2. Results and Discussion
During the course of our continuous studies on bioactive constituents from a 95% EtOH eluate of D101 CC and CHCl
3 layer [
4,
5,
6], obtained from the whole plants of
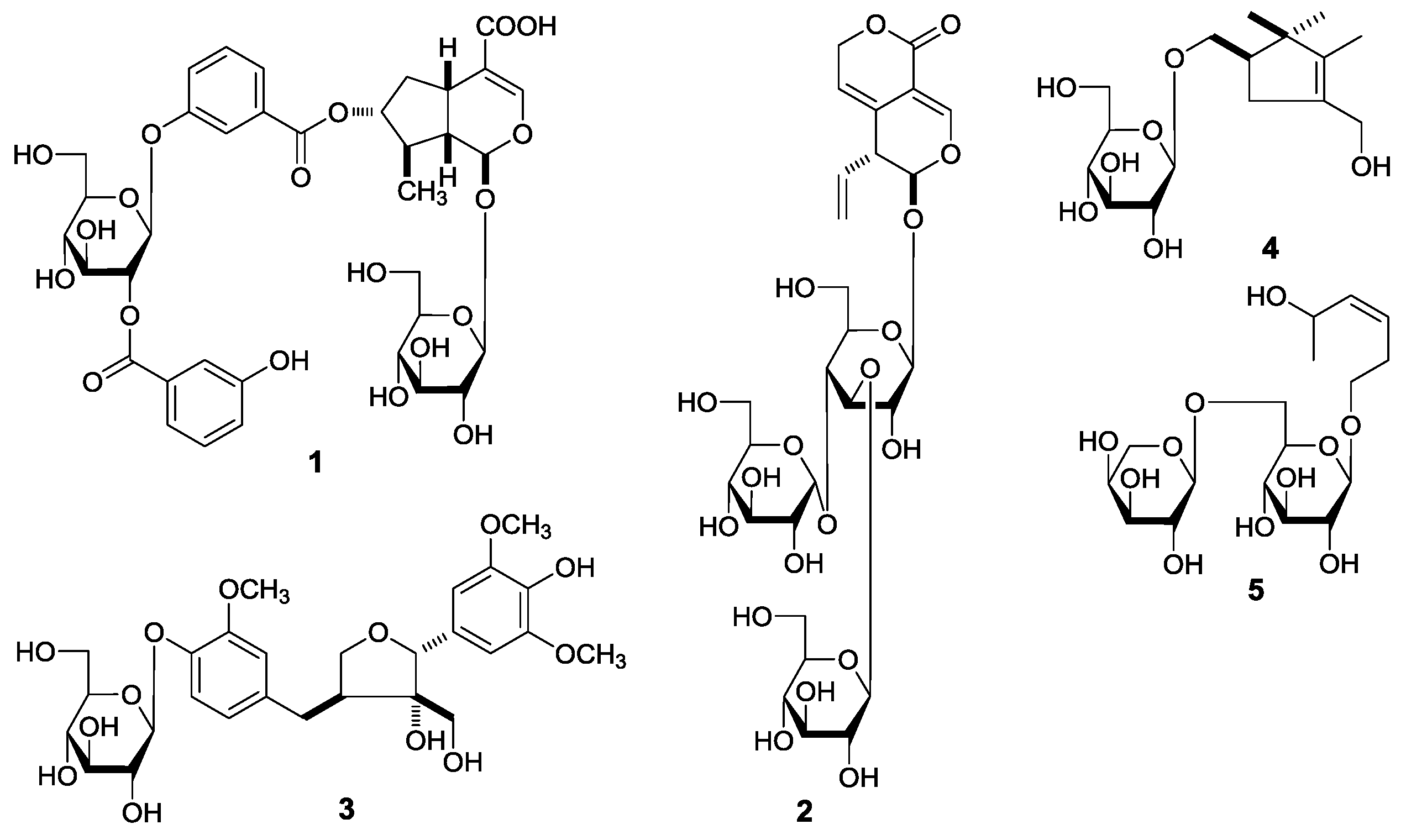
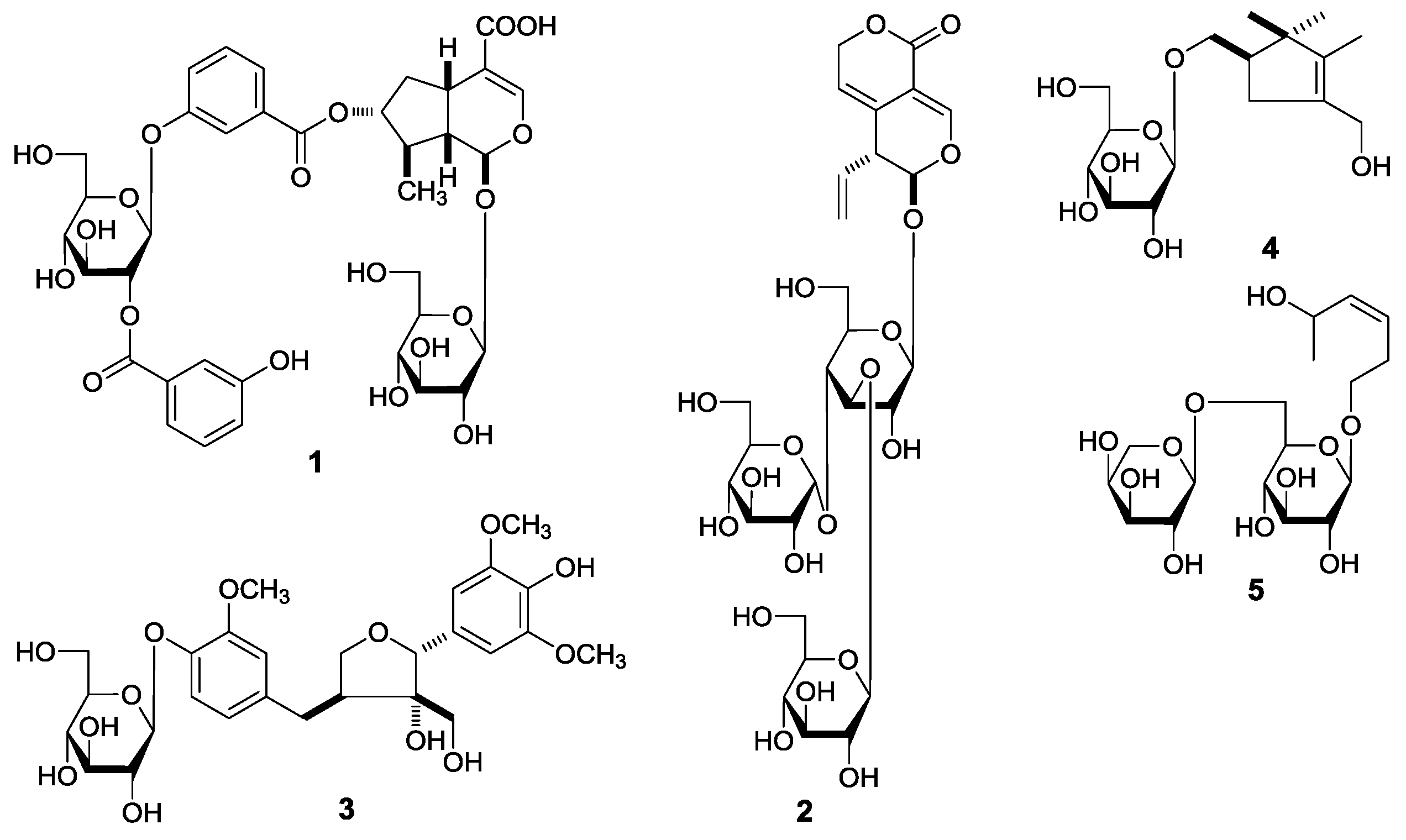
G. acuta, nineteen constituents, including five new compounds, named as gentiiridosides A (
2), B (
2), gentilignanoside A (
3), (1
R)-2,2,3-trimethyl-4-hydroxymethylcyclopent-3-ene-1-methyl-
O-β-
d-glucopyranoside (
4), and (3
Z)-3-hexene-1,5-diol 1-
O-α-
l-arabinopyranosyl(1→6)-β-
d-glucopyranoside (
5) (
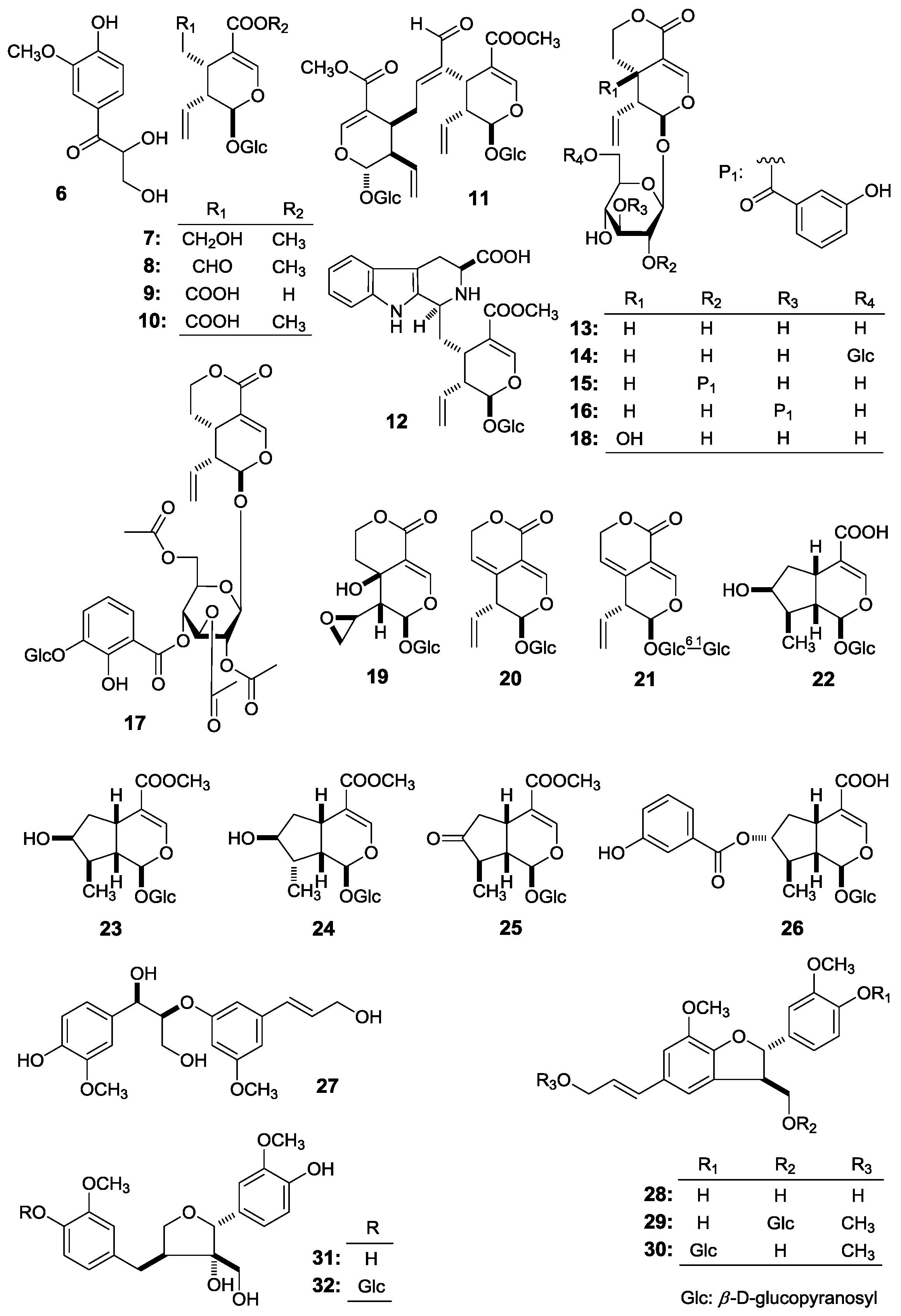
Figure 1), together with fourteen known ones, 2,3-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-propan-1-one (
6) [
7] (
E)-aldosecologanin (
11) [
8], 5α-carboxystrictosidine (
12) [
9], trifloroside (
17) [
10], gentiopicroside (
20) [
11], loganin (
23) [
12], 8-epiloganin (
24) [
13], swertiaside (
26) [
14], (7
R,8
S)-
erythro-4,7,9,9′-tetrahydroxy-3,3′-dimethoxy-8-
O-4′-neolignan (
27) [
15,
16], (7
S,8
R)-dehydrodiconiferyl alcohol (
