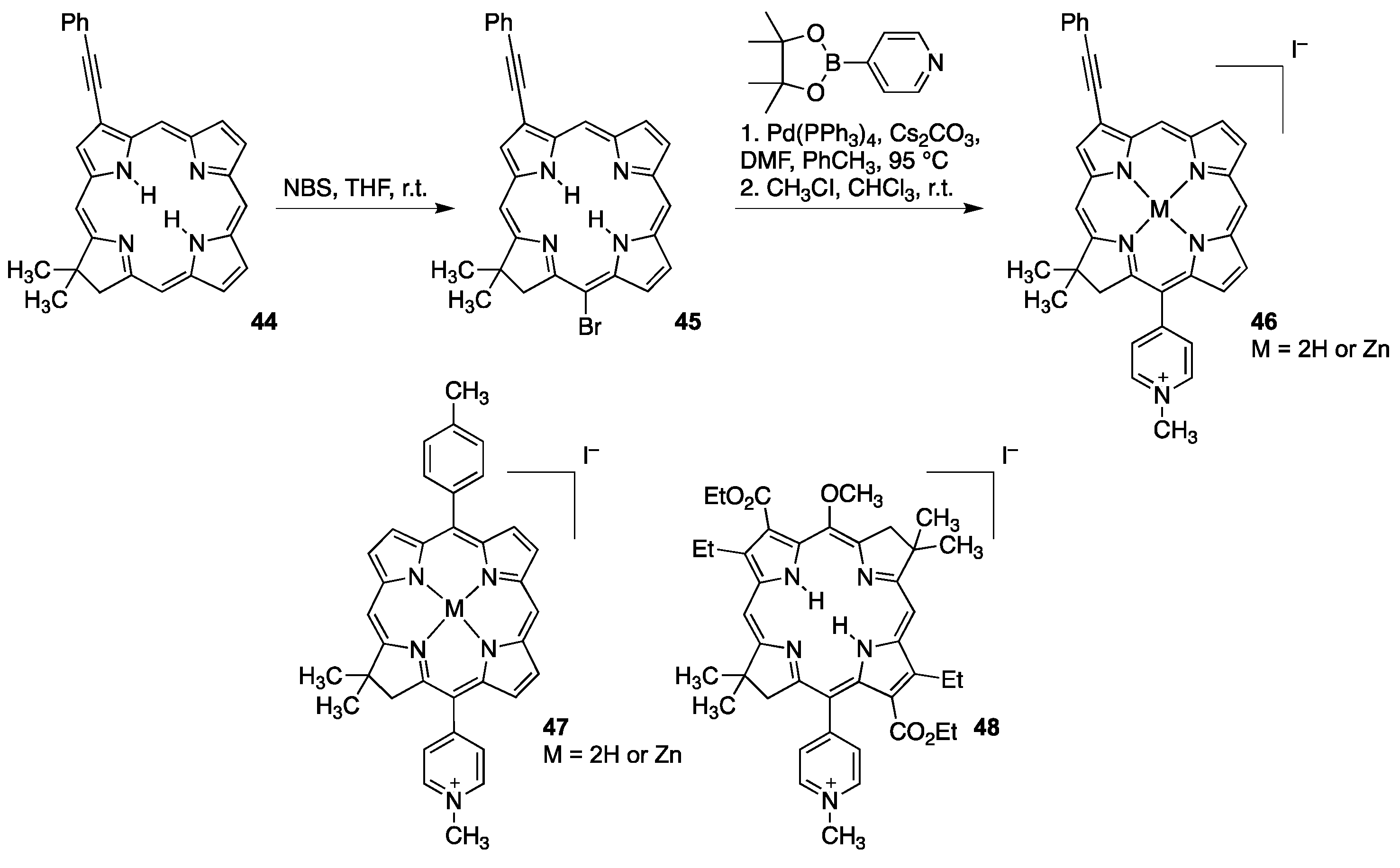{kind=link}
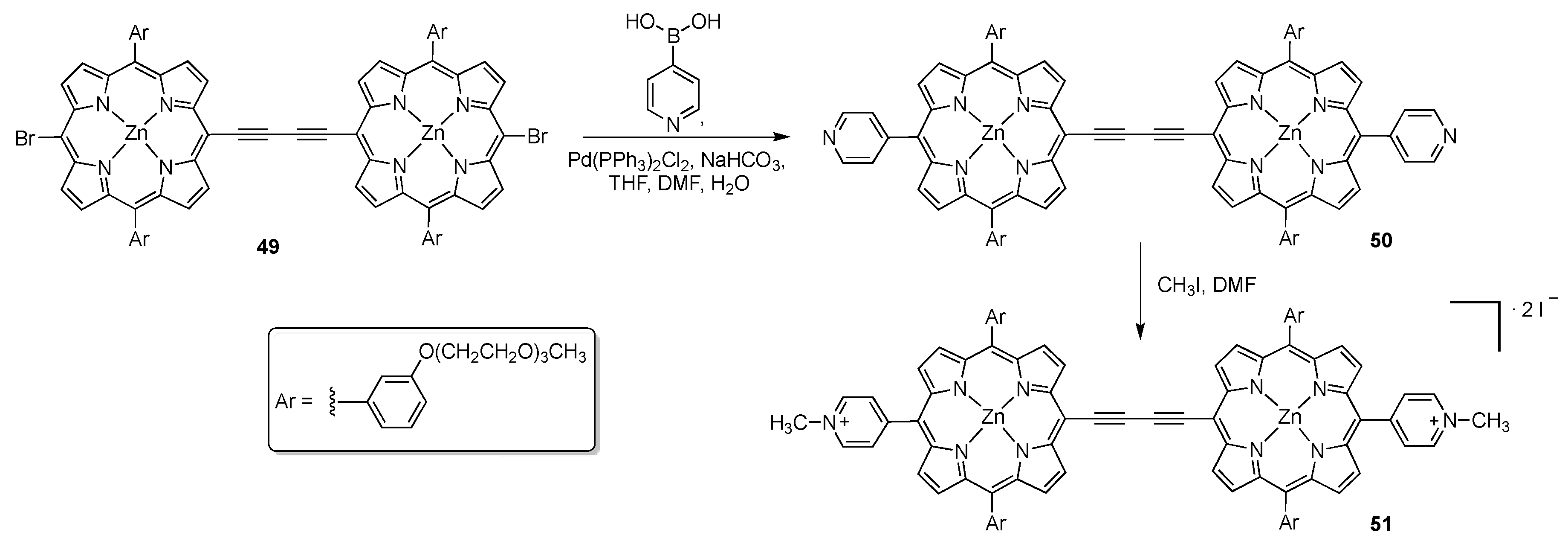{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
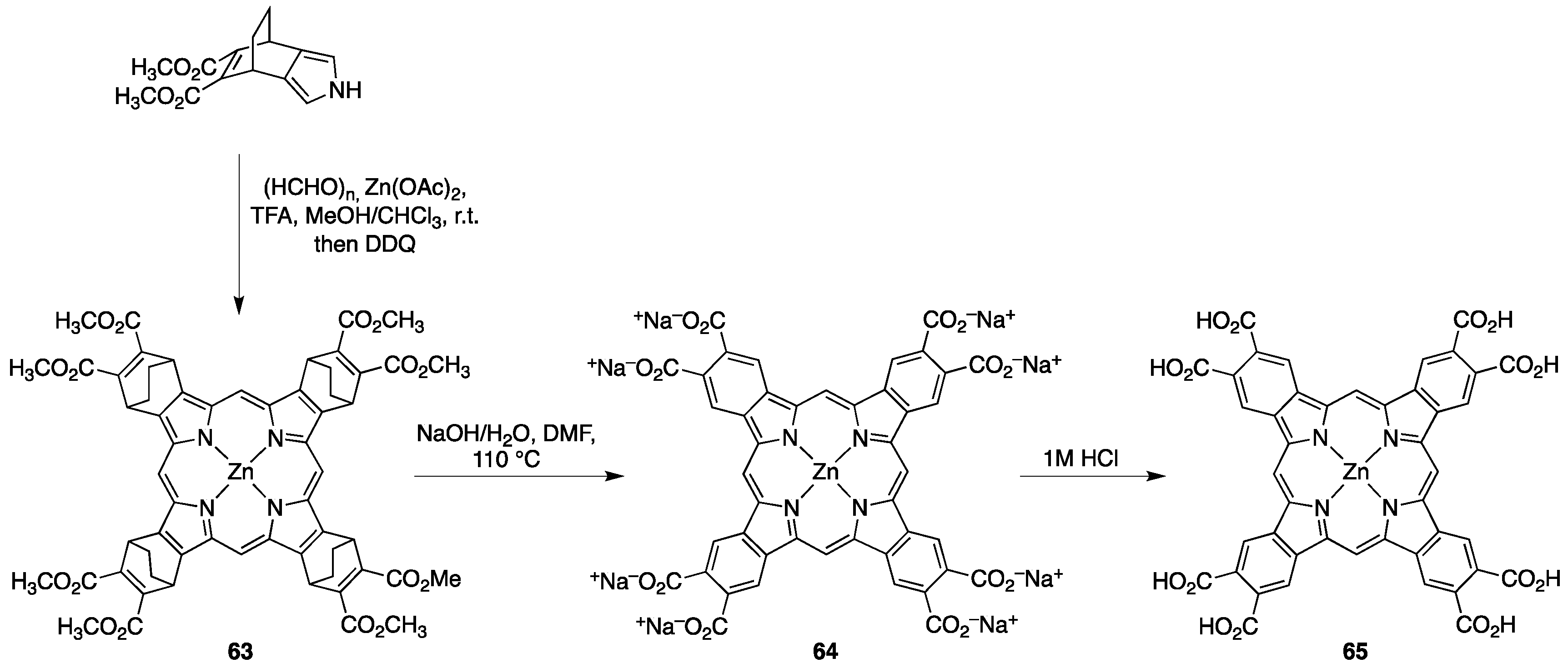{kind=link}
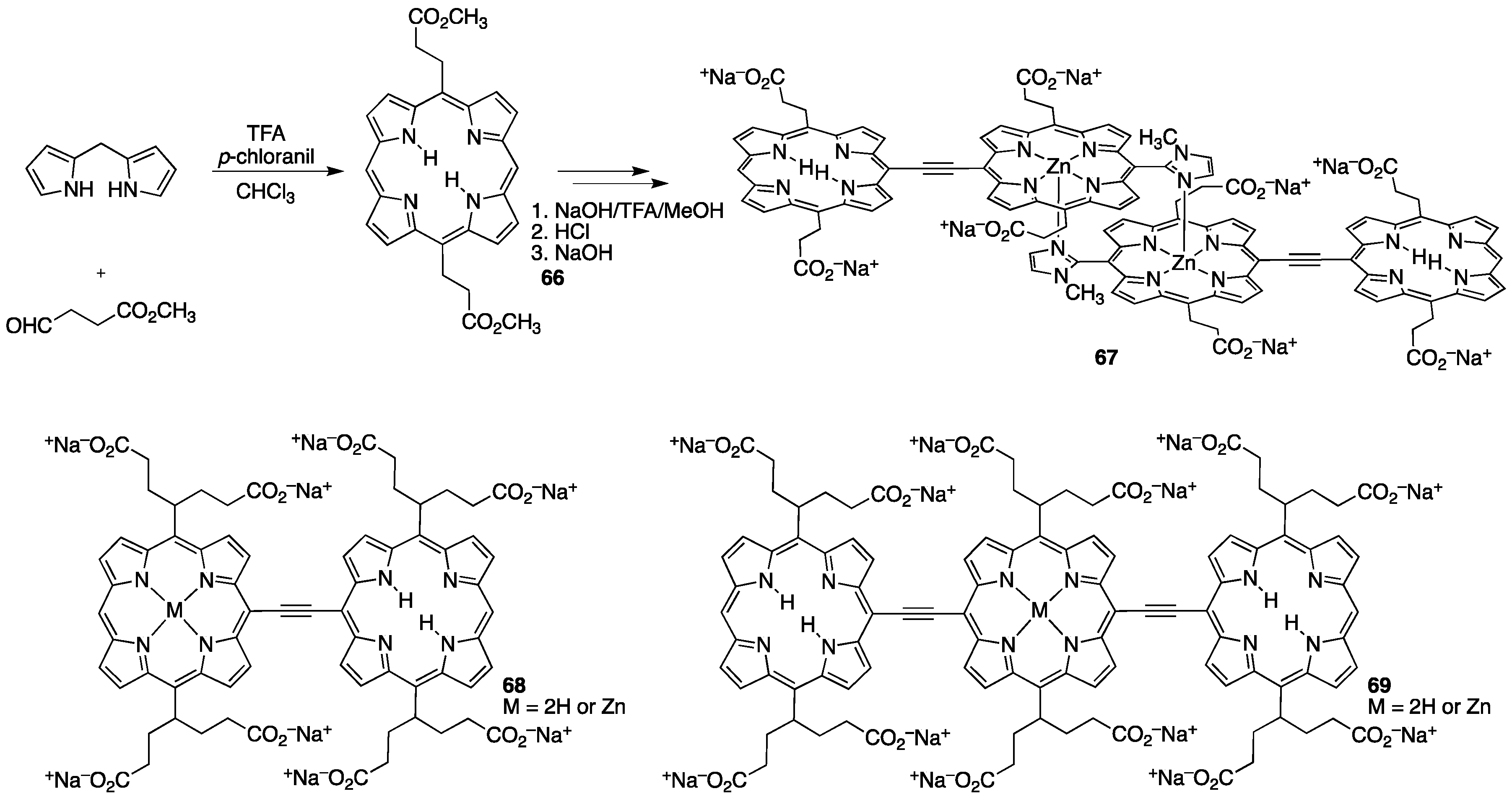{kind=link}
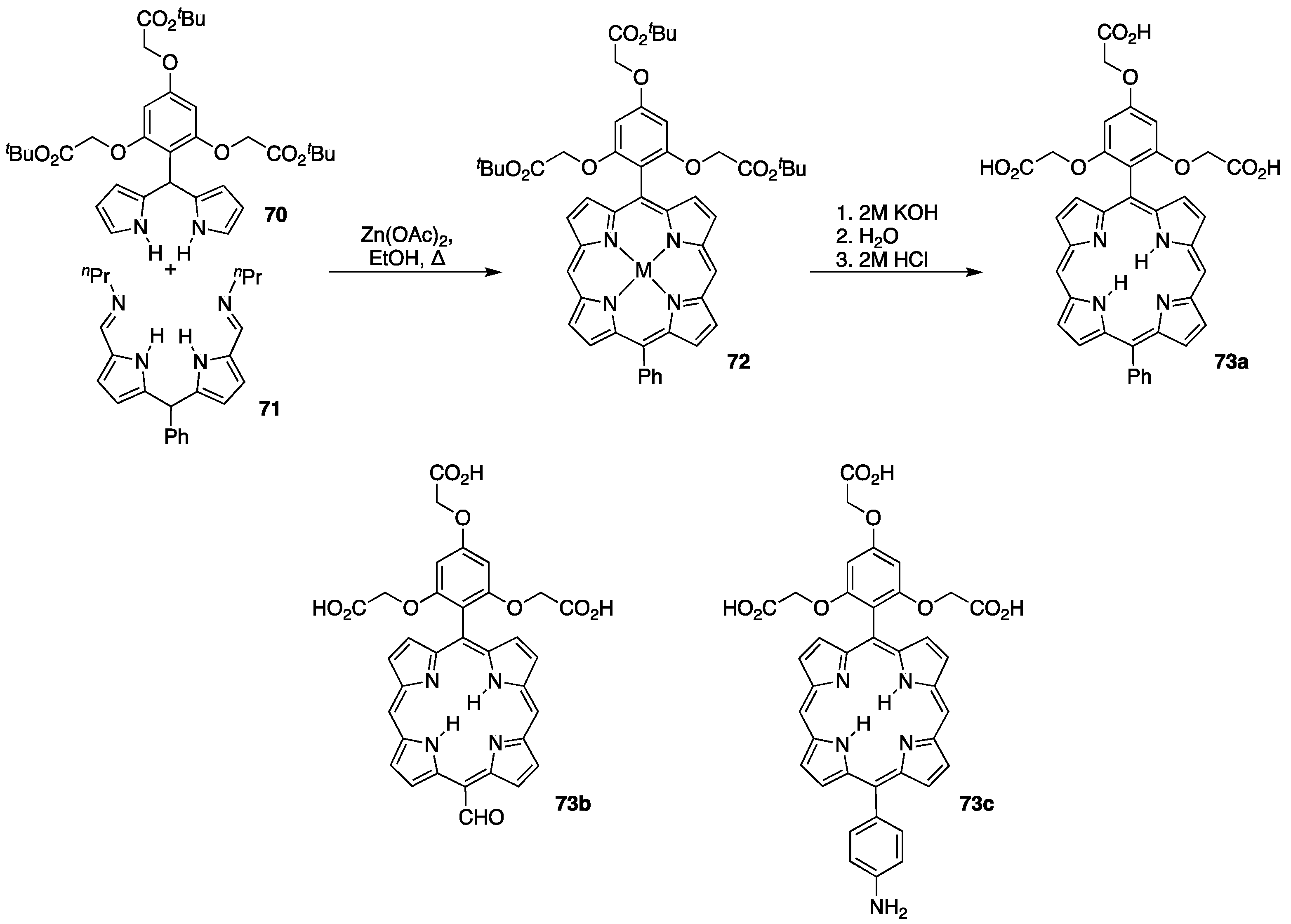{kind=link}
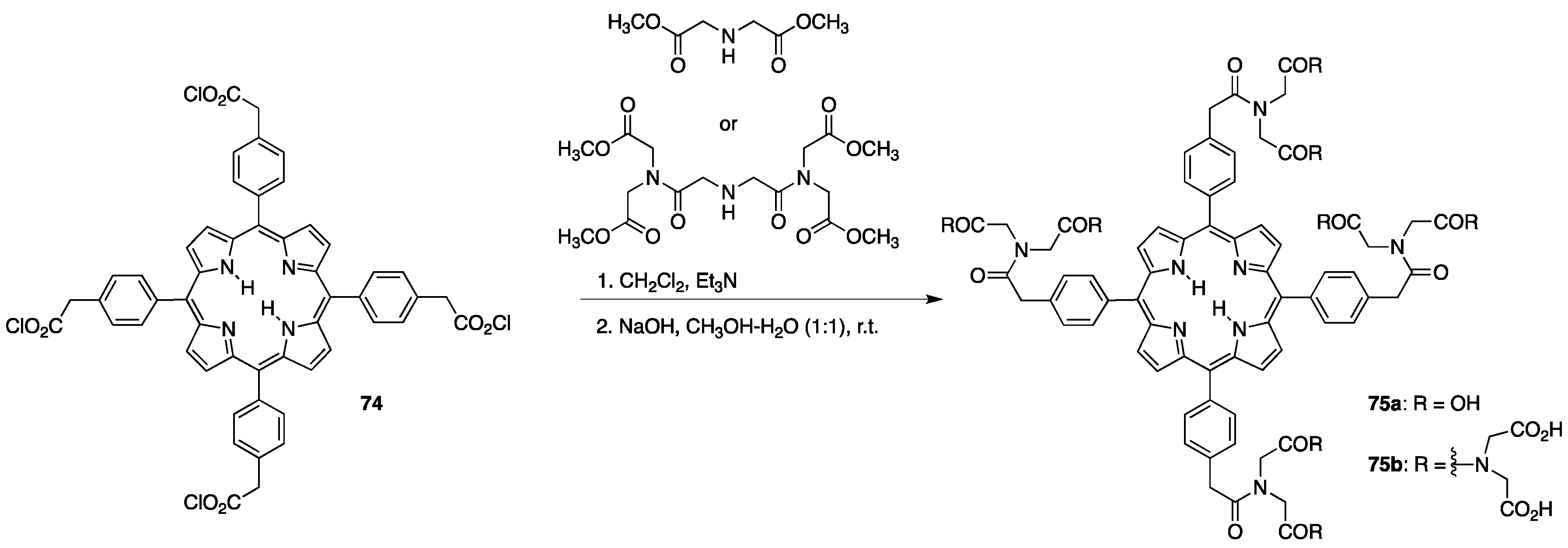{kind=link}
