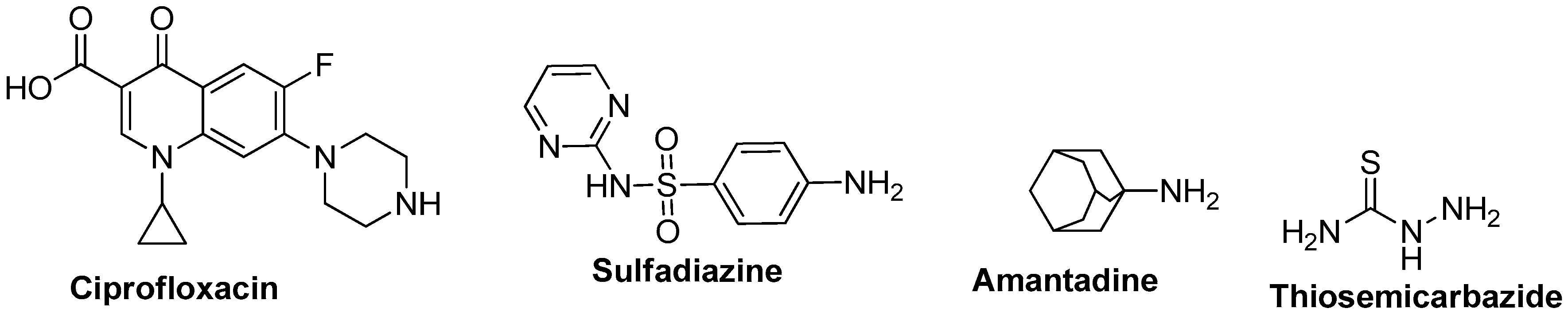
Sulfonamide-Linked Ciprofloxacin, Sulfadiazine and Amantadine Derivatives as a Novel Class of Inhibitors of Jack Bean Urease; Synthesis, Kinetic Mechanism and Molecular Docking

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Jack Bean Urease Inhibition Assay and Structure Activity Relationship (SAR)
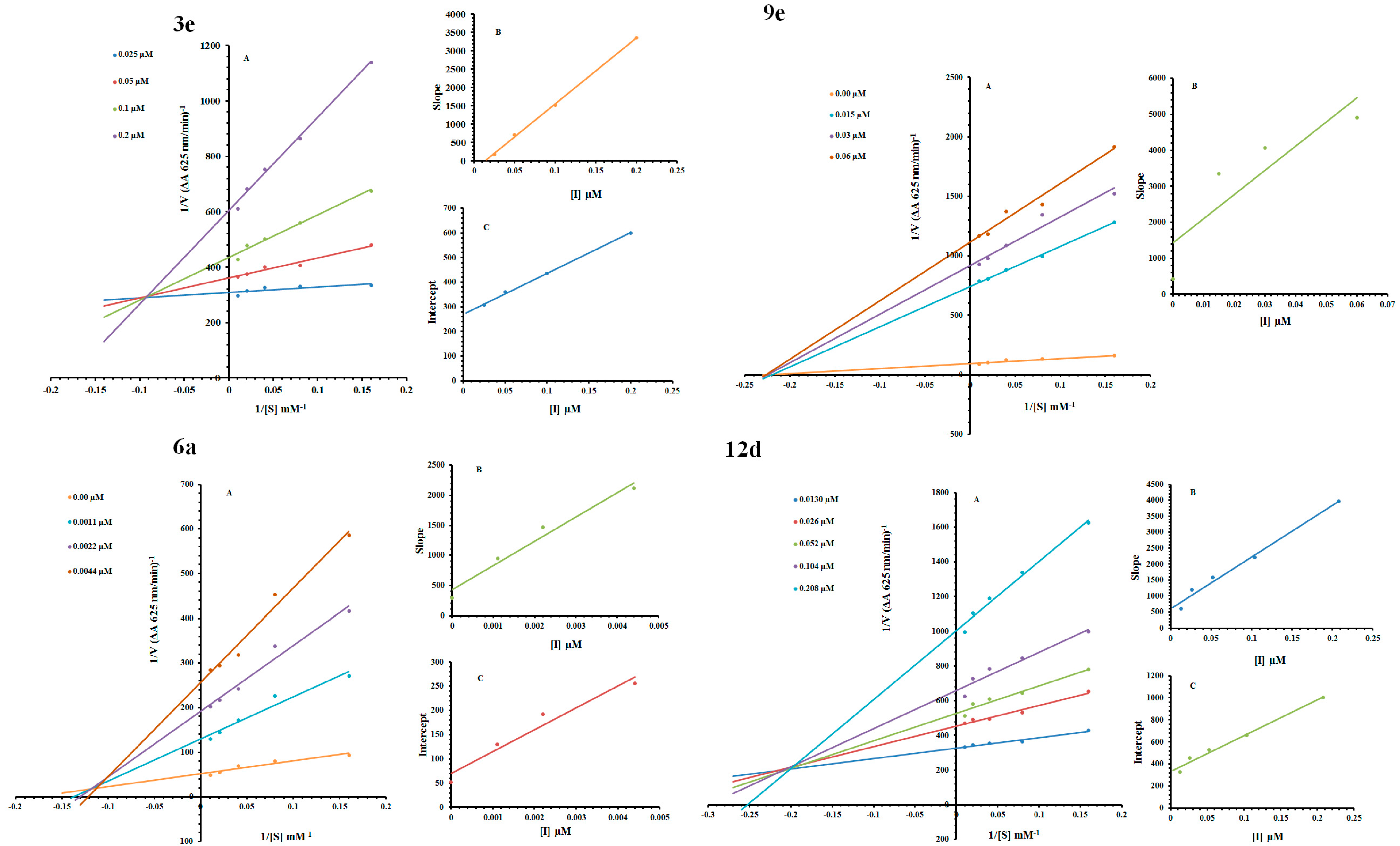
2.2. Kinetic Mechanism
3. Discussion
3.1. Structural Evaluation of the Target Protein
3.2. Biochemical Properties and Rule of Five (RO5) Validation of Synthesized Compounds
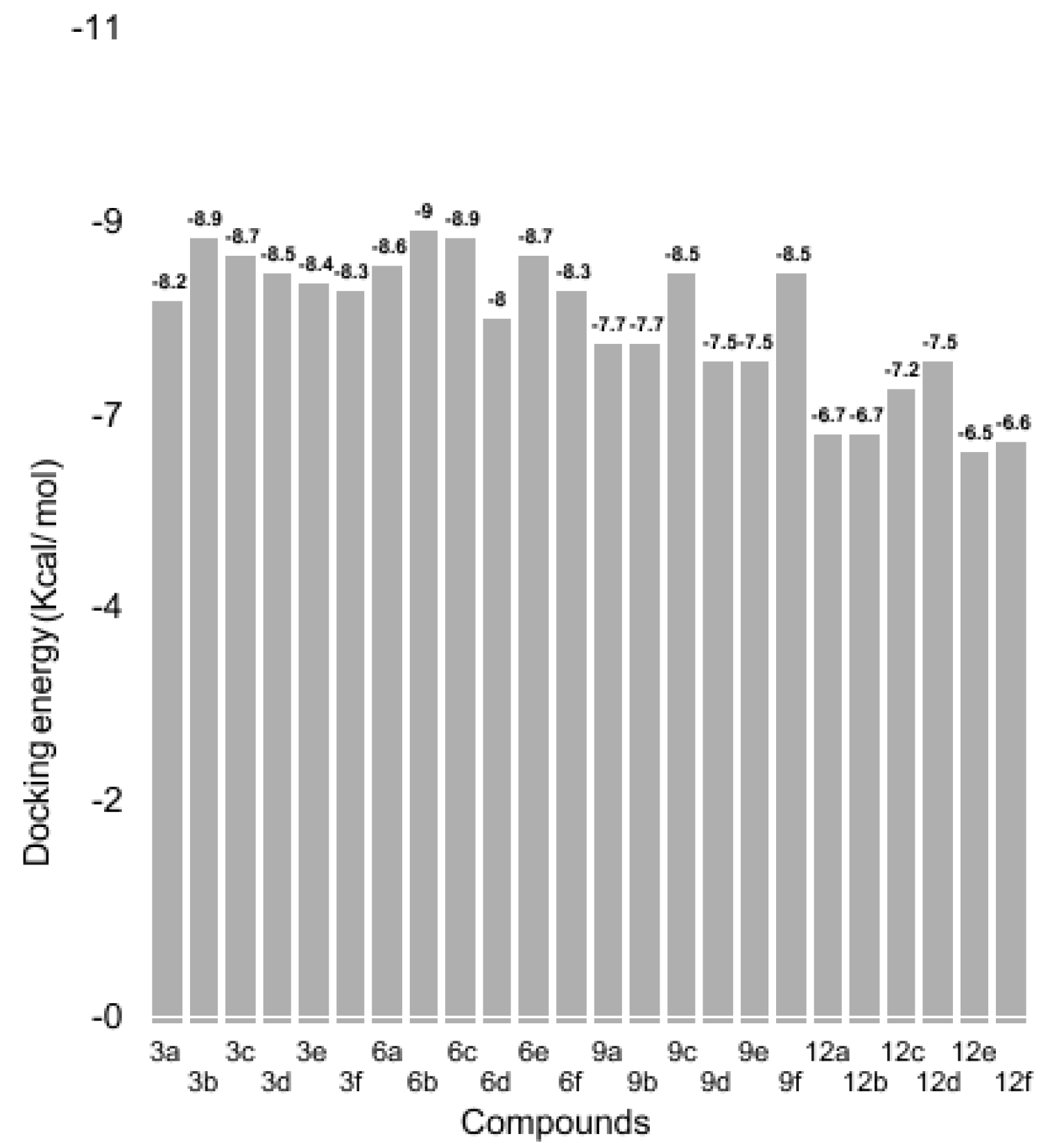
3.3. Binding Energy Evaluation of Synthesized Compounds
3.4. Urease Binding Pocket and Ligand Pose Analysis
3.5. Structure Activity Relationship and Interacting Residues
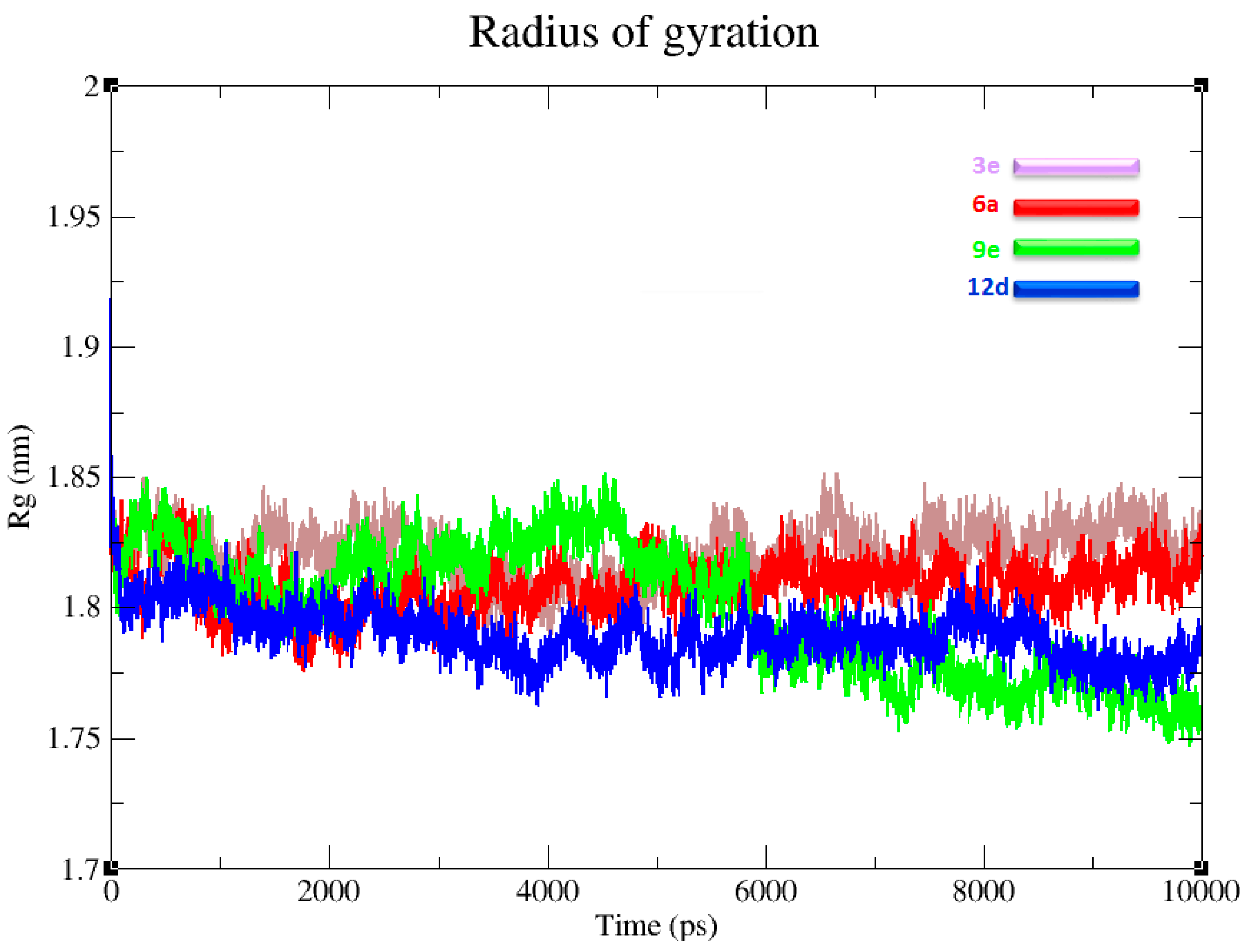
3.6. Radius of Gyration and Protein Stability
4. Materials and Methods
4.1. Chemicals and Instruments
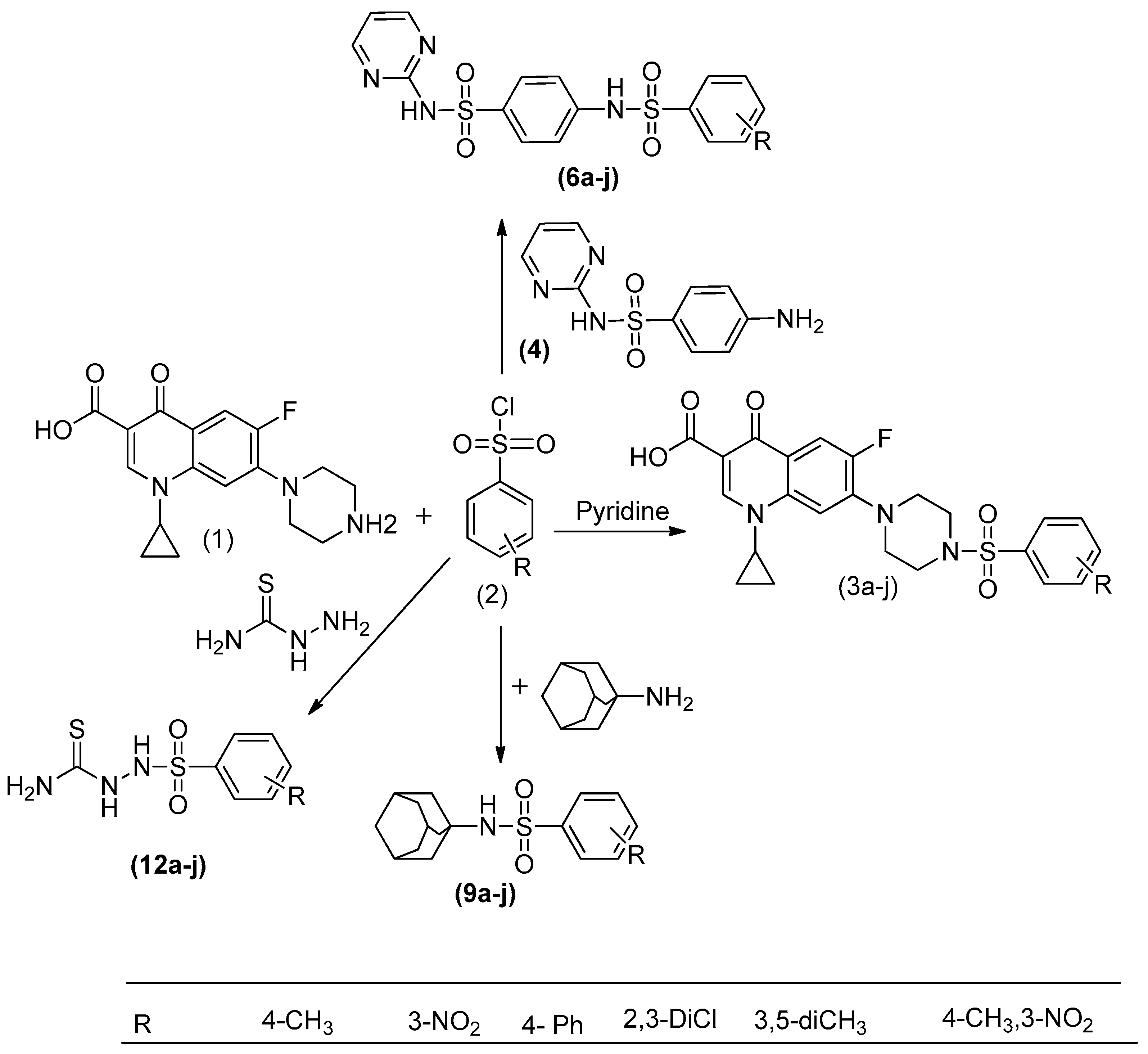
4.2. Synthetic Procedures
4.3. Characterization Data
4.4. Urease Inhibition Assay
4.5. Kinetic Mechanism Study
4.6. Free Radical Scavenging Assay
4.7. Retrieval of Jack Bean Urease Structure from Protein Data Bank (PDB)
4.8. Molecular Docking of Synthesized Compounds Using AutoDock
4.9. Molecular Dynamics Simulations Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khan, K.M.; Iqbal, S.; Lodhi, M.A.; Maharvi, G.M.; Choudhary, M.I.; Perveen, S. Biscoumarin: New class of urease inhibitors; economical synthesis and activity. Bioorg. Med. Chem. 2004, 12, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Arfan, M.; Ali, M.; Ahmad, H.; Anis, I.; Khan, A.; Choudhary, M.I.; Shah, M.R. Urease inhibitors from Hypericum oblongifolium WALL. J. Enzym. Inhibit. Med. Chem. 2010, 25, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Amtul, Z.; Rasheed, M.; Choudhary, M.I.; Rosanna, S.; Khan, K.M. Kinetics of novel competitive inhibitors of urease enzymes by a focused library of oxadiazoles/thiadiazoles and triazoles. Biochem. Biophys. Res. Commun. 2004, 319, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Ullah, Z.; Lodhi, M.A.; Ali, M.; Choudhary, M.I.; ur Rahman, A.; ul Haq, Z. Successful computer guided planned synthesis of (4R)-thiazolidine carboxylic acid and its 2-substituted analogues as urease inhibitors. Mol. Divers. 