
The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequencing and Assembly
2.2. Genome Annotation and Comparative Analysis
2.3. Repeat Analysis
2.4. Phylogenic and Divergence Analysis
3. Results
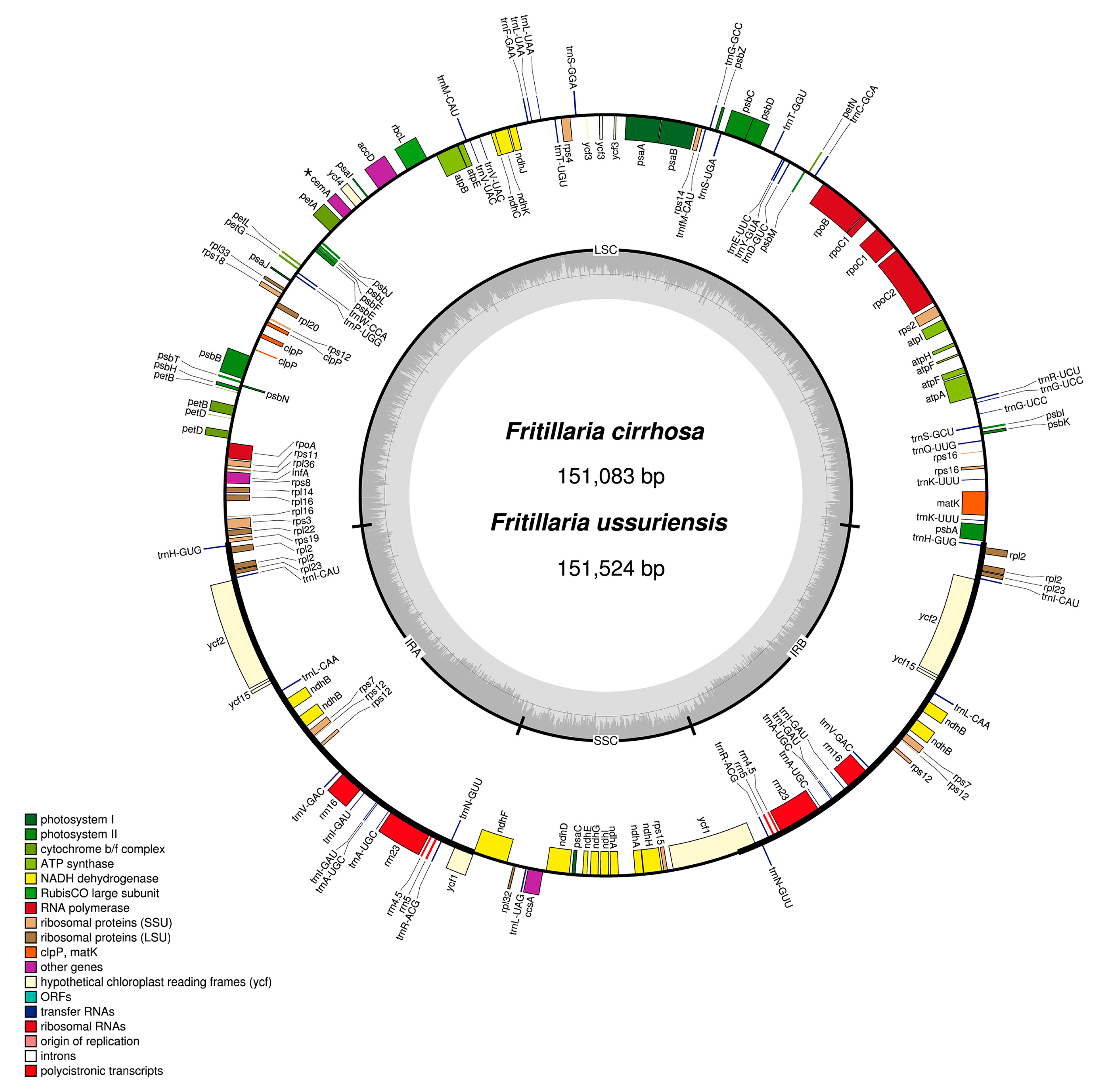
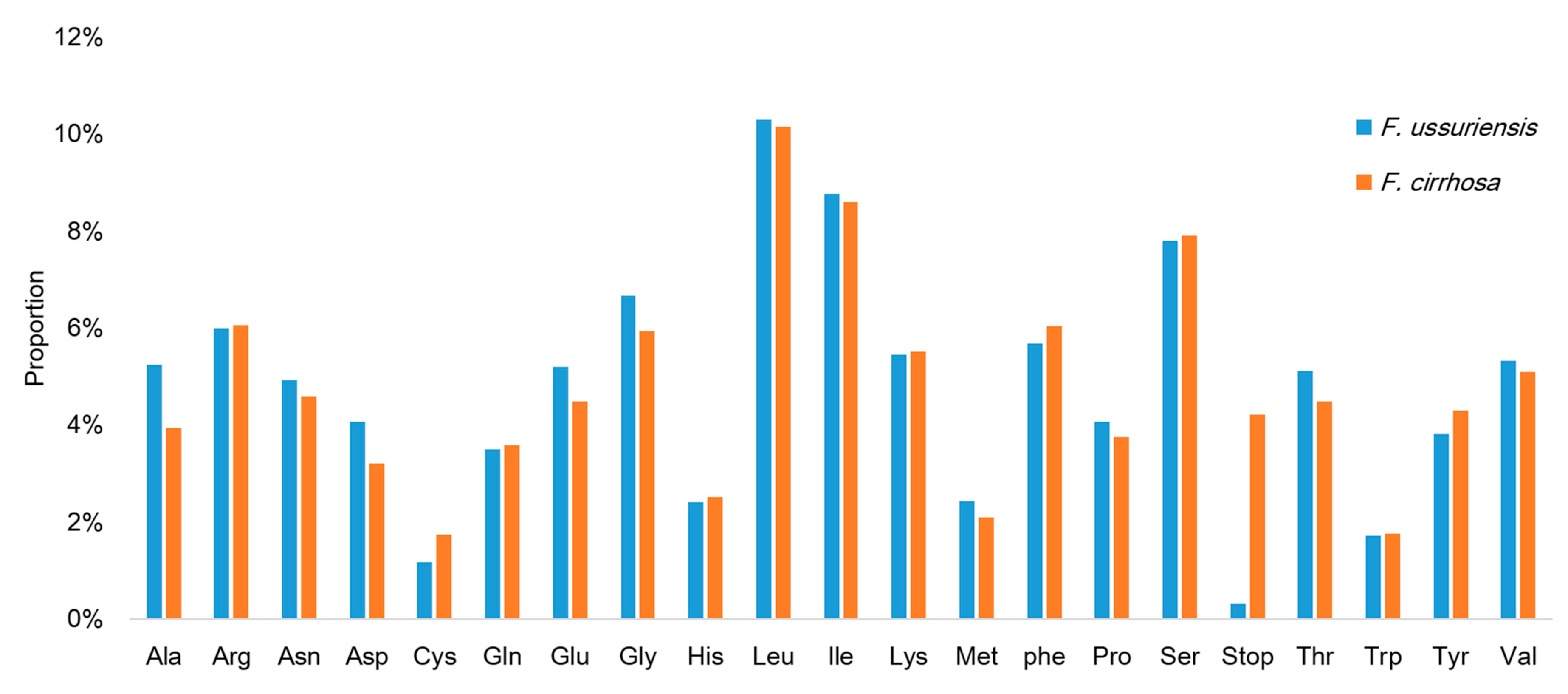
3.1. Chloroplast Genome Organization of Two Fritillaria Species