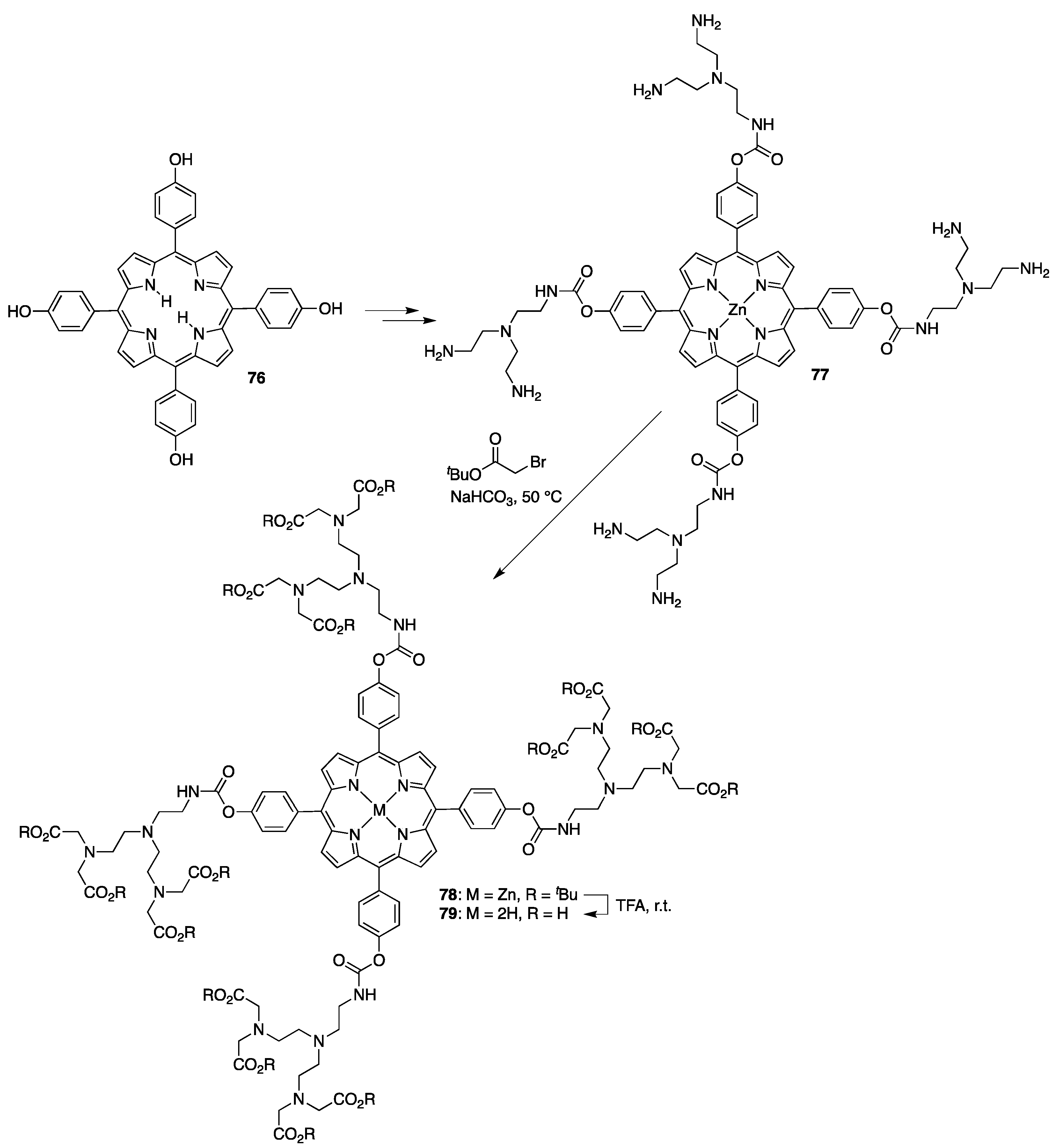{kind=link}
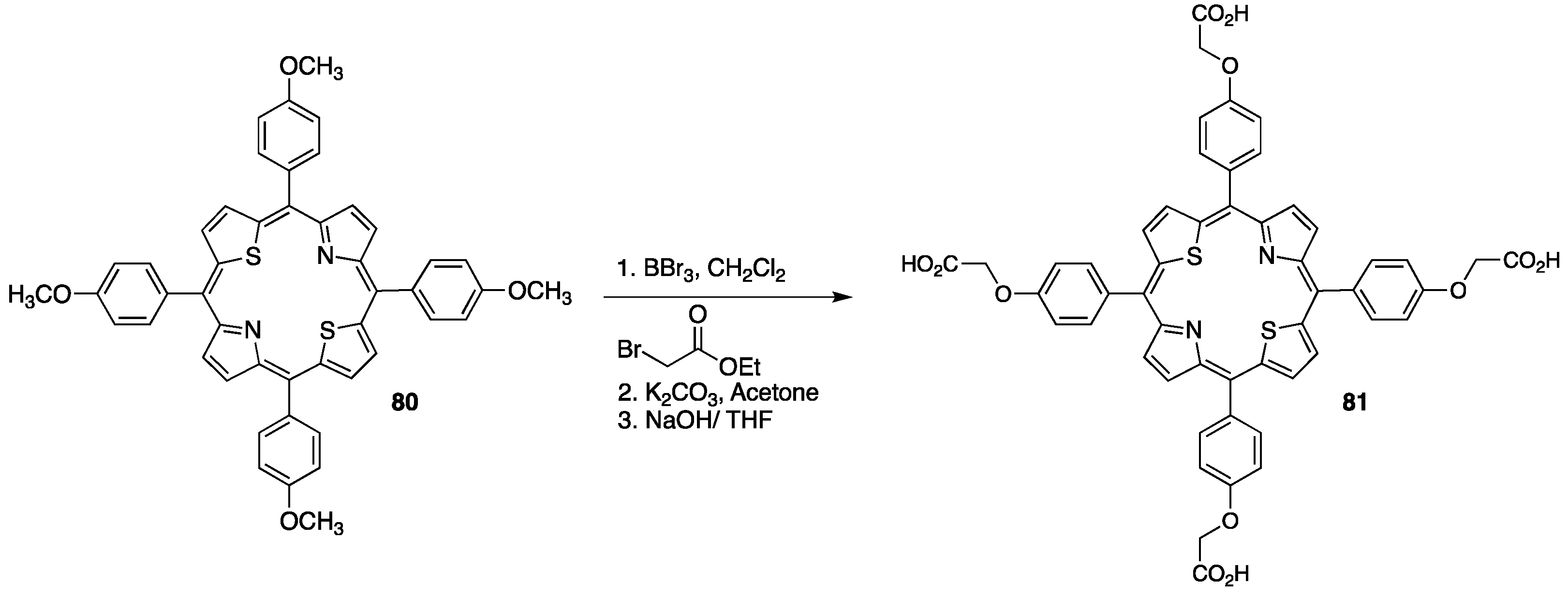{kind=link}
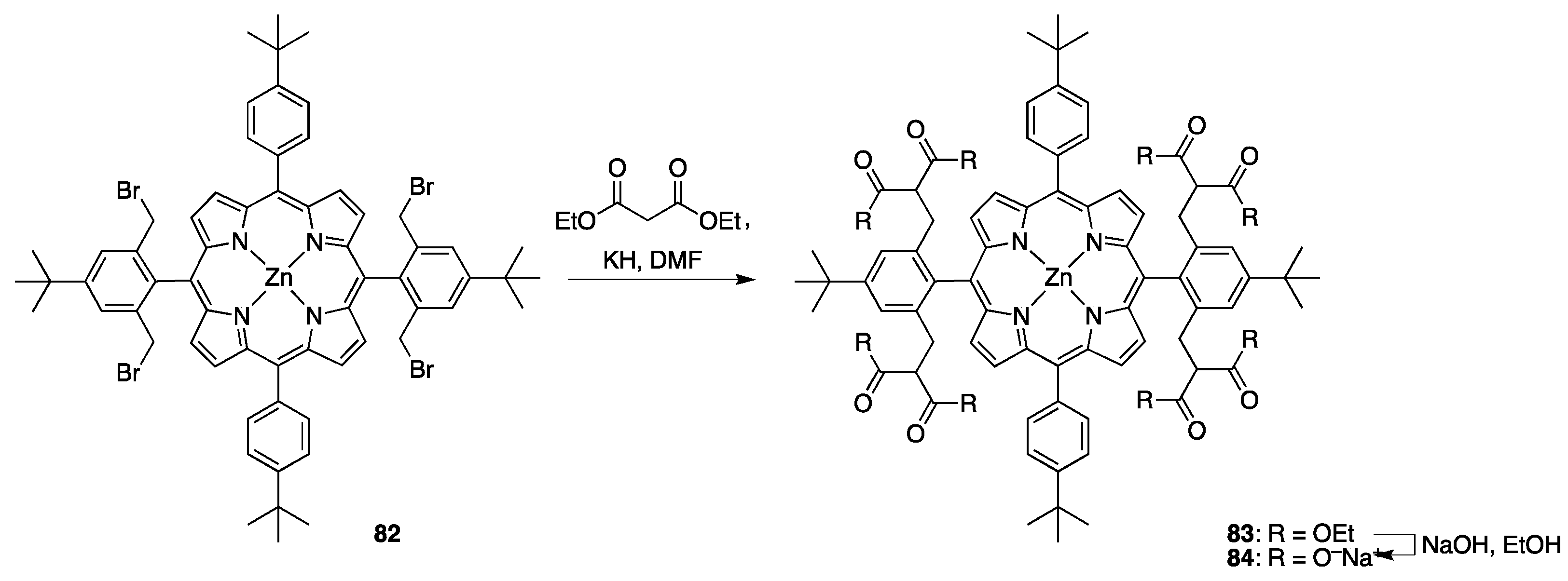{kind=link}
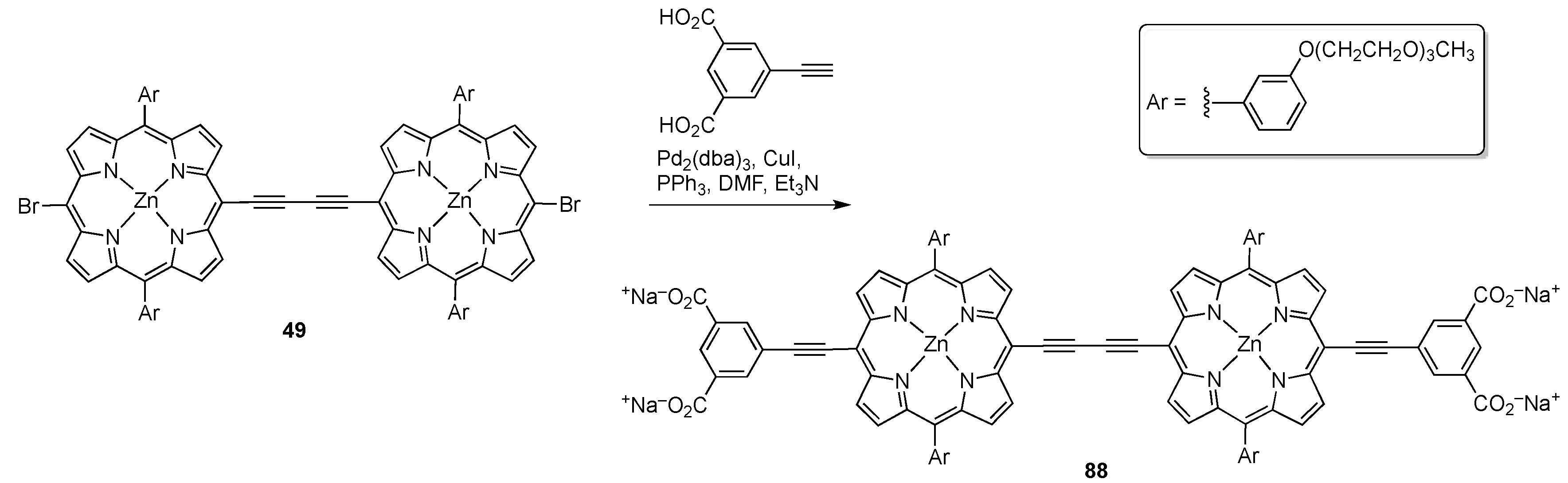{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
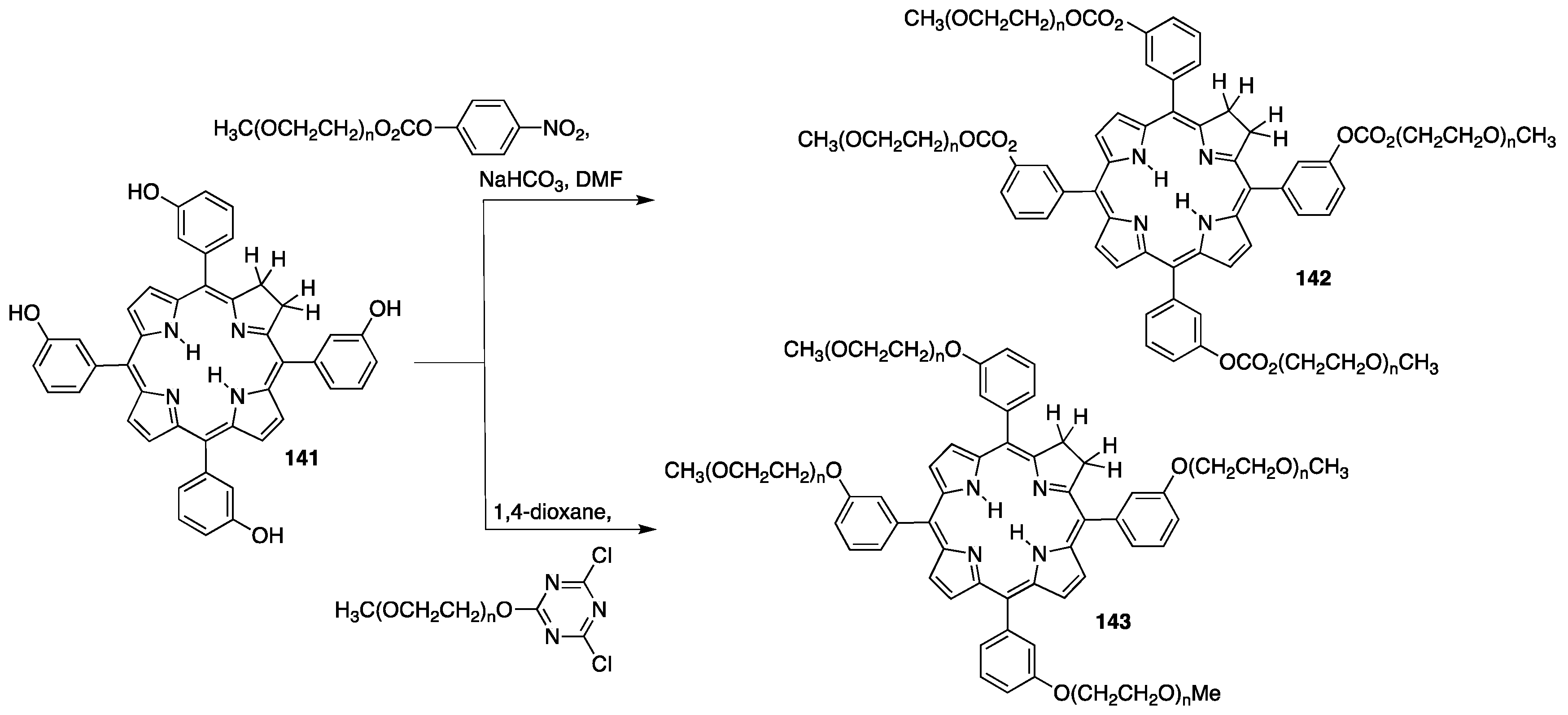{kind=link}
{kind=link}