2006, 10, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Amtul, Z.; Siddiqui, R.A.; Choudhary, M.I. Chemistry and mechanism of urease inhibition. Curr. Med. Chem. 2002, 9, 1323–1348. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Musiani, F.; Benini, S.; Ciurli, S. Chemistry of Ni2+ in urease: Sensing, trafficking, and catalysis. Acc. Chem. Res. 2011, 44, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Marzadori, C.; Miletti, S.; Gessa, C.; Ciurli, S. Immobilization of jack bean urease on hydroxyapatite: Urease immobilization in alkaline soils. Soil Biol. Biochem. 1998, 30, 1485–1490. [Google Scholar] [CrossRef]
- Aslam, M.A.S.; Mahmood, S.U.; Shahid, M.; Saeed, A.; Iqbal, J. Synthesis, biological assay in vitro and molecular docking studies of new Schiff base derivatives as potential urease inhibitors. Eur. J. Med. Chem. 2011, 46, 5473–5479. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Zaib, S.; Pervez, A.; Mumtaz, A.; Shahid, M.; Iqbal, J. Synthesis, molecular docking studies, and in vitro screening of sulfanilamide-thiourea hybrids as antimicrobial and urease inhibitors. Med. Chem. Res. 2013, 22, 3653–3662. [Google Scholar] [CrossRef]
- Saeed, A.; Khan, M.S.; Rafique, H.; Shahid, M.; Iqbal, J. Design, synthesis, molecular docking studies and in vitro screening of ethyl 4-(3-benzoylthioureido) benzoates as urease inhibitors. Bioorg. Chem. 2014, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- O’connor, A.; Gisbert, J.; O’Morain, C. Treatment of Helicobacter pylori infection. Helicobacter 2009, 14, 46–51. [Google Scholar] [CrossRef] [PubMed]
- de Boer, W.A.; Tytgat, G.N. Regular review: Treatment of Helicobacter pylori infection. BMJ Br. Med. J. 2000, 320, 31–34. [Google Scholar] [CrossRef]
- Carlini, C.R.; Ligabue-Braun, R. Ureases as multifunctional toxic proteins: A review. Toxicon 2016, 110, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Mahmood, S.U.; Rafiq, M.; Ashraf, Z.; Jabeen, F.; Seo, S.Y. Iminothiazoline-Sulfonamide Hybrids as Jack Bean Urease Inhibitors; Synthesis, Kinetic Mechanism and Computational Molecular Modeling. Chem. Biol. Drug Des. 2016, 87, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, L.S.B. Urease inhibitors: A review. Indian J. Biotechnol. 2012, 11, 381–388. [Google Scholar]