28) [
17,
18,
19], plucheoside D
3 (
29) [
17,
18,
20], (7
S,8
R)-9′-methoxy-dehydrodiconiferyl alcohol 4-
O-β-
d-glucopyranoside (
30) [
17,
18,
21], (–)-berchemol (
31) [
22,
23], and berchemol-4′-
O-β-
d-glucoside (
32) [
22,
24] (
Figure 2), were further obtained. Among the known isolates,
6,
11,
12,
17,
23, and
27–
32 were isolated from the genus firstly.
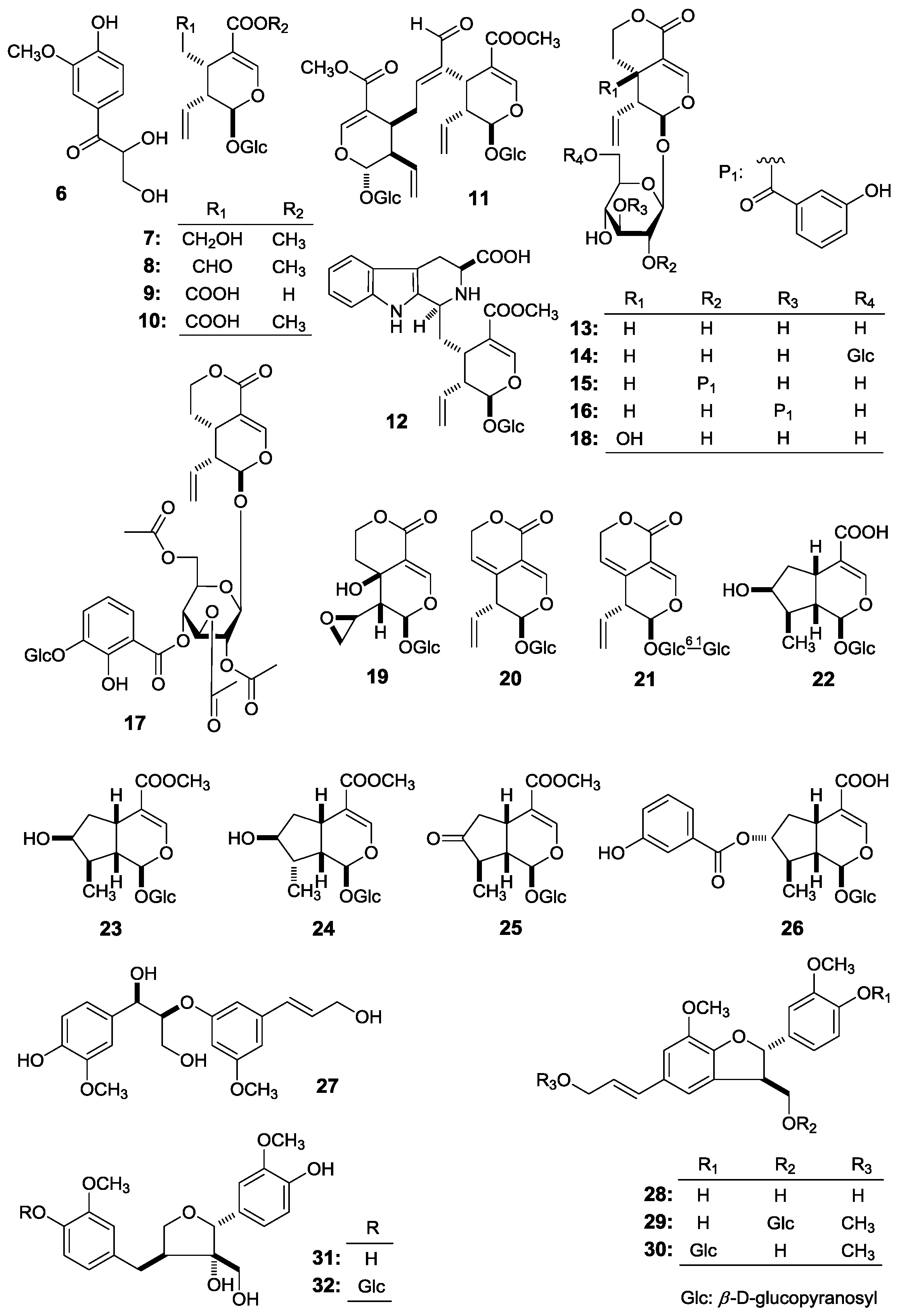
This paper will elucidate the isolation and structure of the new compounds. Meanwhile, the effects of the abovementioned compounds and previously-isolated thirteen iridoid- and secoiridoid-type monoterpenes, secologanol (
7) [
5], secologanin [
6] (
8), secologanoside (
9) [
5], secoxyloganin (
10) [
5], sweroside (
13) [
6], swertiapunimarin (
14) [
5], deacetylcentapicrin (
15) [
6], decentapicrin A (
16) [
6], swertiamarin (
18) [
5], eustomoside (
19) [
5], 6′-
O-β-
d-glucopyranosyl gentiopicroside (
21) [
5], loganic acid (
22) [
6], 7-ketologanin (
25) [
6] (
Figure 2) on the motility of mouse isolated intestine tissue were determined.