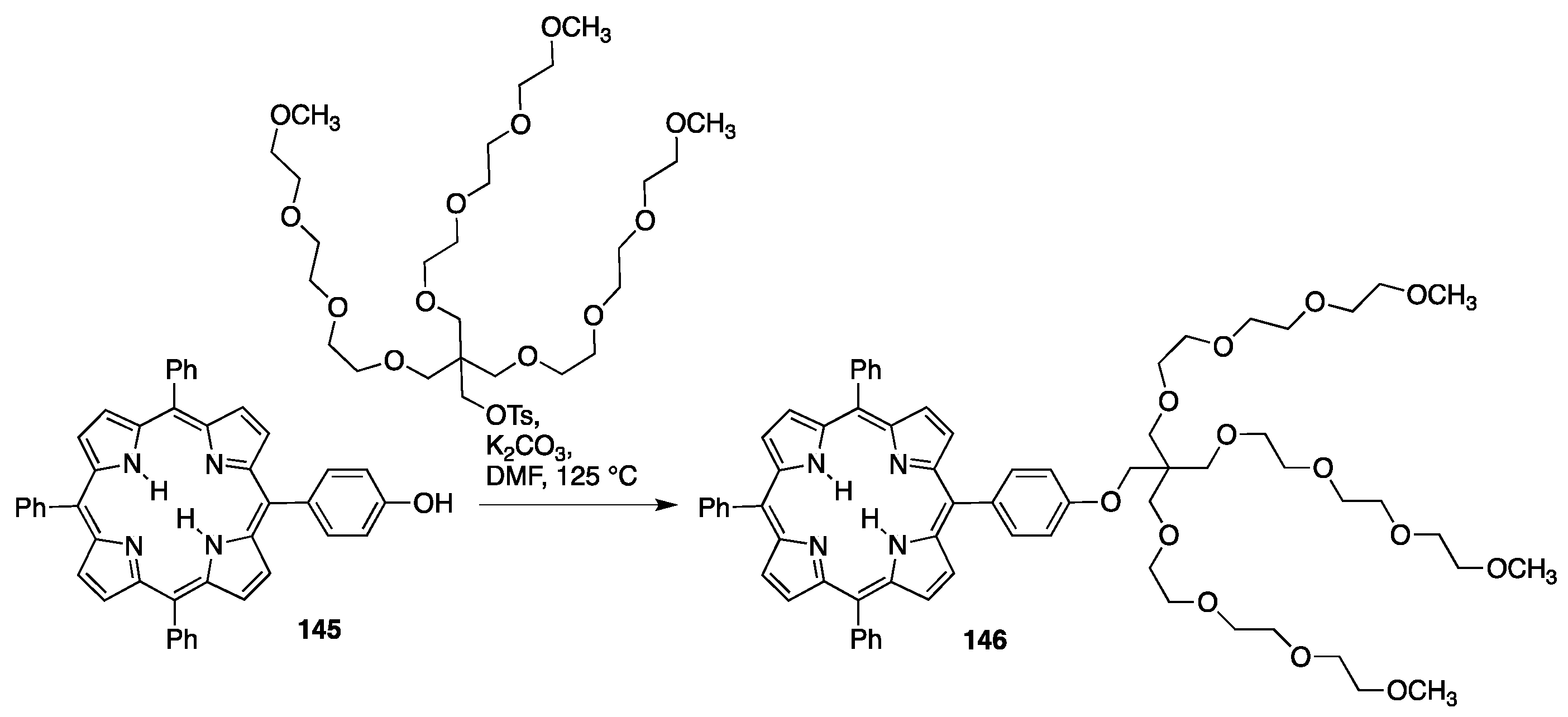{kind=link}
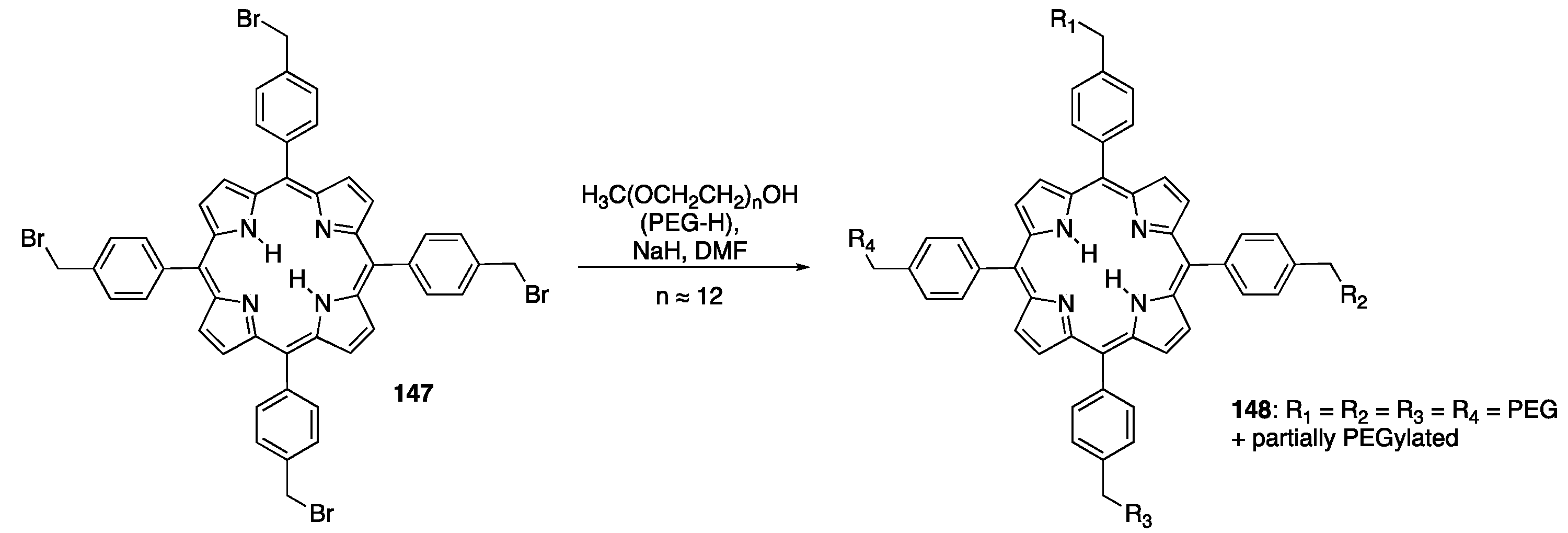{kind=link}
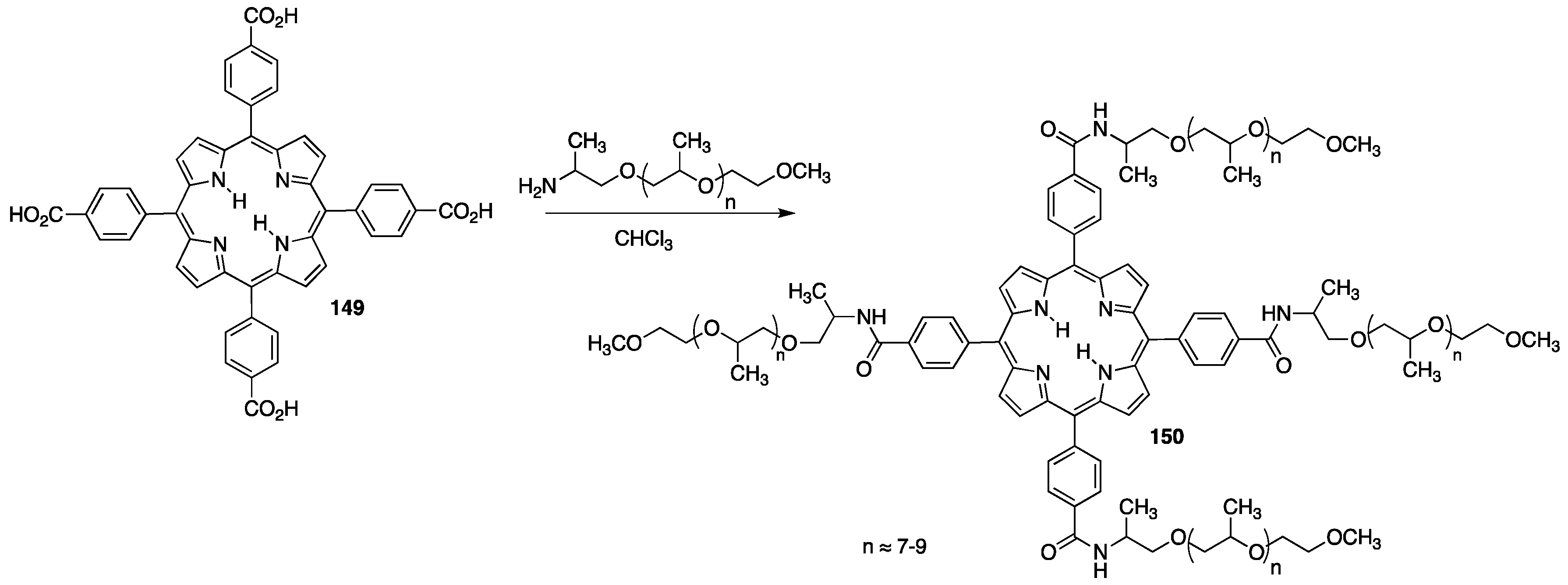{kind=link}
{kind=link}
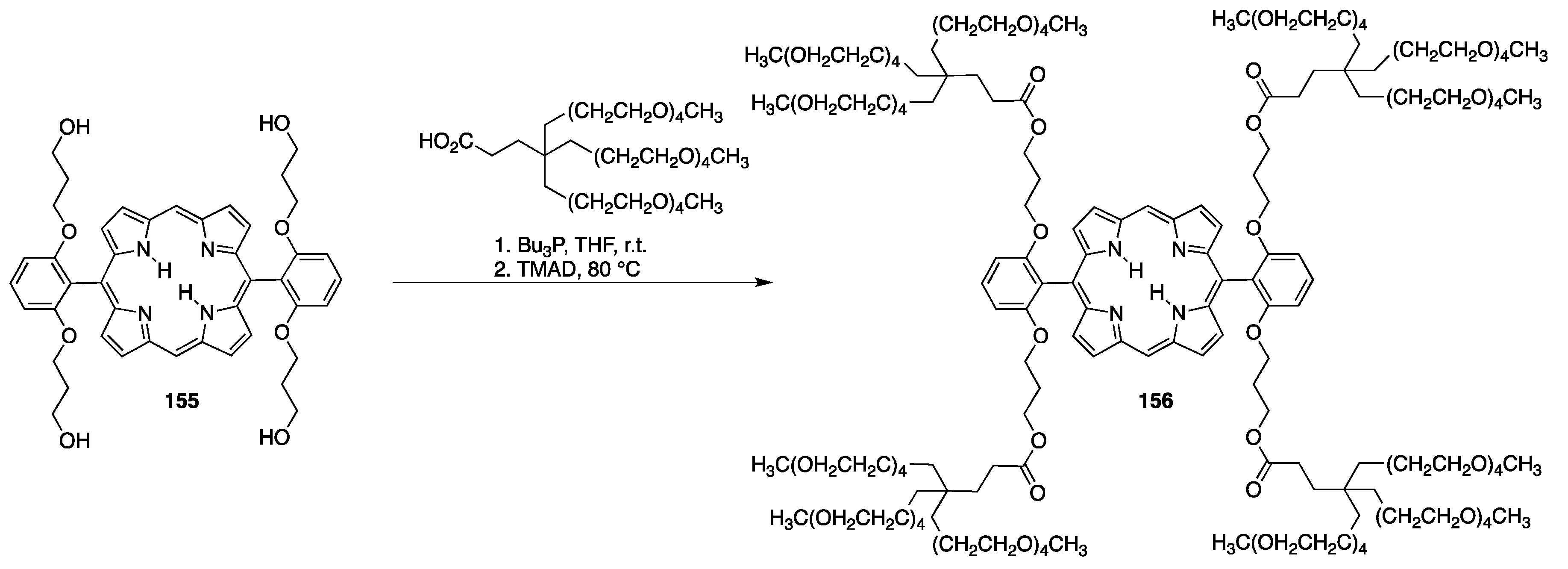{kind=link}
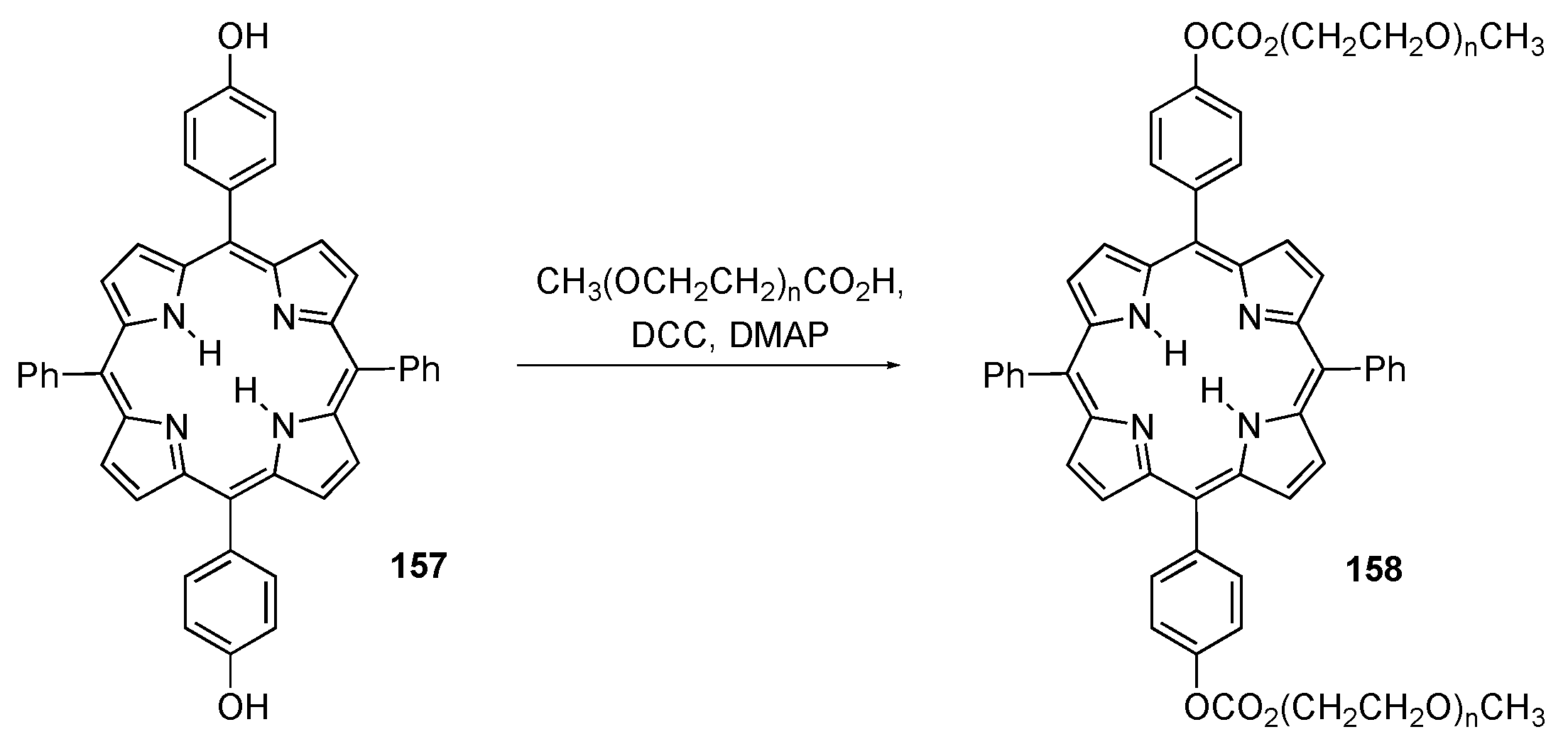{kind=link}
{kind=link}
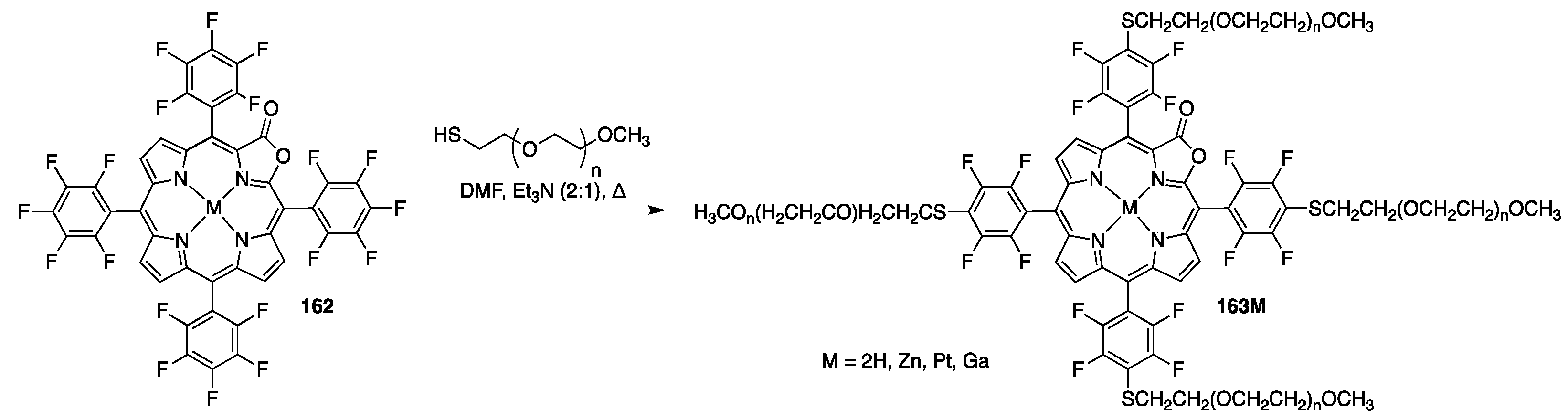{kind=link}
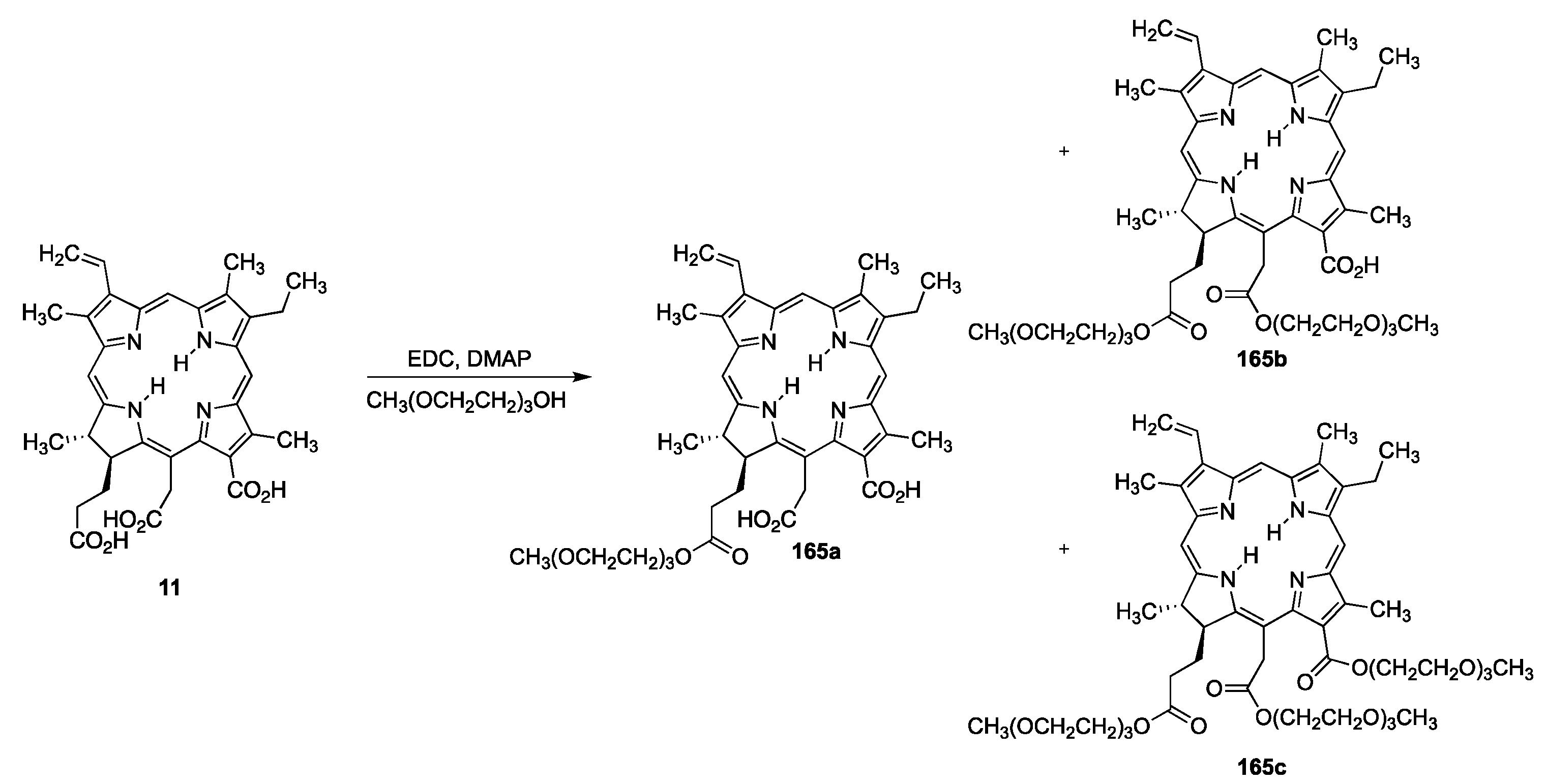{kind=link}