- Kosikowska, P.; Berlicki, Ł. Urease inhibitors as potential drugs for gastric and urinary tract infections: A patent review. Exp. Opin. Ther. Pat. 2011, 21, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Farid, A.; Larik, F.A.; Abbas, Q.; Channar, P.A.; Hassan, M.; Seo, S.Y.; Shehzadi, S.A.; Tehrani, K.A. Novel N-(Substituted benzoyl)-N′-(1-naphthyl)-N″-(substituted phenyl)guanidines as Jack Bean Urease Inhibitors and Free-Radical Scavengers, Synthesis, Kinetics, and Computational Molecular Modeling Studies. ACS Omega 2017. [Google Scholar] [CrossRef]
- Channar, P.A.; Saeed, A.; Larik, F.A.; Sajid Rashid, S.; Iqbal, Q.; Rozi, M.; Younis, S.; Mahar, J. Design and synthesis of 2,6-di(substituted phenyl)thiazolo[3,2-b]-1,2,4-triazoles as a-glucosidase and a-amylase inhibitors, co-relative Pharmacokinetics and 3D QSAR and risk analysis. Biomed. Pharmacother. 2017, 94, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Khan, K.M.; Zehra, S.T.; Ahmed, R.; Shafiq, Z.; Bakht, S.M.; Yaqub, M.; Hussain, M.; de León, A.D.L.V.; Furtmann, N.; et al. Synthesis, biological evaluation and molecular docking of N-phenyl thiosemicarbazones as urease inhibitors. Bioorg. Chem. 2015, 61, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Muri, E.M.F.; Mishra, H.; Avery, M.A.; Williamson, J.S. Design and synthesis of heterocyclic hydroxamic acid derivatives as inhibitors of Helicobacter pylori urease. Synth. Commun. 2003, 33, 1977–1995. [Google Scholar] [CrossRef]
- Gadad, A.K.; Mahajanshetti, C.S.; Nimbalkar, S.; Raichurkar, A. Synthesis and antibacterial activity of some 5-guanylhydrazone/thiocyanato-6-arylimidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide derivatives. Eur. J. Med. Chem. 2000, 9, 853–857. [Google Scholar] [CrossRef]
- Mojzych, M.; Bielawska, A.; Bielawski, K.; Ceruso, M.; Supuran, C.T. Pyrazolo [4,3-e][1,2,4] triazine sulfonamides as carbonic anhydrase inhibitors with antitumor activity. Bioorg. Med. Chem. 2014, 22, 2643–2647. [Google Scholar] [CrossRef] [PubMed]