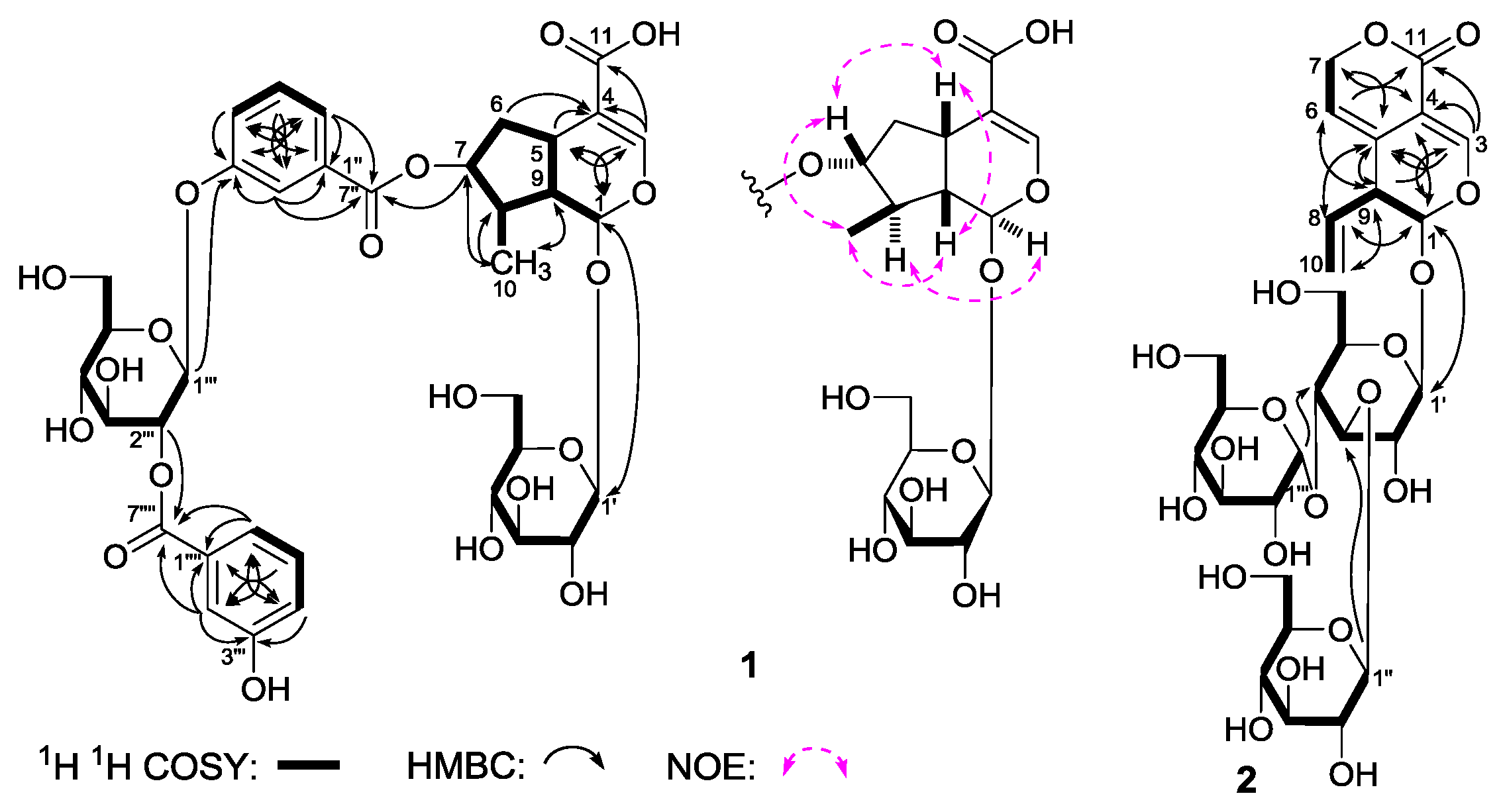
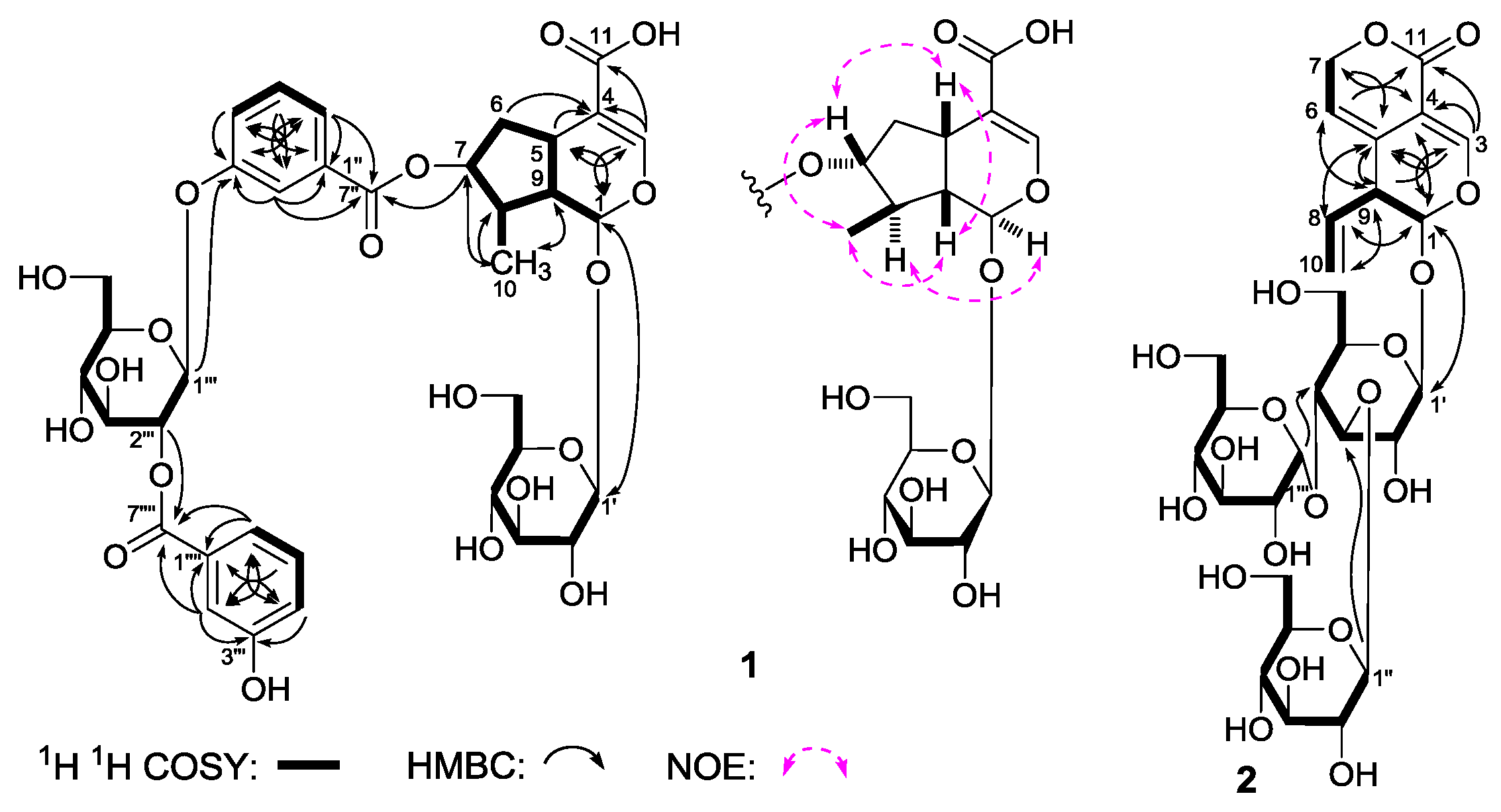
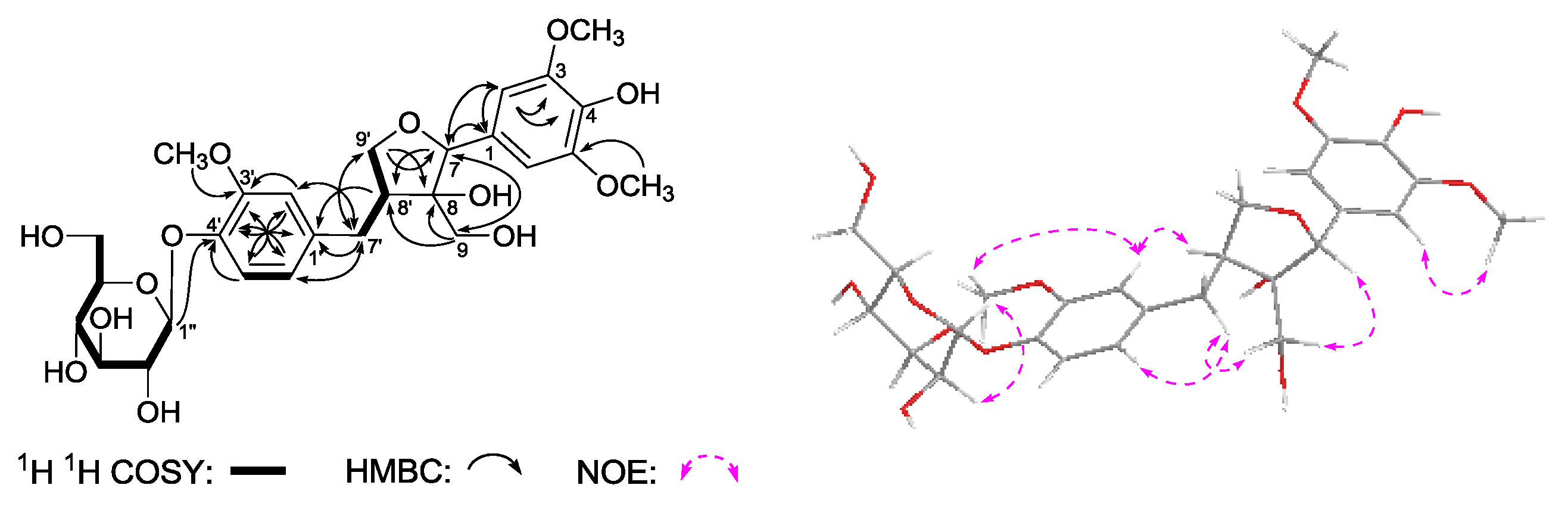
Gentiiridoside A (
1) was isolated as a white powder with negative optical rotation [[α
−90.0° (
c 0.14, MeOH)]. Negative high resolution electrospray ionization-time of flight-mass spectra (HRESI-TOF-MS) afforded [M – H]
− at
m/
z 777.2261 (calcd for C
36H
41O
19, 777.2248), supporting a molecular formula of C
36H
42O
19 for
1. The absorption bands showed in the infrared (IR) spectrum suggested the presence of hydroxyl (3372 cm
−1), α,β-unsaturated carbonyl (1712 cm
−1), aromatic ring (1635, 1588, 1486 cm
−1), and
O-glycosidic linkage (1078 cm
−1). The sugars in
1 were found to be
d-glucose by acid hydrolysis with 1 M HCl [
4]. The
1H,
13C-nuclear magnetic resonance (NMR) spectra (
Table 1) and various two-dimensional (2D) NMR spectra, including
1H
1H chemical-shift correlation spectroscopy (
1H
1H COSY), heteronuclear single quantum correlation (HSQC), heteronuclear multiple bond correlation (HMBC) displayed signals assignable to two 3-hydroxy benzoyl {[δ 7.42 (1H, br. d,
ca.