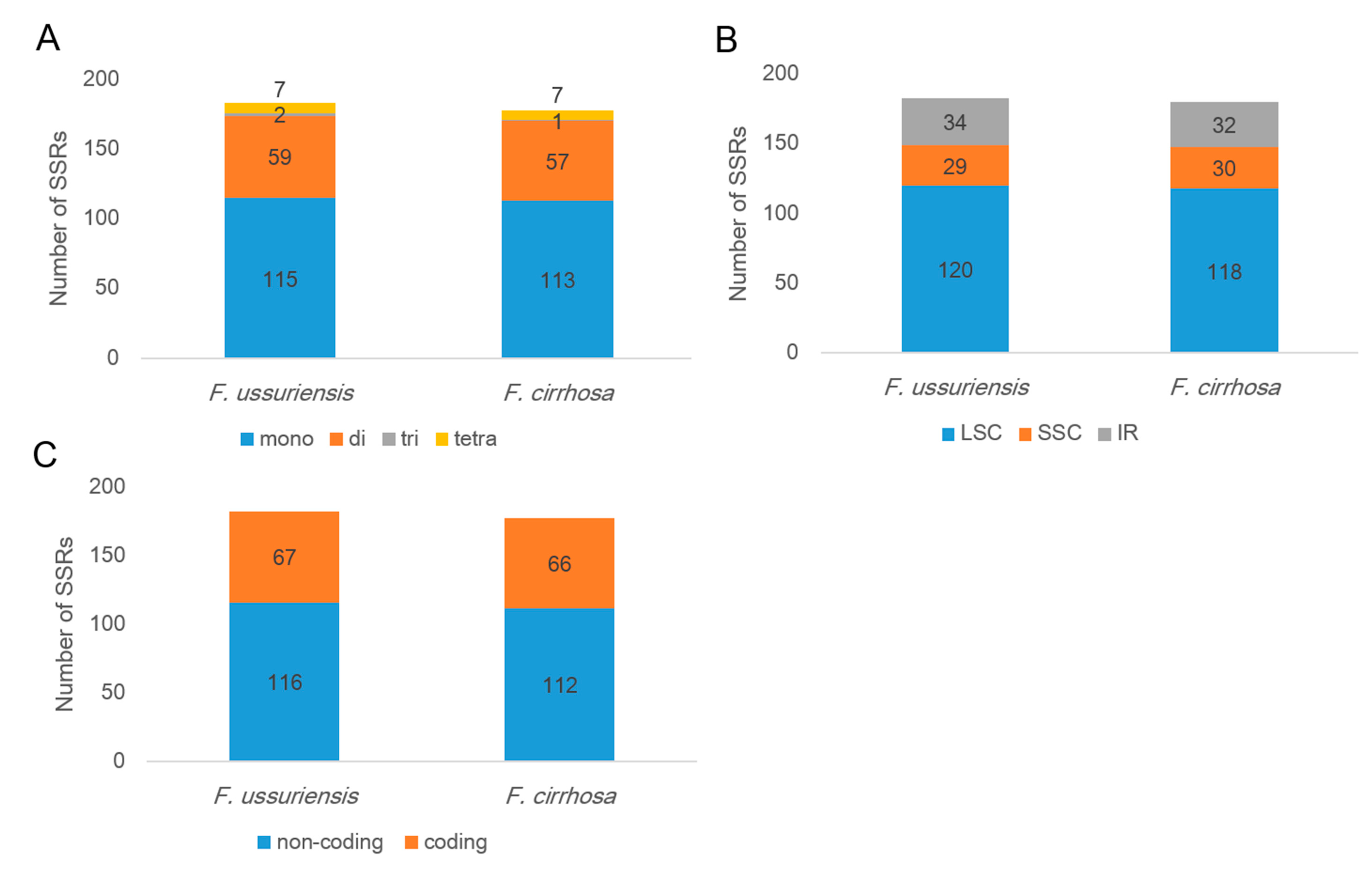
3.2. Repeat Analysis in Two Fritillaria Chloroplast Genomes
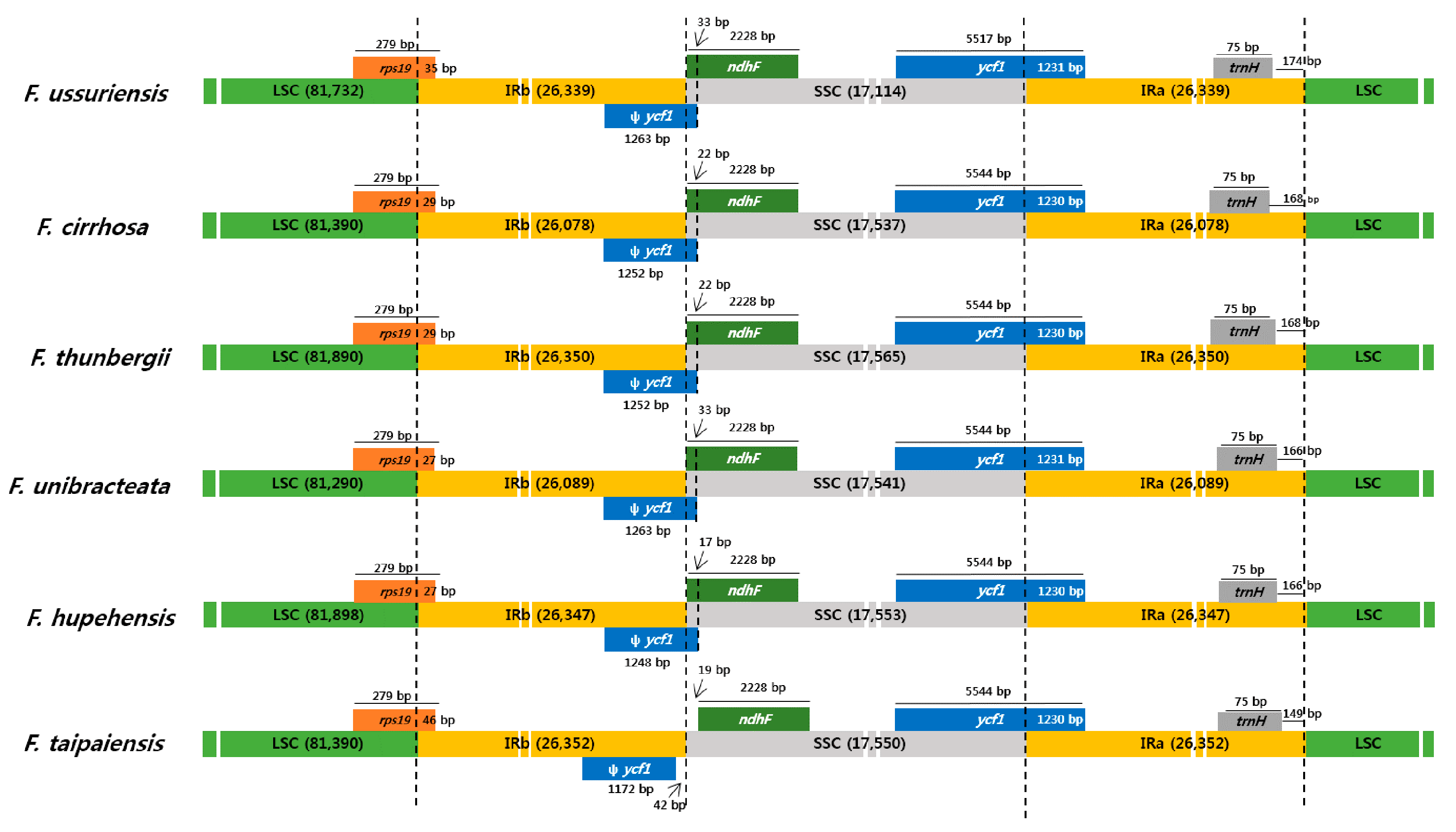
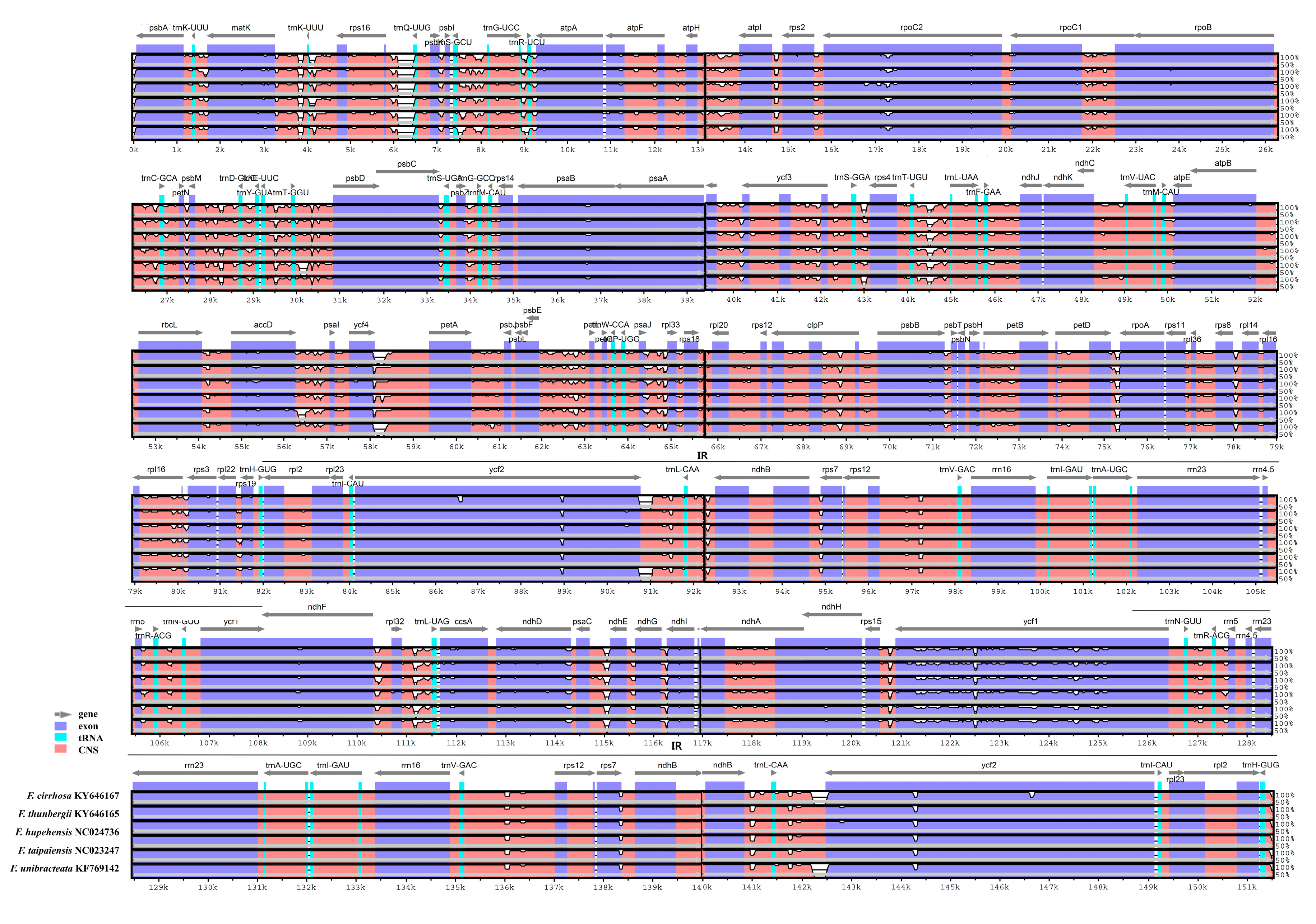
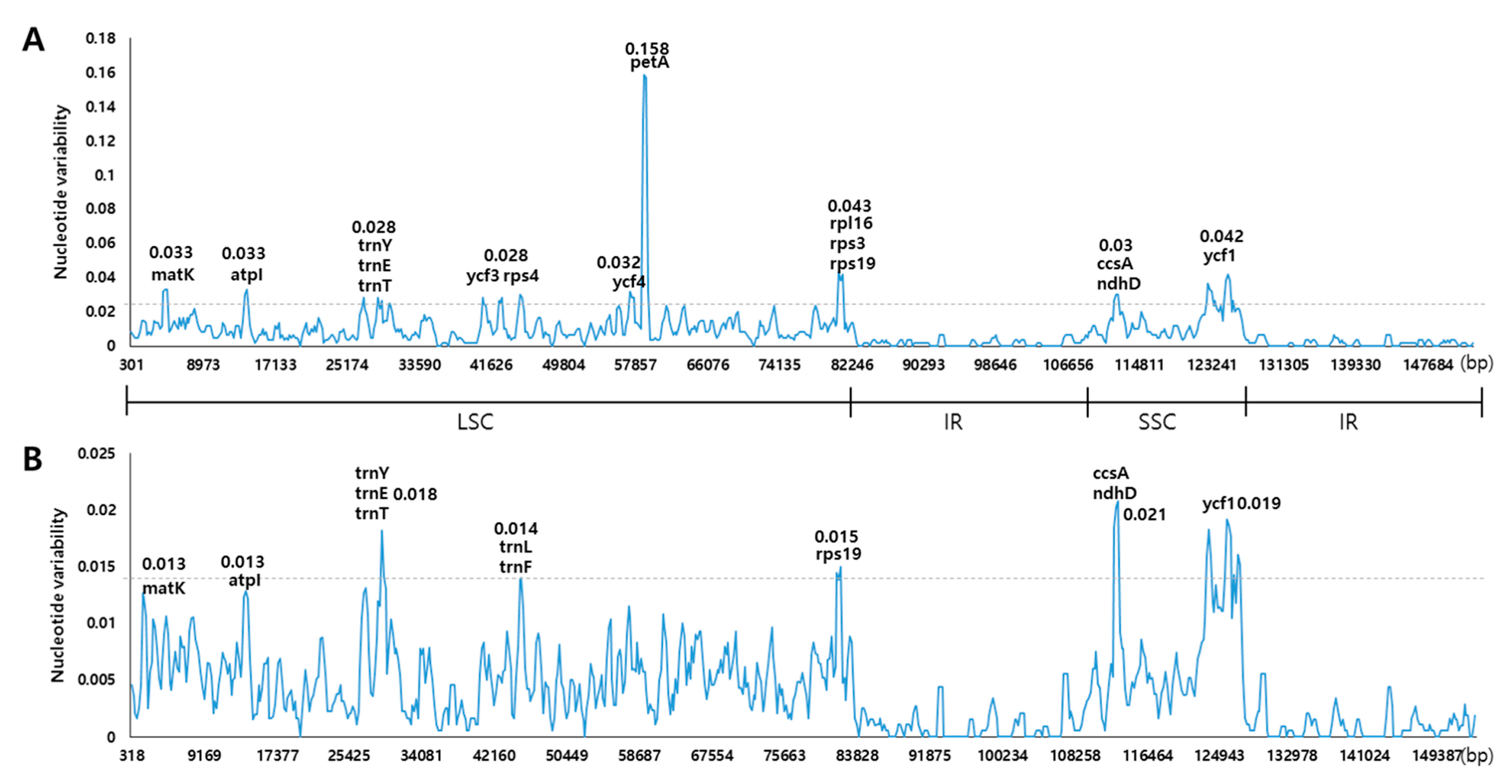
3.3. Comparison of the Chloroplast Genomes with Those of Other Fritillaria Species
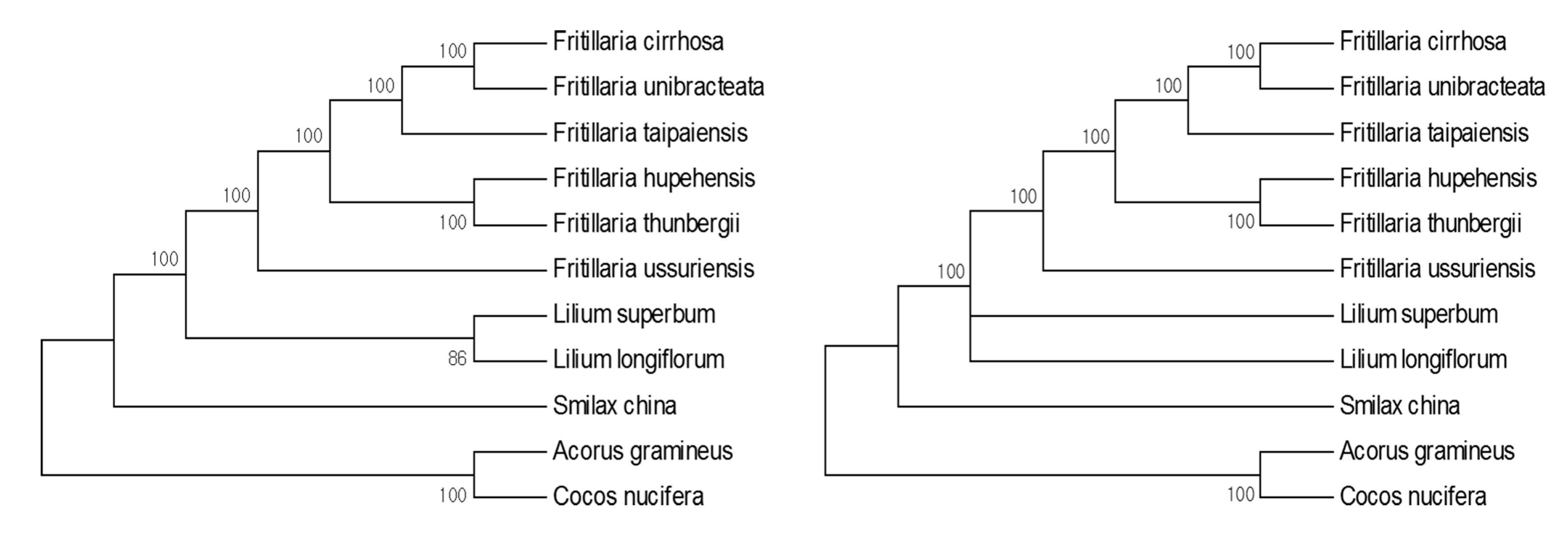