- Zani, F.; Vicini, P. Antimicrobial activity of some 1,2-benzisothiazoles having a benzenesulfonamide moiety. Arch. Pharm. 1998, 331, 219–223. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sławinski, J.; Saczewski, F.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur. J. Med. Chem. 2010, 45, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Renzi, G.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Topical sulfonamide antiglaucoma agents incorporating secondary amine moieties. Bioorg. Med. Chem. Lett. 2000, 10, 673–676. [Google Scholar] [CrossRef]
- Noreen, M.; Rasool, N.; Gull, Y.; Zubair, M.; Mahmood, T.; Ayub, K.; Nasim, F.U.H.; Yaqoob, A.; Zia-Ul-Haq, M.; ade Feo, V. Synthesis, density functional theory (DFT), urease inhibition and antimicrobial activities of 5-aryl thiophenes bearing sulphonylacetamide moieties. Molecules 2015, 20, 19914–19928. [Google Scholar] [CrossRef] [PubMed]
- Noreen, M.; Rasool, N.; Gull, Y.; Zahoor, A.F.; Yaqoob, A.; Kousar, S.; Zubair, M.; Bukhari, I.H.; Rana, U.A. A facile synthesis of new 5-aryl-thiophenes bearing sulfonamide moiety via Pd (0)-catalyzed Suzuki–Miyaura cross coupling reactions and 5-bromothiophene-2-acetamide: As potent urease inhibitor, antibacterial agent and hemolytically active compounds. J. Saudi Chem. Soc. 2017, 21, 403–414. [Google Scholar] [CrossRef]
- Mojzych, M.; Tarasiuk, P.; Kotwica-Mojzych, K.; Rafiq, M.; Seo, S.Y.; Nicewicz, M.; Fornal, E. Synthesis of chiral pyrazolo [4,3-e][1,2,4] triazine sulfonamides with tyrosinase and urease inhibitory activity. J. Enzyme Inhibit. Med. Chem. 2017, 32, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L.; Hausinger, R.P. Microbial ureases: Significance, regulation, and molecular characterization. Microbiol. Rev. 1989, 53, 85–108. [Google Scholar] [PubMed]
- Krajewska, B.; Ureases, I. Functional, catalytic and kinetic properties: A review. J. Mol. Catal. B Enzym. 2009, 59, 9–21. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev. 2003, 23, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.C.; Mackey, Z.; Hansell, E.; Doyle, P.; Gut, J.; Caffrey, C.R.; Lehrman, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and structure—Activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 2004, 47, 3212–3219. [Google Scholar] [CrossRef] [PubMed]