J = 8 Hz, H-4′′), 7.48 (1H, dd,
J = 7.5, 7.5 Hz, H-5′′), 7.84 (1H, br. d,
ca.
J = 8 Hz, H-6′′), 7.89 (1H, br. s, H-2′′);
δC 166.1 (C-7′′)]; [
δ 7.45 (1H, br. d,
ca.
J = 8 Hz, H-4′′′′), 7.54 (1H, dd,
J = 7.5, 7.5 Hz, H-5′′′′), 7.82 (1H, br. s, H-2′′′′), 7.88 (1H, br. d,
ca.
J = 8 Hz, H-6′′′′);
δC 166.8 (C-7′′′′)]}, and two β-
d-glucopyranosyl [δ 4.69 (1H, d,
J = 7.5 Hz, H-1′), 5.02 (1H, d,
J = 8.0 Hz, H-1′′′)]. On the other hand, thirty-six carbon signals were shown in its
13C-NMR spectrum, in addition to twenty-six carbon signals occupied by the abovementioned fragments, the other 10 carbon signals, together with the relative proton signals [δ 5.50 (1H, d,
J = 3.0 Hz, H-1), 7.46 (1H, s, H-3)] indicated the aglycon of
1 was iridoid. As shown in
Figure 3, the
1H
1H COSY experiment on
1 suggested the existence of five partial structures. Furthermore, in the HMBC experiment, long-range correlations from δ
H 5.50 (H-1) to δ
C 152.4 (C-3); δ
H 7.46 (H-3) to δ
C 32.5 (C-5), 96.2 (C-1), 112.8 (C-4), 170.9 (C-11); δ
H 4.96 (H-7) to δ
C 166.1 (C-7′′); δ
H 4.69 (H-1′) to δ
C 96.2 (C-1); δ
H 7.89 (H-2′′), 7.84 (H-6′′) to δ
C 166.1 (C-7′′); δ
H 5.02 (H-1′′′) to δ
C 159.2 (C-3′′); δ
H 3.51 (H-2′′′) to δ
C 166.8 (C-7′′′′); δ
H 7.82 (H-2′′′′), 7.88 (H-6′′′′) to δ
C 166.8 (C-7′′′′) were observed. Therefore, the planar structure of
1 was constructed. The relative configuration of
1 was determined by a nuclear Overhauser effect spectroscopy (NOESY) experiment, and NOE correlations were observed between H-1 and H-8; H-5 and H-7; H
3-10 and H-7, H-9. The
1H and
13C-NMR sepctra of
1 were found very similar to those of swertiaside (
26) [
14], except that a 2-(3-hydroxybenzoyl)-β-
d-glucopyranosyl appeared at the 3′′-position in
1. Consequently, the structure of gentiiridoside A (
1) was determined.
Gentiiridoside B (
2) was obtained as a white powder with negative optical rotation [[α
−36.7° (
c 0.12, MeOH)]. HRESI-TOF-MS exhibited a molecular ion peak at
m/z 679.1023 [M – H]
−, and revealed the molecular formula C
28H
40O
18 (calcd for C
28H
39O
18, 679.1033) for it. Acid hydrolysis of
2 with 1 M HCl afforded
d-glucose, whose absolute configuration was determined by HPLC analysis [
4]. The
1H and
13C-NMR (
Table 2) spectra of
2 indicated the presence of two β-
d-glucopyranosyl [δ 4.27 (1H, d,
J = 7.5 Hz, H-1′′), 4.49 (1H, d,
J = 8.0 Hz, H-1′)], and one α-
d-glucopyranosyl [δ 4.91 (1H, d,
J = 3.5 Hz, H-1′′′)]. Twenty-eight carbon signals were displayed in its
13C-NMR spectrum, except for the above mentiond moieties, the other ten signals as well as their relative
1H-NMR signals [δ 5.21 (2H, m, H
2-10), 5.59 (1H, d,
J = 3.0 Hz, H-1), 5.72 (1H, ddd,
J = 6.5, 10.5, 17.5 Hz, H-8), 7.41 (1H, s, H-3)] suggested the aglycon of
2 was the same as that of gentiopicroside (
20) [
11]. Finally, the long-range correlations from δ
H 5.59 (H-1) to δ
C 124.9 (C-5), 148.8 (C-3); δ
H 7.41 (H-3) to δ
C 96.4 (C-1), 103.2 (C-4), 124.9 (C-5), 162.7 (C-11); δ
H 5.65 (H-6) to δ
C 44.3 (C-9), 103.2 (C-4); δ
H 4.97, 5.04 (H
2-7) to δ
C 124.9 (C-5), 162.7 (C-11); δ
H 5.72 (H-8) to δ
C 96.4 (C-1), 124.9 (C-5); δ
H 3.31 (H-9) to δ
C 103.2 (C-4), 116.1 (C-6), 124.9 (C-5); δ
H 4.49 (H-1′) to δ
C 96.4 (C-1); δ
H 4.27 (H-1′′) to δ
C 76.7 (C-1′); δ
H 4.91 (H-1′′′) to δ
C 69.9 (C-1′′) were observed in the HMBC spectrum. Then, the structure of gentiiridoside B (
2) was clarified.