3.4. Phylogenic Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CP | Chloroplast |
| LSC | Large Single Copy |
| SSC | Small Single Copy |
| IR | Inverted Repeat |
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
| NGS | Next-Generation Sequencing |
| KIOM | Korea Institute of Oriental Medicine |
| ML | Maximum Likelihood |
| MP | Maximum Parsimony |
| WGS | Whole-Genome Sequencing |
| IRb | Inverted Repeat b |
| SSRs | Simple Sequence Repeats |
| SNPs | Single-Nucleotide Polymorphisms |
References
- Day, P.D.; Berger, M.; Hill, L.; Fay, M.F.; Leitch, A.R.; Leitch, I.J.; Kelly, L.J. Evolutionary relationships in the medicinally important genus Fritillaria L. (liliaceae). Mol. Phylogenet. Evol. 2014, 80, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Xingi, C.; Mordak, H. Tulipa L. In Flora China Flagellariaceae Marantaceae; Science Press: Beijing, China, 2000; pp. 123–136. [Google Scholar]
- The State Pharmacopoeia Commission of the People’s Republic of China. Pharmacopoeia of the People’s Republic of China, 2010 ed.; Chemical Industry Press: Beijing, China, 2010; Volume 1, p. 191. [Google Scholar]
- Korea Institute of Oriental Medicine (KIOM). Defining Dictionary for Medicinal Herbs [Korean, ‘hanyak giwon sajeon’]. 2016. Available online: http://boncho.kiom.re.kr/codex/ (accessed on 15 March 2017).