- Gürsoy, A.; Terzioglu, N.; Ötük, G. Synthesis of some new hydrazide-hydrazones, thiosemicarbazides and thiazolidinones as possible antimicrobials. Eur. J. Med. Chem. 1997, 32, 753–757. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. NeuroSci. 2011, 3, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2625. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.; Roy, N. Recent trends in drug-likeness prediction: A comprehensive review of in silico methods. Indian. J. Pharm. Sci. 2007, 69, 609–615. [Google Scholar]
- Bakht, M.A.; Yar, M.S.; Abdel-Hamid, S.G.; Al Qasoumi, S.I.; Samad, A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5862–5869. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized Hydrophobic Interactions and Hydrogen Bonding at the Target-Ligand Interface Leads the Pathways of Drug-Designing. PLoS ONE 2010, 5, 12029. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M.A. Medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Mahesar, P.A.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Seo, S.Y. Hybrid Pharmacophoric Approach in the Design and Synthesis of Coumarin Linked Pyrazolinyl as Urease Inhibitors, Kinetic Mechanism and Molecular Docking. Chem. Biodivers. 2017. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Seo, S.Y.; Upadhyay, L.B.; Han, Y.; Huo, G.F.; et al. Jack Bean Urease Inhibitors, and Antioxidant Activity Based on Palmitic acid Derived 1-acyl-3-Arylthioureas: Synthesis, Kinetic Mechanism and Molecular Docking Studies. Drug Res. 2012, 11, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Larik, F.A.; Channar, P.A.; Mehfooz, H.; Ashraf, M.H.; Abbas, Q.; Hassan, M.; Seo, S.Y. An Expedient Synthesis of N-(1-(5-mercapto-4-((substituted benzylidene)amino)-4H-1,2,4-triazol-3-yl)-2-phenylethyl) benzamides as Jack bean Urease inhibitors and free radical scavengers; kinetic mechanism and molecular docking studies. Chem. Biol. Drug Des. 2017. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Sajid, R.; Channar, P.A.; Larik, F.A.; Qamar, A.; Hussain, M.; Raza, U.; Flörke, U.; Sung, Y.S. Long Chain 1-Acyl-3-arylthioureas as Jack Bean Urease Inhibitors, Synthesis, Kinetic Mechanism and Molecular Docking Studies. J. Taiwan Inst. Chem. Eng. 2017, 77, 54–63. [Google Scholar] [CrossRef]