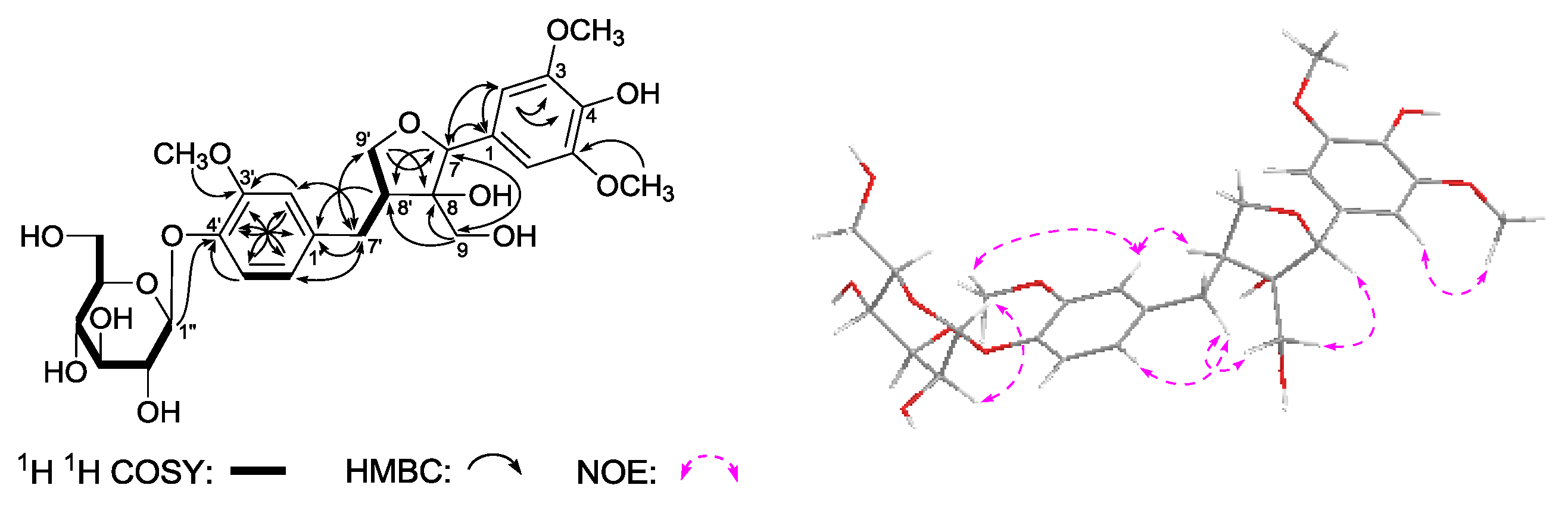
Gentilignanoside A (
3) was obtained as a white powder that exhibited negative optical rotation [[α
−36.0° (
c 0.10, MeOH)]. The molecular formula, C
27H
34O
13, of
3 was determined from Q-TOF-ESI-MS analysis (
m/
z 567.2083 [M – H]
−, calcd for C
27H
33O
13, 567.2083). Its IR spectrum showed absorption bands due to hydroxyl (3368 cm
−1), aromatic ring (1613, 1513, 1463 cm
−1), and
O-glycosidic linkage (1073 cm
−1). The
1H,
13C-NMR spectra (
Table 3) and kinds of 2D NMR spectra (
1H
1H COSY, HSQC, HMBC) showed signals ascribable to one ABX-type aromatic protons [δ 6.77 (1H, br. d,
ca. J = 8 Hz, H-6′), 6.90 (1H, br. s, H-2′), 7.10 (1H, d,
J = 8.0 Hz, H-5′)], one 1,3,4,5-symmetrical substituted phenyl group [δ 6.64 (2H, s, H-2,6)], two methylene bearing oxygen funtion {δ [3.64, 3.80 (1H each, both m, overlapped, H
2-9)], [3.64 (1H, m, overlapped), 4.06 (1H, dd, J = 7.5, 7.5 Hz), H
2-9′]]}, three methoxyl [δ 3.84 (6H, s, 3,5-OCH
3), 3.86 (3H, s, 3′-OCH
3)], and one β-
d-glucopyranosyl [δ 4.88 (1H, d,
J = 7.5 Hz, H-1′′)]. The planar structure of
3 was constructed by the assignment of the
1H
1H COSY and HMBC experiments as shown in
Figure 4. The
1H
1H COSY experiment indicated the presence of three partial moieties. On the other hand, in the HMBC experiment, long-range correlations were found from the following proton and carbon pairs: δ
H 6.64 (H-2,6) to δ
C 129.9 (C-1), 136.2 (C-4), 148.9 (C-3,5); δ
H 4.84 (H-7) to δ
C 51.8 (C-8′), 64.6 (C-9), 106.2 (C-2,6), 129.9 (C-1); δ
H 3.64, 3.80 (H
2-9) to δ
C 51.8 (C-8′), 83.3 (C-8), 85.8 (C-7); δ
H 6.90 (H-2′) to δ
C 35.1 (C-7′), 122.4 (C-6′), 146.5 (C-4′), 150.9 (C-3′); δ
H 7.10 (H-5′) to δ
C 136.9 (C-1′), 146.5 (C-4′), 150.9 (C-3′); δ
H 6.77 (H-6′) to δ
C 35.1 (C-7′), 114.4 (C-2′), 146.5 (C-4′); δ
H 2.54, 3.13 (H
2-7′) to δ
C 72.0 (C-9′), 114.4 (C-2′), 122.4 (C-6′), 136.9 (C-1′); δ
H 2.59 (H-8′) to δ
C 136.9 (C-1′); δ
H 3.64, 4.06 (H
2-9′) to δ
C 35.1 (C-7′), 83.3 (C-8), 85.8 (C-7); δ
H 3.84 (3,5-OCH
3) to δ
C 148.9 (C-3,5); δ
H 3.86 (3′-OCH
3) to δ
C 150.9 (C-3′); δ
H 4.88 (H-1′′) to δ
C 146.5 (C-4′). Furthermore, the relative configuration of
3 was determined by the NOE correlations between δ
H 4.84 (H-7) and δ
H 3.64, 3.80 (H
2-9); δ
H 3.64, 3.80 (H
2-9) and δ
H 2.54, 3.13 (H
2-7′) observed in its NOESY spectrum. Finally,
3 showed negative Cotton effect at 278 and 232 nm, which indicated the absolute configuration of it was 7
R,8
S,8′
S [
22].
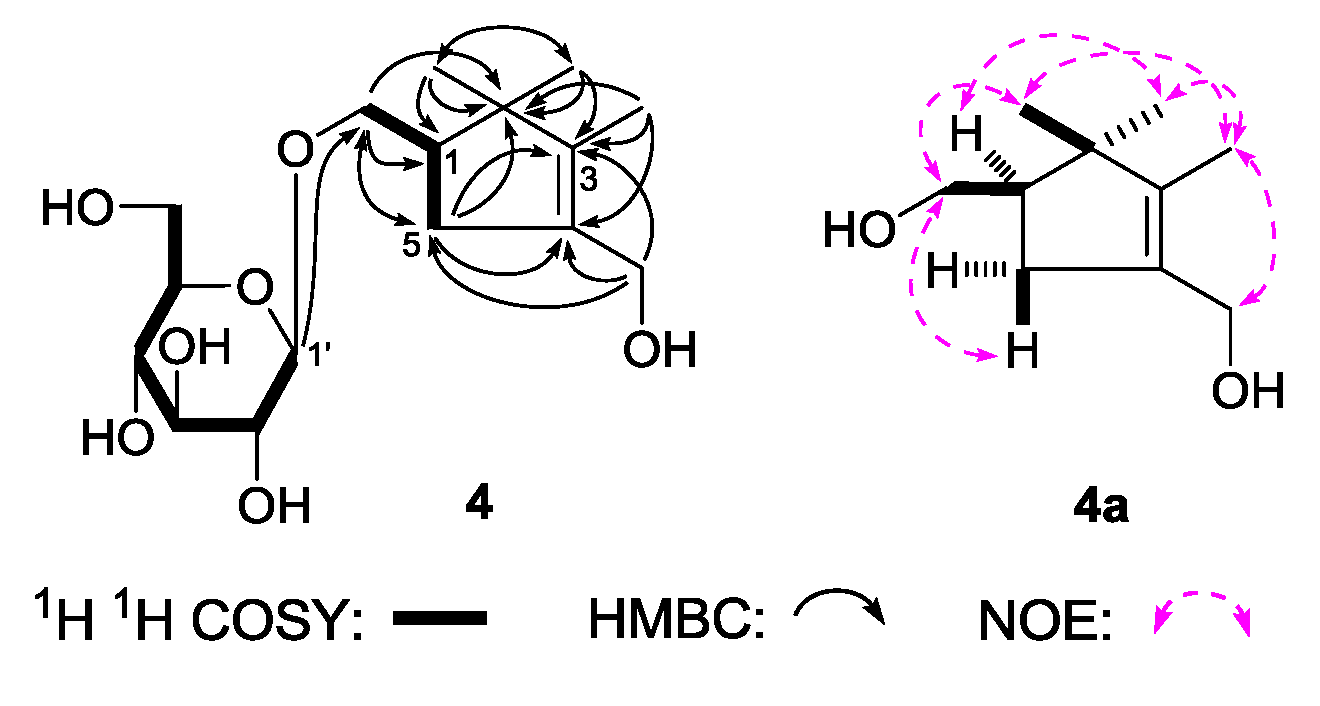
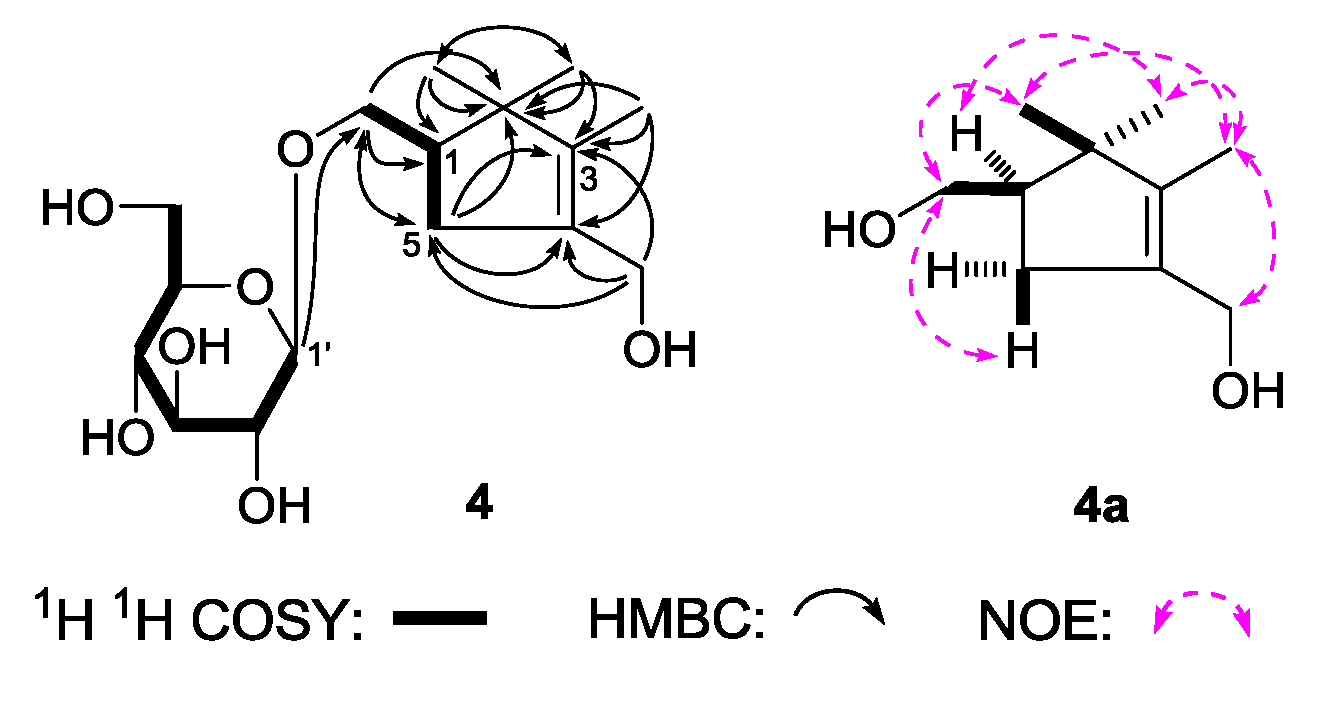
(1R)-2,2,3-Trimethyl-4-hydroxymethylcyclopent-3-ene-1-methyl-O-β-
d-glucopyranoside (
4), was obtained as a white powder. It had the molecular formula C
16H
28O
7, determined by negative-ion HRESI-TOF-MS (
m/z 377.1814 [M + COOH]
−, calcd for C
17H
29O
9, 377.1817). Its IR spectrum showed absorption bands at 3367, 1636, and 1076 cm
−1 ascribable to hydroxyl, olefin, and
O-glycosidic linkages, respectively. It was reated with 1 M HCl to give
d-glucose [
4]. The
1H,
13C-NMR (
Table 4) spectra showed signals assignable to three methyl [δ 0.87, 1.09, 1.56 (3H each, all s, 2β, 2α, 3-CH
3)], two methylene with oxygen function {[δ 3.66 (1H, m, overlapped), 3.95 (1H, dd,