- Lee, S.; Ju, Y. Identification of 11 species of paemo through each original plant and medicines. Korea J. Herbol. 2014, 29, 133–140. [Google Scholar] [CrossRef]
- Xiang, L.; Su, Y.; Li, X.; Xue, G.; Wang, Q.; Shi, J.; Wang, L.; Chen, S. Identification of Fritillariae bulbus from adulterants using ITS2 regions. Plant Gene 2016, 7, 42–49. [Google Scholar] [CrossRef]
- Xin, G.Z.; Lam, Y.C.; Maiwulanjiang, M.; Chan, G.K.; Zhu, K.Y.; Tang, W.L.; Dong, T.T.; Shi, Z.Q.; Li, P.; Tsim, K.W. Authentication of Bulbus Fritillariae Cirrhosae by RAPD-derived DNA markers. Molecules 2014, 19, 3450–3459. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare iii. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Muller, K.F.; de Pamphilis, C.W.; Quandt, D.; Wickett, N.J.; Zhang, Y.; Renner, S.S.; Schneeweiss, G.M. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the Broomrape family. Plant Cell 2013, 25, 3711–3725. [Google Scholar] [CrossRef] [PubMed]
- Umesono, K.; Inokuchi, H.; Ohyama, K.; Ozeki, H. Nucleotide sequence of Marchantia polymorpha chloroplast DNA: A region possibly encoding three trnas and three proteins including a homologue of E. coli ribosomal protein S14. Nucleic Acids Res. 1984, 12, 9551–9565. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Rapp, B.A.; Wheeler, D.L. Genbank. Nucleic Acids Res. 2000, 28, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Yun, B.K.; Yoon, Y.H.; Hong, S.Y.; Mekapogu, M.; Kim, K.H.; Yang, T.J. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 2015, 10, e0125332. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using mummer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Song, J.; Xu, H.; Xu, J.; Zhu, Y.; Li, X.; Gao, H.; Dong, L.; Qian, J.; et al. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 2014, 204, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. Trnascan-se: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. Vista: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T. Misa—Microsatellite Identification Tool. 2003. Available online: http://pgrc.ipk-gatersleben. de/misa/ (accessed on 15 March 2017).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Warburton, P.E.; Giordano, J.; Cheung, F.; Gelfand, Y.; Benson, G. Inverted repeat structure of the human genome: The X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004, 14, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. Mafft: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acid Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Matzke, A.J.; Matzke, M. Complete sequence and comparative analysis of the chloroplast genome of Coconut palm (Cocos nucifera). PLoS ONE 2013, 8, e74736. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.Y.; Lin, Z.; Li, G.; Wang, Y.Y.; Qiu, J.; Fu, F.; Zhang, H.; Chen, L.; Ye, S.; Song, W.; et al. Echinochloa chloroplast genomes: Insights into the evolution and taxonomic identification of two weedy species. PLoS ONE 2014, 9, e113657. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puerta, M.V.; Abbona, C.C. The chloroplast genome of Hyoscyamus niger and a phylogenetic study of the tribe Hyoscyameae (Solanaceae). PLoS ONE 2014, 9, e98353. [Google Scholar] [CrossRef] [PubMed]
- Kahlau, S.; Aspinall, S.; Gray, J.C.; Bock, R. Sequence of the Tomato chloroplast DNA and evolutionary comparison of Solanaceous plastid genomes. J. Mol. Evol. 2006, 63, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Yukawa, Y.; Miyamoto, T.; Obokata, J.; Sugiura, M. Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of Tobacco (Nicotiana tabacum): Comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol. Biol. Evol. 2003, 20, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yi, X.; Yang, Y.X.; Su, Y.J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Do, H.D.; Kim, J.S.; Kim, J.H. A trnI_CAU triplication event in the complete chloroplast genome of paris verticillata m.Bieb. (melanthiaceae, liliales). BMC Evol. Biol. 2014, 6, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6, 447. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xue, Q. Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 2005, 84, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, H.-L. Complete chloroplast genome sequences from Korean ginseng (panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Kim, J.; Lee, J.; Kim, S.; Cho, O.; Yang, K.; Ahn, J.; Nahm, S.; Kim, H. Development of SSR markers by next-generation sequencing of Korean landraces of chamoe (Cucumis melo var. makuwa). Mol. Bio. Rep. 2013, 40, 6855–6862. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dong, W.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 9040. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Dhingra, A.; Soltis, P.S.; Shaw, R.; Farmerie, W.G.; Folta, K.M.; Soltis, D.E. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2006, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.H.; Park, H.S.; Yang, T.J. Comprehensive survey of genetic diversity in chloroplast genomes and 45s nrDNAs within panax ginseng species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef] [PubMed]