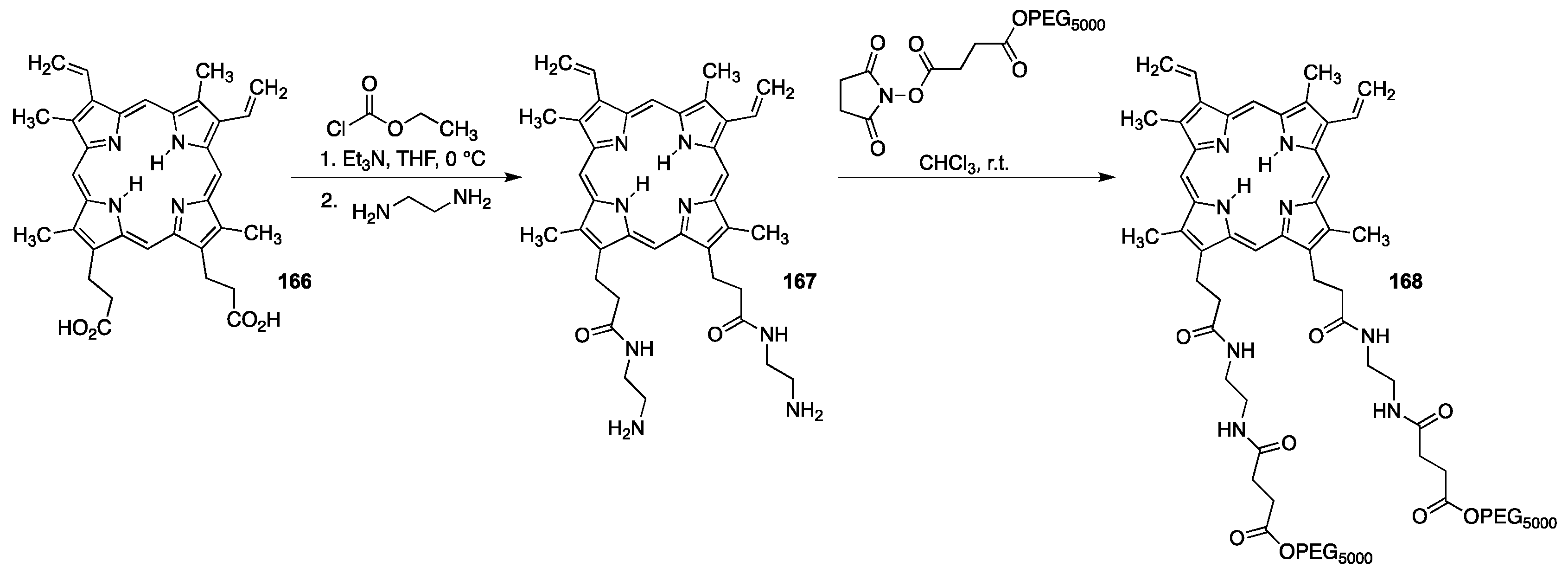{kind=link}
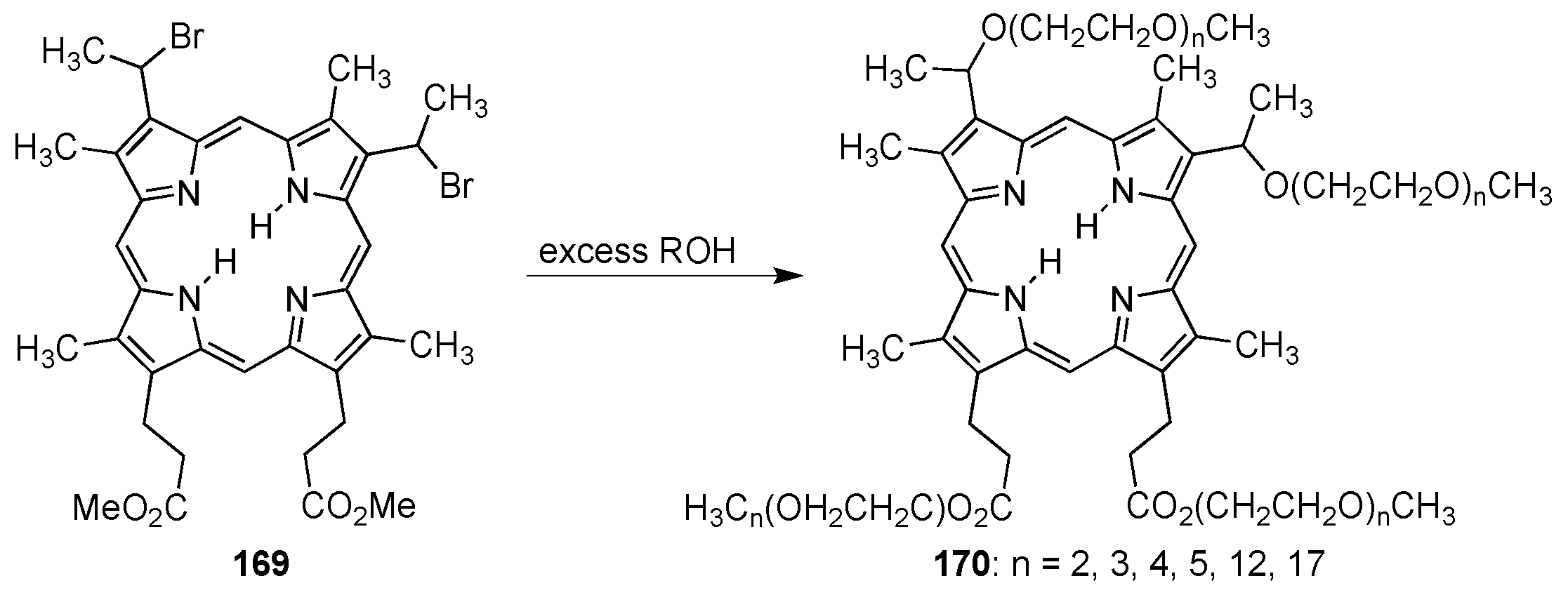{kind=link}
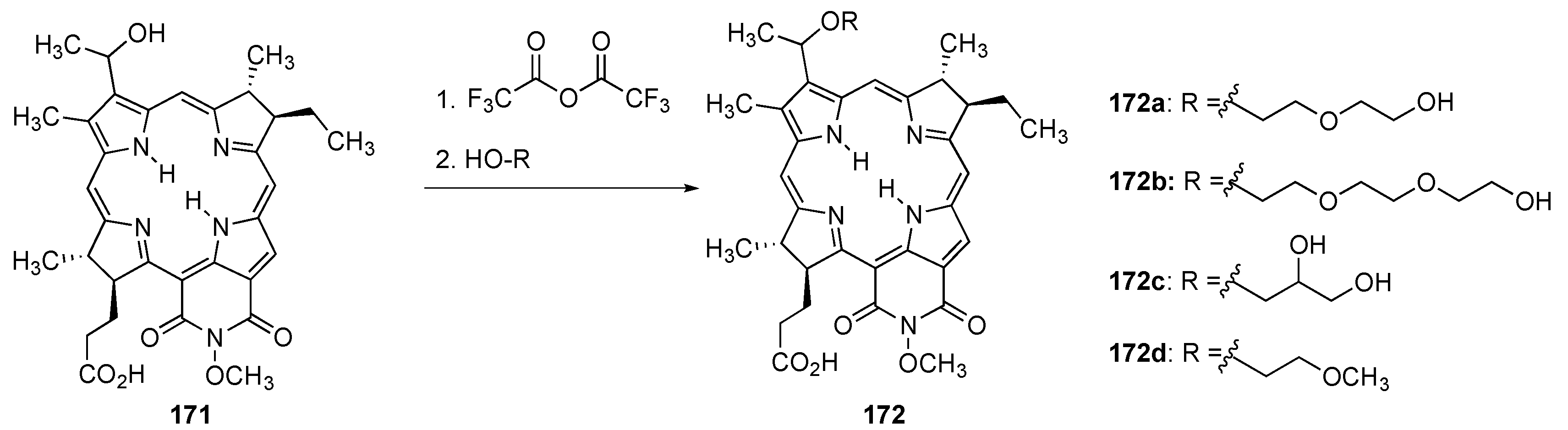{kind=link}
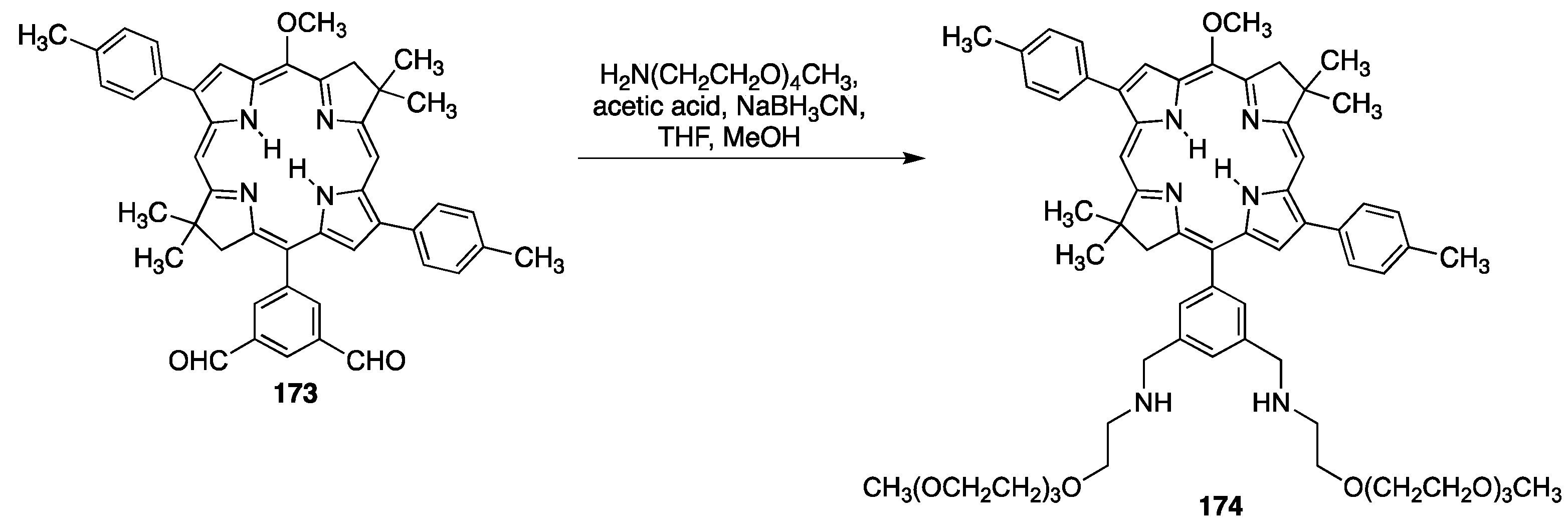{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porphyrin Dimer | Retention Time/min a |
|---|---|
| 51 | 6.2 |
| 29 | 6.8 |