- Arshad, N.; Perveen, F.; Saeed, A.; Channar, P.A.; Farooqi, S.I.; Larik, F.A.; Ismail, H.; Mirza, B. Spectroscopic, molecular docking and structural activity studies of (E)-N′-(substituted benzylidene/methylene) isonicotinohydrazide derivatives for DNA binding and their biological screening. J. Mol. Struct. 2017, 1139, 371–380. [Google Scholar] [CrossRef]
- Mehfooz, H.; Saeed, A.; Sharma, A.; Albericio, F.; Larik, F.A.; Jabeen, F.; Channar, P.A.; Flörke, U. Dual Inhibition of AChE and BChE with the C-5 Substituted Derivative of Meldrum’s Acid: Synthesis, Structure Elucidation, and Molecular Docking Studies. Crystals 2017, 7. [Google Scholar] [CrossRef]
- Gioiello, A.; Rosatelli, E.; Teofrasti, M.; Filipponi, P.; Pellicciari, R. Building a sulfonamide library by eco-friendly flow synthesis. ACS Comb. Sci. 2013, 15, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Ismail, H.; Dilshad, E.; Mirza, B. New 1-octanoyl-3-aryl thiourea derivatives: Solvent-free synthesis, characterization and multi-target biological activities. Bangladesh J. Pharmacol. 2016, 11, 894–902. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Accelrys. Studio D; Version 2.1; Accelrys: San Diego, CA, USA, 2008. [Google Scholar]
- Willard, L.; Ranjan, A.; Zhang, H.; Monzavi, H.; Boyko, R.F.; Sykes, B.D.; Wishart, D.S. VADAR: A web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31, 3316–3319. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.W.; Pandit, S.A.; Scott, H.L.; Jakobsson, E. An improved united atom force field for simulation of mixed lipid bilayers. J. Phys. Chem. B 2009, 113, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Kleywegt, G.J. Crystallographic refinement of ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dommert, F.; Holm, C. Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. J. Chem. Phys. 2010, 133, 034117. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Sansom, M.S.; Biggin, P.C. Molecular dynamics studies of AChBP with nicotine and carbamylcholine: The role of water in the binding pocket. Protein Eng. Des. Sel. 2007, 20, 353–359. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Urease Activity | Compound | Urease Activity |
|---|---|---|---|
| IC50 ± SEM (µM) | IC50 ± SEM (µM) | ||
| 3a | 0.081 ± 0.003 | 9a | 0.2888 ± 0.015 |
| 3b | 0.078 ± 0.003 | 9b | 0.02757 ± 0.001 |