J = 6.5, 11.0 Hz), 1-C
H2OH], 4.07 (2H, d,
J = 9.0 Hz, 4-C
H2OH)}, one β-
d-glucopyranosyl [δ 4.26 (1H, d,
J = 7.5 Hz, H-1′)], one methylene [δ 2.09 (1H, dd,
J = 8.0, 9.0 Hz), 2.49 (1H, dd,
J = 8.0, 8.0 Hz), H
2-5], together with one methine [δ 2.15 (1H, m, overlapped, H-1)]. The
1H
1H COSY experiment suggested the presence of two partial fragments shown in bold lines (
Figure 5). Then, the planar structure of
4 was further elucidated by the long-range correlations from δ
H 0.87 (2β-CH
3) to δ
C 27.2 (2α-CH
3), 49.0 (C-1), 49.3 (C-2), 143.5 (C-3); δ
H 1.09 (2α-CH
3) to δ
C 20.1 (2β-CH
3), 49.0 (C-1), 49.3 (C-2), 143.5 (C-3); δ
H 1.56 (3-CH
3) to δ
C 49.3 (C-2), 133.1 (C-4), 143.5 (C-3); δ
H 4.07 (4-C
H2OH) to δ
C 36.6 (C-5), 133.1 (C-4), 143.5 (C-3); δ
H 2.15, 2.48 (H
2-5) to δ
C 49.3 (C-2), 72.1 (1-CH
2OH), 133.1 (C-4), 143.5 (C-3); δ
H 4.26 (H-1′) to δ
C 72.1 (1-CH
2OH) observed in the HMBC spectrum. To determine the sterostructure of it,
4 was treated with β-glucosidase, to give the aglycon, (1
R)-2,2,3-trimethyl-4-hydroxymethylcyclopent-3-ene-1-methanol (
4a), which was obtained with negative optical rotation ([α]
D −6.5°, CHCl
3) and had only one chiral carbon. Using the the same method reported in literatures [
25,
26], compared optical rotation of
4a with that of its simialr compound, (–)-(
R)-
γ-necrodol ([α]
D −21.2°, CHCl
3) [
27], the absolute configuration of
4 was elucidated to be 1
R. Finally, the chemical shift of two methyl at the 2-position was determined by NOE correlations displayed in NOESY experiment. On the basis of above mentioned evidences, the structure of
4 was identified as (1
R)-2,2,3-trimethyl-4-hydroxymethylcyclopent-3-ene-1-methyl-
O-β-
d-glucopyranoside.
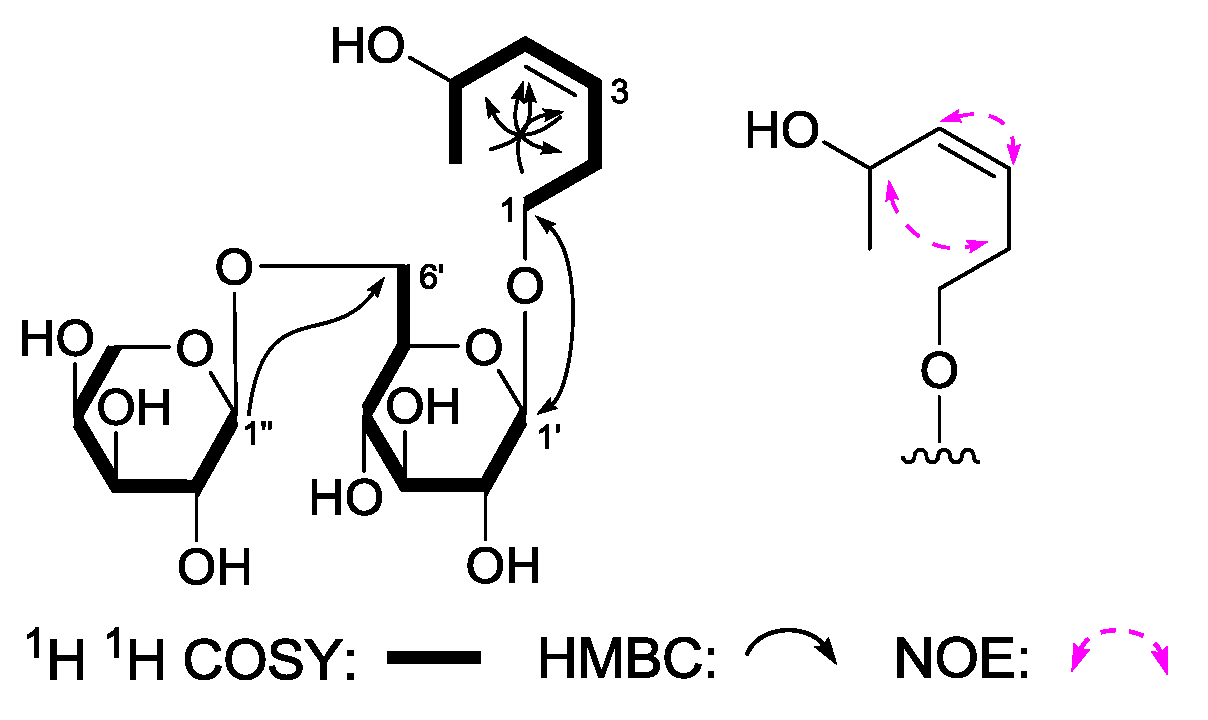
(3Z)-3-Hexene-1,5-diol 1-O-α-
l-arabinopyranosyl(1→6)-β-
d-glucopyranoside (
5) was obtained as a white powder with negative optical rotation [[α
−14.5° (
c 0.11, MeOH)]. Its molecular formula, C
17H
30O
11 (
m/
z 455.1773 [M + COOH]
−; calcd for C
18H
31O
13, 455.1770), was recorded by Q-TOF-ESI-MS. Furthermore, using acid hydrolysis and HPLC analysis, the presence of
d-glucose and
l-arabinose in
5 was revealed [
4]. The
1H,
13C-NMR spectra (
Table 5) and 2D NMR (
1H
1H COSY, HSQC, HMBC) spectra indicated the presence of two olefinic protons [δ 5.45, 5.47 (1H each, both m, H-3 and 4)], one methoxyl [δ 1.20 (3H, d,
J = 6.0 Hz, H
3-6)], one β-
d-glucopyranosyl [δ 4.27 (1H, d,
J = 7.5 Hz, H-1′)], along with one α-
l-arabinopyranosyl [δ 4.31 (1H, d,
J = 6.5 Hz, H-1′′)]. The planar structure of
5 was constructed on the basis of
1H
1H COSY and HMBC experiments. Namely, the
1H
1H COSY experiment suggested the existence of three partial structures, as shown as bold lines in
Figure 6. Meanwhile, in its HMBC spectrum, long-rang correlations from δ
H 4.27 (H-1′) to δ
C 70.4 (C-1); δ
H 4.31 (H-1′′) to δ
C 69.6 (C-6′) were observed. Finally, the NOE correlation between δ
H 2.39, 2.46 (H
2-2), and δ
H 4.61 (H-5); 5.45 (H-3) and δ
H 5.47 (H-5) found in the NOESY spectrum indicated the configuration in the 3-position was
Z. Consequently, the structure of
5 was elucidated to be (3
Z)-3-hexene-1,5-diol 1-
O-α-
l-arabinopyranosyl(1→6)-β-
d-glucopyranoside.