- Garaycochea, S.; Speranza, P.; Alvarez-Valin, F. A strategy to recover a high-quality, complete plastid sequence from low-coverage whole-genome sequencing. Appl. Plant Sci. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.C.; Cooper, J.D.; Mian, M.A.; Chekhovskiy, K.; May, G.D. Tall fescue genomic SSR markers: Development and transferability across multiple grass species. Theor. Appl. Genet. 2006, 113, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.Y.; Rohner, T.; Greenfield, A.; Van Der Merwe, M.; McPherson, H.; Glenn, W.; Kornfeld, G.; Marendy, E.; Pan, A.Y.; Wilton, A.; et al. Complete chloroplast genome of the Wollemi pine (Wollemia nobilis): Structure and evolution. PLoS ONE 2015, 10, e0128126. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhao, Z.; Dorje, G.; Ma, M. The complete chloroplast genome of Ye-Xing-Ba (Scrophularia dentata; Scrophulariaceae), an alpine Tibetan herb. PLoS ONE 2016, 11, e0158488. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Yang, J. The complete chloroplast genome sequence of Morus mongolica and a comparative analysis within the Fabidae clade. Curr. Genet. 2016, 62, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.C.; Yang, T.J.; et al. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Arif, I.A.; Bakir, M.A.; Khan, H.A.; Al Farhan, A.H.; Al Homaidan, A.A.; Bahkali, A.H.; Sadoon, M.A.; Shobrak, M. A brief review of molecular techniques to assess plant diversity. Int. J. Mol. Sci. 2010, 11, 2079–2096. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; Munhoz, C.D.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Moose, S.P.; Mumm, R.H. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, Crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Jheng, C.F.; Chen, T.C.; Lin, J.Y.; Chen, T.C.; Wu, W.L.; Chang, C.C. The comparative chloroplast genomic analysis of photosynthetic orchids and developing DNA markers to distinguish Phalaenopsis orchids. Plant Sci. 2012, 190, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Q.; Li, X.; Song, J.; Sun, C. Complete chloroplast genome sequence of Fritillaria unibracteata var. wabuensis based on SMRT sequencing technology. Mitochondrial DNA 2016, 27, 3757–3758. [Google Scholar] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Fritillaria ussuriensis | Fritillaria cirrhosa |
|---|---|---|
| Total CP genome size (bp) | 151,524 | 151,083 |
| LSC (bp) | 81,732 | 81,390 |
| IR (bp) | 52,678 | 52,156 |
| SSC (bp) | 17,114 | 17,537 |
| GC content (%) | 36.95 | 36.96 |
| LSC (%) | 34.71 | 34.79 |
| IR (%) | 42.40 | 42.60 |
| SSC (%) | 30.63 | 30.50 |
| Total number of genes | 111 | 112 |
| Protein-coding gene | 77 | 78 |
| rRNA | 4 | 4 |
| tRNA | 30 | 30 |
| Gene Products of Two Fritillaria Species | |
|---|---|
| Photosystem I | psaA, B, C, I, J |
| Photosystem II | psbA, B, C, D, E, F, H, I, J, K, L, M, N, T, Z |
| Cytochrome b6/f | petA, B (1), D (1), G, L, N |
| ATP synthase | atpA, B, E, F (1), H, I |
| Rubisco | rbcL |
| NADH oxidoreductase | ndhA (1), B (1,3), C, D, E, F, G, H, I, J, K |
| Large subunit ribosomal proteins | rpl2 (1,3), 14, 16 (1), 20, 22, 23 (3), 32, 33, 36 |
| Small subunit ribosomal proteins | rps2, 3, 4, 73 (3), 8, 11, 12 (2–4), 14, 15, 16, 18, 19 |
| RNA polymerase | rpoA, B, C1 (1), C2 |
| Unknown function protein-coding gene | ycf1 (3), 2 (3), 3 (2), 4 |
| Other genes | accD, ccsA, cemA (5), clpP (2), matK |
| Ribosomal RNAs | rrn16 (3), 23 (3), 4.5 (3), 5 (3) |
| Transfer RNAs | trnA-UGC (1,3), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-UCC (1), trnG-GCC, trnH-GUG, trnI-CAU (3), trnI-GAU (1,3), trnK-UUU1 (3), trnL-UAA1 (3), trnL-UAG, trnL-CAA (3), trnM-CAU, trnfM-CAU, trnN-GUU (3), trnP-UGG, trnQ-UUG, trnR-ACG (3), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-UAC (1), trnV-GAC (3), trnW-CCA, trnY-GUA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, I.; Kim, W.J.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B.C. The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species. Molecules 2017, 22, 982. https://doi.org/10.3390/molecules22060982
Park I, Kim WJ, Yeo S-M, Choi G, Kang Y-M, Piao R, Moon BC. The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species. Molecules. 2017; 22(6):982. https://doi.org/10.3390/molecules22060982
Chicago/Turabian StylePark, Inkyu, Wook Jin Kim, Sang-Min Yeo, Goya Choi, Young-Min Kang, Renzhe Piao, and Byeong Cheol Moon. 2017. "The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species" Molecules 22, no. 6: 982. https://doi.org/10.3390/molecules22060982
APA StylePark, I., Kim, W. J., Yeo, S.-M., Choi, G., Kang, Y.-M., Piao, R., & Moon, B. C. (2017). The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species. Molecules, 22(6), 982. https://doi.org/10.3390/molecules22060982