| 3c | 0.110 ± 0.004 | 9c | 0.01776 ± 0.000 |
| 3d | 0.082 ± 0.003 | 9d | 0.0533 ± 0.001 |
| 3e | 0.045 ± 0.001 | 9e | 0.0250 ± 0.000 |
| 3f | 0.073 ± 0.004 | 9f | 0.0558 ± 0.001 |
| 6a | 0.002 ± 0.000 | 12a | 0.2254 ± 0.006 |
| 6b | 0.233 ± 0.011 | 12b | 0.141 ± 0.004 |
| 6c | 0.141 ± 0.007 | 12c | 0.186 ± 0.007 |
| 6d | 0.095 ± 0.004 | 12d | 0.026 ± 0.002 |
| 6e | 0.174 ± 0.009 | 12e | 0.096 ± 0.0040 |
| 6f | 0.127 ± 0.0076 | 12f | 0.067 ± 0.0029 |
| Thiourea | 17.814 ± 0.096 |
| Code | Dose (µM) | Vmax (ΔA/Min) | Km (mM) | Inhibition Type | Ki (µM) | Ki′ (µM) |
|---|---|---|---|---|---|---|
| 3e | 0.025 | 0.00337 | 0.606 | |||
| 0.05 | 0.00273 | 1.923 | Mixed-inhibition | 0.016 | 0.195 | |
| 0.01 | 0.00233 | 3.389 | ||||
| 0.02 | 0.001632 | 5.263 | ||||
| 0 | 0.0204 | 5 | ||||
| 6a | 0.0011 | 0.00769 | 6.666 | Mixed-inhibition | 0.0011 | 0.0016 |
| 0.0022 | 0.00493 | 6.896 | ||||
| 0.0044 | 0.00351 | 7.518 | ||||
| 0 | 0.0114 | 4.166 | ||||
| 9e | 0.015 | 0.00127 | 4.166 | Noncompetitive | 0.0225 | --- |
| 0.03 | 0.00108 | 4.166 | ||||
| 0.06 | 0.00085 | 4.166 | ||||
| 0.013 | 0.00299 | 1.754 | ||||
| 12d | 0.026 | 0.00213 | 2.439 | Mixed-inhibition | 0.053 | 0.125 |
| 0.052 | 0.00194 | 2.857 | ||||
| 0.104 | 0.0016 | 3.174 | ||||
| 0.208 | 0.001 | 3.773 |
| Properties | 3a | 3b | 3c | 3d | 3e | 3f | 6a | 6b | 6c | 6d | 6e | 6f | 9a | 9b | 9c | 9d | 9e | 9f | 12a | 12b | 12c | 12d | 12e | 12f |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mol. weight (g/mol) | 485 | 517 | 547 | 539 | 499 | 531 | 404 | 436 | 466 | 466 | 457 | 418 | 305 | 337 | 367 | 359 | 319 | 351 | 245 | 277 | 307 | 298 | 259 | 291 |
| No. HBA | 6 | 8 | 6 | 6 | 6 | 8 | 6 | 8 | 6 | 6 | 6 | 6 | 3 | 5 | 3 | 3 | 3 | 5 | 4 | 6 | 4 | 4 | 4 | 6 |
| No. HBD | 1 | 2 | 1 | 1 | 1 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 1 | 1 | 2 | 4 | 5 | 4 | 4 | 4 | 5 |
| Mol. LogP | 3.5 | 2.7 | 4.9 | 4.3 | 3.9 | 2.9 | 2.3 | 1.4 | 3.7 | 3.7 | 3.1 | 2.7 | 4.3 | 3.5 | 5.7 | 5.1 | 4.7 | 3.8 | 1.1 | 0.29 | 2.5 | 1.8 | 1.50 | 0.5 |
| Mol. PSA (A2) | 78.3 | 116 | 78 | 78 | 78 | 116 | 102 | 140 | 101 | 101 | 102 | 102 | 41 | 80 | 41 | 41 | 41 | 79 | 73 | 111 | 72 | 73 | 73 | 111 |