Furthermore, inhibitory effects of all fractions obtained from 70% EtOH extract of
G. acuta and the abovementioned isolates on motility of mouse isolated intestine tissue were determined by using the same method as reported previously [
4,
28]. As results, all of the test samples showed no significant changing on isolated intestinal tissue contraction frequency, while 70% EtOH extract of
G. acut, 95% EtOH eluate from D101 macroporous resin CC, CHCl
3 layer, as well as compounds
5,
7,
10,
12–
14,
16,
17,
31, and
32 displayed significant inhibitory effects on contraction tension (
Table 6).
From the whole plants of G. acuta, two types of monoterpenes, iridoid- (1, 22–26) and secoiridoid-type (7–21) monoterpenes were obtained. Structure-activity relationship analysis revealed that iridoid-type monoterpenes showed no significant effect on contraction tension. However, secoiridoid-type monoterpenes, such as 7, 10, 12–14, 16, 17 had strong inhibitory effect. Furthermore, when an olefin functional existed between the 5- and 6-positions (2, 20, 21), or H-5 was substituted by hydroxyl (18, 19), the bioactivity disappeared.
Meanwhile, comparing the inhibitory effect on contraction tension of 8-O-4′- (27), 7-O-4′- (28–30) with those of 7-O-9′-type (31, 32) lignan, we found that 7-O-9′-type (31, 32) lignan displayed strong inhibitory bioactivity on contraction tension.
3. Experimental
3.1. General
Physical data was obtained by using the following instruments: UV and IR spectra were determined on a Varian Cary 50 UV-VIS (Varian, Inc., Hubbardsdon, MA, USA) and Varian 640-IR FT-IR spectrophotometer (Varian Australia Pty Ltd., Mulgrave, Australia), respectively. Optical rotations were obtained on a Rudolph Autopol® IV automatic polarimeter (l = 50 mm) (Rudolph Research Analytical, 55 Newburgh Road, Hackettstown, NJ, 07840 USA). NMR spectra were run on a Bruker 500 MHz NMR spectrometer (Bruker BioSpin AG Industriestrasse 26 CH-8117, Fällanden, Switzerland) at 500 MHz for 1H and 125 MHz for 13CNMR (internal standard: TMS). Negative-ion HRESI-TOF-MS were recorded on an Agilent 6520 Accurate-Mass Q-Tof LC/MS spectrometer (Agilent Corp., Santa Clara, CA, USA). Column chromatographies (CC) were performed on macroporous resin D101 (Haiguang Chemical Co., Ltd., Tianjin, China), silica gel (74–149 μm, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), and Sephadex LH-20 (Ge Healthcare Bio-Sciences, Uppsala, Sweden). Preparative high-performance liquid chromatography (PHPLC) column, cosmosil 5C18-MS-II (20 mm i.d. × 250 mm, Nakalai Tesque, Inc., Tokyo, Japan) were used to isolate the compounds.
3.2. Plant Material
The whole plants of Gentianella acuta (Michx.) Hulten were collected from Alxa Youqi, Inner Mongolia Autonomous region, China in September 2013, and identified by Dr. Li Tianxiang (Experiment Teaching Department, Tianjin University of Traditional Chinese Medicine). The voucher specimen was deposited at the Academy of Traditional Chinese Medicine of Tianjin University of TCM.
3.3. Extraction and Isolation
The whole plants of G. acuta (3.0 kg) were cut and refluxed with 70% ethanol–water. Then the 70% EtOH extract (868.5 g) was partitioned in a CHCl3–H2O mixture (1:1, v/v). The H2O layer (670.0 g) was subjected to D101 macroporous resin CC (H2O → 95% EtOH → acetone). As a result, H2O (332.4 g), 95% EtOH (294.9 g), and acetone (5.1 g) eluates were obtained.
The 95% EtOH eluate (200.0 g) was subjected to silica gel CC [CHCl3 → CHCl3-MeOH (100:1 → 100:5, v/v) → CHCl3–MeOH–H2O (10:3:1 → 7:3:1 → 6:4:1, v/v/v, lower layer)] to give 16 fractions (Fr. 1–Fr. 16). Fraction 7 (25.7 g) was centrifugated (MeOH), and two fractions (Fr. 7-1–Fr. 7-2) were yielded. Fraction 7-2 (9.8 g) was separated by PHPLC [CH3CN–H2O (18:82 → 35:65 → 42:58, v/v) + 1% HAc] to give 27 fractions (Fr. 7-2-1–Fr. 7-2-27). Fraction 7-2-2 (70.6 mg) was purified by PHPLC [MeOH–H2O (22:78, v/v)], and 2,3-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-propan-1-one (6, 5.9 mg) was given. Fraction 7-2-8 (46.3 mg) was isolated by PHPLC [CH3CN-H2O (20:80, v/v) + 1% HAc] to gain (7R,8S)-erythro-4,7,9,9′-tetrahydroxy-3,3′-dimethoxy-8-O-4′-neolignan (27, 6.0 mg). Fraction 9 (15.0 g) was subjected to Sephadex LH-20 CC [CHCl3–MeOH (1:1, v/v)] to yield seven fractions (Fr. 9-1–Fr. 9-7). Fraction 9-4 (7.3 g) was separated by PHPLC [CH3CN–H2O (22:78 → 30:70 → 45:55, v/v) + 1% HAc], as a result, 12 fractions (Fr. 9-4-1–Fr. 9-4-12) were obtained. Fraction 9-4-2 (962.8 mg) was purified by PHPLC [MeOH–H2O (23:77, v/v) + 1% HAc] to give gentiiridoside B (2, 14.8 mg). Fraction 9-4-9 (277.1 mg) was centrifuged (MeOH), and two fractions (Fr. 9-4-9-1–Fr. 9-4-9-2) were gained. Fraction 9-4-9-2 (199.7 mg) was isolated by PHPLC [MeOH–H2O (45:55, v/v) + 1% HAc] to yield plucheoside D3 (29, 8.8 mg). Fraction 11 (20.0 g) was subjected to PHPLC [CH3CN–H2O (15:85 → 25:75 → 42:58, v/v) + 1% HAc], and 29 fractions (Fr. 11-1–Fr. 11-29) were given. Fraction 11-5 (631.3 mg) was separated by PHPLC [CH3CN–H2O (10:90, v/v) + 1% HAc], and 8-epiloganin (24, 29.7 mg) was yielded. Fraction 11-6 (388.0 mg) was isolated by PHPLC [CH3CN–H2O (11:89, v/v) + 1% HAc] to yield gentilignanoside A (3, 9.2 mg), loganin (23, 143.5 mg), and berchemol-4′-O-β-d-glucoside (32, 84.5 mg). Fraction 11-12 (660.5 mg) was centrifugated (MeOH) and further purified by PHPLC [MeOH–H2O (35:65, v/v) + 1% HAc] to give (1R)-2,2,3-trimethyl-4-hydroxymethylcyclopent-3-ene-1-methyl-O-β-d-glucopyranoside (4, 11.5 mg). Fraction 11-17 (790.7 mg) was isolated by PHPLC [MeOH–H2O (42:58, v/v) + 1% HAc] to obtain six fractions (Fr. 11-17-1–Fr. 11-17-6). Fraction 11-17-1 (97.1 mg) was purified by PHPLC [CH3CN-H2O (24:76, v/v) + 1% HAc] to gain swertiaside (26, 79.0 mg). Fraction 11-21 (376.1 mg) was separated by PHPLC [MeOH–H2O (42:58, v/v) + 1% HAc] to afford (7S,8R)-9′-methoxy-dehydrodiconiferyl alcohol 4-O-β-d-glucopyranoside (30, 17.4 mg). Fraction 13 (20.0 g) was subjected to PHPLC [MeOH-H2O (35:65 → 45:55 → 55:45, v/v) + 1% HAc], and 20 fractions (Fr. 13-1–Fr. 13-20) were yielded. Fraction 13-11 (1.9 g) was centrifuged (MeOH) to obtain two fractions (Fr. 13-11-1–Fr. 13-11-2). Fraction 13-11-1 (1.0 g) was isolated by PHPLC [CH3CN–H2O (16:84, v/v) + 1% HAc], as a result, eleven fractions (Fr. 13-11-1-1–Fr. 13-11-1-11) were given. Fraction 13-11-1-9 (117.1 mg) were purified by Sephadex LH-20 CC (MeOH) and PHPLC [MeOH–H2O (35:65, v/v) + 1% HAc] to afford 5α-carboxystrictosidine (12, 26.2 mg). Fraction 13-12 (501.4 mg) was isolated by PHPLC [CH3CN–H2O (18:82, v/v) + 1% HAc] and to yield (E)-aldosecologanin (11, 38.2 mg). Fraction 13-17 (504.0 mg) was purified by PHPLC [CH3CN–H2O (22:78, v/v) + 1% HAc] to yield gentiiridoside A (1, 265.0 mg). Fraction 14 (15.3 g) was subjected to PHPLC [CH3CN–H2O (15:85 → 25:75, v/v) + 1% HAc], and 20 fractions (Fr. 14-1–Fr. 14-20) were obtained. Fraction 14-1 (1.4 g) was separated by PHPLC [CH3CN–H2O (9:91, v/v) + 1% HAc] to gain 14 fractions (Fr. 14-1-1–Fr. 14-1-14). Fraction 14-1-2 (32.1 mg) was purified by Sephadex LH-20 CC (MeOH) and finally by PHPLC [CH3CN–H2O (7:93, v/v)] to give (3Z)-3-hexene-1,5-diol 1-O-α-l-arabinopyranosyl(1→6)-β-d-glucopyranoside (5, 2.8 mg).