| 135 | 7.6 |
| 136 | 15.6 |
| 88 | 18.0 |
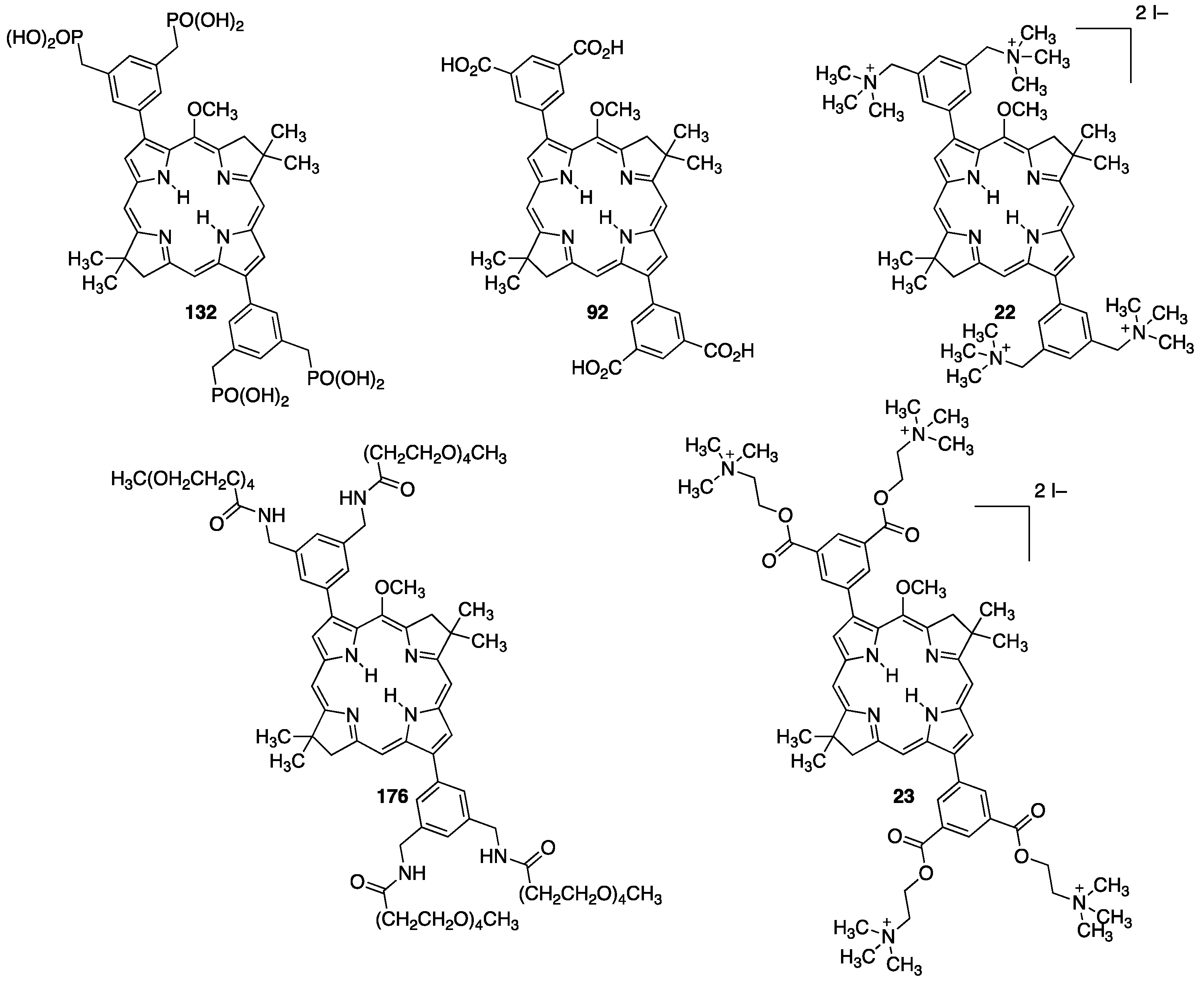
| Compounds | Purification Method a | Water-Solubility b | FWHM (abs, fl) (nm) c | Stability in Light (%) d | Stability in Dark (%) e |
|---|---|---|---|---|---|
| 132 | reverse phase chromatography | 0.46 μM–460 μM | 26, 30 | 85 | >95 |
| 92 | ppt, washing with hexanes/MeOH (49:1) | 0.32 μM–320 μM | 26, 26 | 96 | >95 |
| 22 | ppt, washing with Et2O/THF (1:1) | 0.24 μM–240 μM | 26, 26 | 82 | >95 |
| 23 | ppt.,washing with Et2O/THF (1:1) | 0.18 μM–180 μM | 40, 26 | 96 | >95 |
| 176 | ppt, washing with hexanes/CH2Cl2 (19:1) | 0.43 μM–430 μM | 22, 23 | 95 | >95 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luciano, M.; Brückner, C. Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media. Molecules 2017, 22, 980. https://doi.org/10.3390/molecules22060980
Luciano M, Brückner C. Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media. Molecules. 2017; 22(6):980. https://doi.org/10.3390/molecules22060980
Chicago/Turabian StyleLuciano, Michael, and Christian Brückner. 2017. "Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media" Molecules 22, no. 6: 980. https://doi.org/10.3390/molecules22060980
APA StyleLuciano, M., & Brückner, C. (2017). Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media. Molecules, 22(6), 980. https://doi.org/10.3390/molecules22060980