| Stereo centers | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mol. Vol (A3) | 470 | 479 | 525 | 482 | 491 | 500 | 327 | 335 | 382 | 382 | 336 | 348 | 312 | 321 | 363 | 321 | 333 | 342 | 202 | 212 | 258 | 211 | 224 | 232 |
| Molar Refractivity (cm3) | 122 | 124 | 142 | 127 | 127 | 128 | 101 | 102 | 121 | 121 | 106 | 105 | 83 | 85 | 104 | 88 | 88 | 90 | 62 | 64 | 82 | 67 | 67 | 68 |
| Surface tension (dyne/cm) | 69 | 79 | 67 | 73 | 67 | 76 | 74 | 87 | 71 | 71 | 79 | 71 | 51 | 62 | 57 | 58 | 50 | 60 | 65 | 84 | 63 | 73 | 61 | 77 |
| Density (g/cm3) | 1.4 | 1.6 | 1.4 | 1.6 | 1.4 | 1.5 | 1.4 | 1.6 | 1.4 | 1.3 | 1.6 | 1.4 | 1.2 | 1.4 | 1.2 | 1.4 | 1.2 | 1.3 | 1.4 | 1.6 | 1.3 | 1.6 | 1.3 | 1.5 |
| Polarizability (cm3) | 48 | 49 | 56 | 50 | 50 | 51 | 40 | 40 | 48 | 46 | 42 | 42 | 33 | 33 | 41 | 35 | 35 | 35 | 24 | 25 | 32 | 26 | 26 | 27 |
| Drug Score | 0.92 | 0.59 | 1.07 | 0.80 | 1.16 | 0.46 | −0.14 | −0.64 | 0.12 | 0.13 | 0.04 | 0.21 | −0.85 | −0.61 | −0.54 | −0.82 | −0.30 | −1.00 | −0.06 | −0.01 | 0.12 | −0.39 | 0.23 | −0.70 |
| No. Rotatable bonds | 5 | 6 | 6 | 5 | 5 | 6 | 6 | 7 | 7 | 7 | 6 | 6 | 3 | 3 | 4 | 3 | 3 | 4 | 4 | 5 | 5 | 4 | 4 | 4 |
| Lipinski Rule | Yes | No | No | No | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Channar, P.A.; Saeed, A.; Albericio, F.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Seo, S.-Y. Sulfonamide-Linked Ciprofloxacin, Sulfadiazine and Amantadine Derivatives as a Novel Class of Inhibitors of Jack Bean Urease; Synthesis, Kinetic Mechanism and Molecular Docking. Molecules 2017, 22, 1352. https://doi.org/10.3390/molecules22081352
Channar PA, Saeed A, Albericio F, Larik FA, Abbas Q, Hassan M, Raza H, Seo S-Y. Sulfonamide-Linked Ciprofloxacin, Sulfadiazine and Amantadine Derivatives as a Novel Class of Inhibitors of Jack Bean Urease; Synthesis, Kinetic Mechanism and Molecular Docking. Molecules. 2017; 22(8):1352. https://doi.org/10.3390/molecules22081352
Chicago/Turabian StyleChannar, Pervaiz Ali, Aamer Saeed, Fernando Albericio, Fayaz Ali Larik, Qamar Abbas, Mubashir Hassan, Hussain Raza, and Sung-Yum Seo. 2017. "Sulfonamide-Linked Ciprofloxacin, Sulfadiazine and Amantadine Derivatives as a Novel Class of Inhibitors of Jack Bean Urease; Synthesis, Kinetic Mechanism and Molecular Docking" Molecules 22, no. 8: 1352. https://doi.org/10.3390/molecules22081352