The CHCl3 layer (50.0 g, Fr. C) was subjected to Silica gel CC [CHCl3–MeOH (100:2 → 100:3 → 100:5, v/v) → CHCl3–MeOH–H2O (10:3:1, v/v/v, lower layer) → MeOH], and eight fractions (Fr. C-1–Fr. C-8) were yielded. Fraction C-5 (1.1 g) was separated by Sephadex LH-20 CC [MeOH–CH2Cl2 (1:1, v/v)] to gain three fractions (Fr. C-5-1–Fr. C-5-3). Fraction C-5-2 (110.0 mg) was purified by PHPLC [MeOH-H2O (45:55, v/v)] to afford (7S,8R)-dehydrodiconiferyl alcohol (28, 13.3 mg) and (–)-berchemol (31, 14.6 mg). Fraction C-7 (9.0 g) was isolated by ODS CC [MeOH–H2O (20:80 → 30:70 → 40:60 → 50:50, v/v) → MeOH], and eight fractions (Fr. C-7-1–Fr. C-7-8) were obtained. Fraction C-7-2 (351.4 mg) was purified by PHPLC [MeOH–H2O (32:68, v/v) + 1% HAc] to give gentiopicroside (20, 15.9 mg). Fraction C-7-6 (486.4 g) was subjected to Sephadex LH-20 CC [MeOH–CH2Cl2 (1:1, v/v)] and PHPLC [MeOH–H2O (50:50, v/v) + 1% HAc] to afford trifloroside (17, 16.0 mg).
Compounds
7–
10,
13–
16,
18,
19,
21,
22 and
25 were obtained and identified by using the method reported previously [
2,
3].
Gentiiridoside A (
1): White powder; [α
−90.0° (
c 0.14, MeOH); UV
λmax (MeOH) nm (log
ε): 229 (4.46), 285 (3.53); IR
νmax (KBr): 3372, 2929, 1712, 1635, 1588, 1486, 1372, 1264, 1201, 1154, 1078, 1017, 902, 873 cm
−1;
1H-NMR (CD
3OD, 500 MHz) and
13C-NMR (CD
3OD, 125 MHz) data, see
Table 1. HRESI-TOF-MS negative-ion mode
m/z 777.2261 [M – H]
− (calcd for C
36H
41O
19, 777.2248).
Gentiiridoside B (
2): White powder; [α
−36.7° (
c 0.12, MeOH); UV
λmax (MeOH) nm (log
ε): 231 (4.26, sh), 273 (4.08, sh); IR
νmax (KBr) 3357, 2924, 1705, 1609, 1518, 1457, 1418, 1375, 1272, 1209, 1074, 1024 cm
−1;
1H-NMR (DMSO-
d6, 500 MHz) and
13C-NMR (DMSO-
d6, 125 MHz) data see
Table 2. HRESI-TOF-MS negative-ion mode
m/z 679.1023 [M – H]
– (calcd for C
28H
39O
18, 679.1033).
Gentilignanoside A (
3): White powder; [α
−36.0° (
c 0.10, MeOH); CD (
c 0.0018 M, MeOH) mdeg (
λnm): −3.8 (278), −16.9 (232), −27.6 (206); UV
λmax (MeOH) nm (log
ε): 226 (4.32), 275 (3.73); IR
νmax (KBr): 3368, 2937, 1613, 1514, 1463, 1425, 1324, 1266, 1224, 1158, 1115, 1073, 1026 cm
−1;
1H-NMR (CD
3OD, 500 MHz) and
13C-NMR (CD
3OD, 125 MHz) data see
Table 3. HRESI-TOF-MS negative-ion mode
m/z 679.1023 [M – H]
− m/z 567.2083 [M – H]
− (calcd for C
27H
33O
13, 567.2083).
(1R)-2,
2,
3-Trimethyl-4-hydroxymethylcyclopent-3-ene-1-methyl-O-β-d-glucopyranoside (
4): White powder; [α
−43.9° (
c 0.12, MeOH); IR
νmax (KBr): 3367, 2927, 1636, 1576, 1436, 1286, 1161, 1076, 1038 cm
−1.
1H-NMR (CD
3OD, 500 MHz) and
13C-NMR (CD
3OD, 125 MHz) data see
Table 4. HRESI-TOF-MS negative-ion mode
m/z 377.1814 [M + COOH]
– (calcd for C
17H
29O
9, 377.1817).
(3Z)-3-Hexene-1,5-diol 1-O-α-l-arabinopyranosyl(1→6)-β-d-glucopyranoside (
5): White powder; [α
−14.5° (
c 0.11, MeOH); IR
νmax (KBr): 3364, 2966, 2920, 1593, 1419, 1370, 1258, 1166, 1047, 1009 cm
−1;
1H-NMR (CD
3OD, 500 MHz) and
13C-NMR (CD
3OD, 125 MHz) data see
Table 5. HRESI-TOF-MS negative-ion mode
m/z 455.1773 [M + COOH]
– (calcd for C
18H
31O
13, 455.1770).
Enzymatic Hydrolysis of 4 A solution of 4 (6.0 mg) in H2O (2.0 mL) was reacted with β-glucosidase (6.0 mg, Almond, Sigma-Aldrich, Co. 3050 Spruce Street, St. Louis, MO, 63103 USA) at 37 °C for 2.5 h. Then the reaction mixture was extracted with EtOAc. And the residue from EtOAc solvent was subjected to Silica gel CC [CHCl3–MeOH (100:5, v/v)], as a result, the aglycon, (1R)-2,2,3-trimethyl-4-hydroxymethylcyclopent-3-ene-1-methanol (4a, 2.8 mg, 93.33%) was yielded.
(1R)-2,
2,
3-Trimethyl-4-hydroxymethylcyclopent-3-ene-1-methanol (
4a): White powder; [α
−6.5° (
c 0.09, CHCl
3); IR
νmax (KBr): 3318, 2953, 2925, 2867, 1717, 1576, 1462, 1437, 1380, 1240, 1179, 1118, 1087, 999 cm
−1;
1H-NMR (CD
3OD, 500 MHz) and
13C-NMR (CD
3OD, 125 MHz) data see
Table 4. HRESI-TOF-MS negative-ion mode
m/z 170.1423 [M – H]
− (calcd for C
10H
17O
2, 170.1433).
Acid Hydrolysis of 1–
5 The solution of compounds
1–
5 (each 2.0 mg) in 1 M HCl (1.0 mL) was treated by using the same method as described in reference [
4]: They were heated under reflux for 3 h. The reaction mixture was then analyzed by CH
3CN–H
2O (75:25,
v/v; flow rate 0.7 mL/min). As results,
d-glucose was detected from the aqueous phase of
1–
5, and
l-arabinose was found from that of
5 by comparison of its retention time and optical rotation with those of the authentic sample,
d-glucose (
tR 16.8 min (positive)) and
l-arabinose (
tR 13.1 min (positive)), respectively.
3.4. Inhibitory Effects of Fractions and Compounds 1–32 on the Motility of Mouse Isolated Intestine Tissue
Inhibitory effects of fractions and compounds
1–
32 on motility of mice isolated intestine tissue were determined by using the similar method as we reported previously [
4,
28]: Mice were fasted for 12 h before experiments, intestinal tissue were collected immediately. The Maxwell bath was filled with 10 mL of Tyrode’s solution (one liter contains: NaCl 8.0 g, CaCl
2 0.2 g, KCl 0.2 g, MgCl
2 0.1 g, NaHCO
3 1.0 g, KH
2PO
4 0.05 g, glucose 1.0 g, pH 7.4) and maintained at a constant temperature (37.0 ± 0.5°C), and bubbled with 95% O
2 and 5% CO
2 gas. The intestinal tissue was fixed on bottom hook in and the other end was connected to an isometric tension transducer. Samples in DMSO solution were added after 15 min to equilibrate incubation, the final DMSO concentration was 0.1%, and the final concentration of fractions and compounds was 100 μg/mL and 40 μM, respectively. The mean tension and frequency of intestine muscle contractions were recorded for 1 min before and 4 min after drug additions using isolated tissue bath systems (Radnoti Glass Technology Inc., Monrovia, CA, 159901A, USA). Loperamide hydrochloride (Xi’an Janssen Pharmaceutical Ltd., Xi’an, China) was used as a positive control, and the final concentration was 10 μM.
Values are expressed as mean ± S.D. All the grouped data were statistically performed with SPSS 11.0. Significant differences between means were evaluated by one-way analysis of variance (ANOVA) and Tukey’s Studentized range test was used for post hoc evaluations. p < 0.05 was considered to indicate statistical significance.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}