Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Standard Stock Solutions
2.3. HPLC System
2.4. Mobile Phase
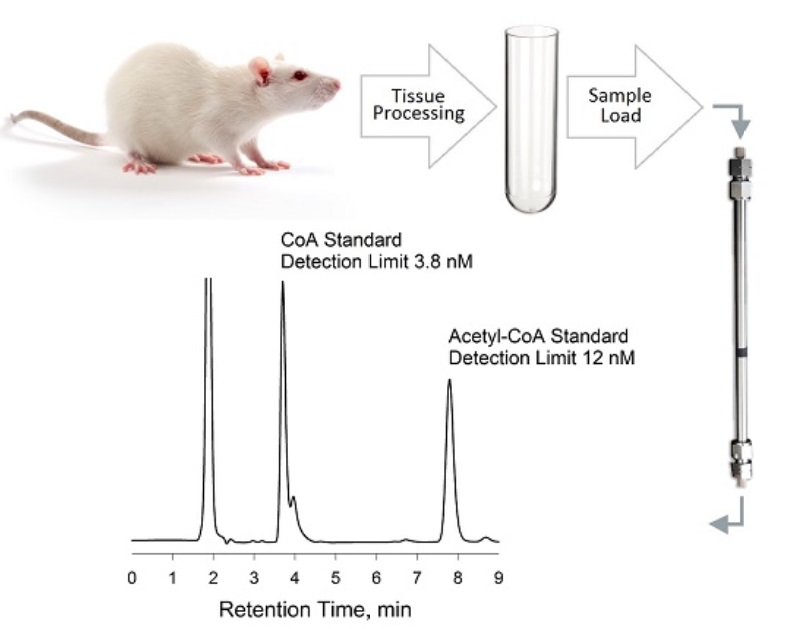
2.5. Preparation of Biological Samples for HPLC Analysis
2.5.1. Cell Cultures
2.5.2. Mouse Cerebral Cortex
2.5.3. Rat Tissues
2.5.4. Rat Plasma
2.5.5. Treatment of Neutralized Tissue Extracts with Citrate Synthase (CS)
2.5.6. Treatment of Neutralized Tissue Extracts with Phosphotransacetylase (PTA)
2.6. Protein Analysis
2.7. Statistical Analysis
3. Results
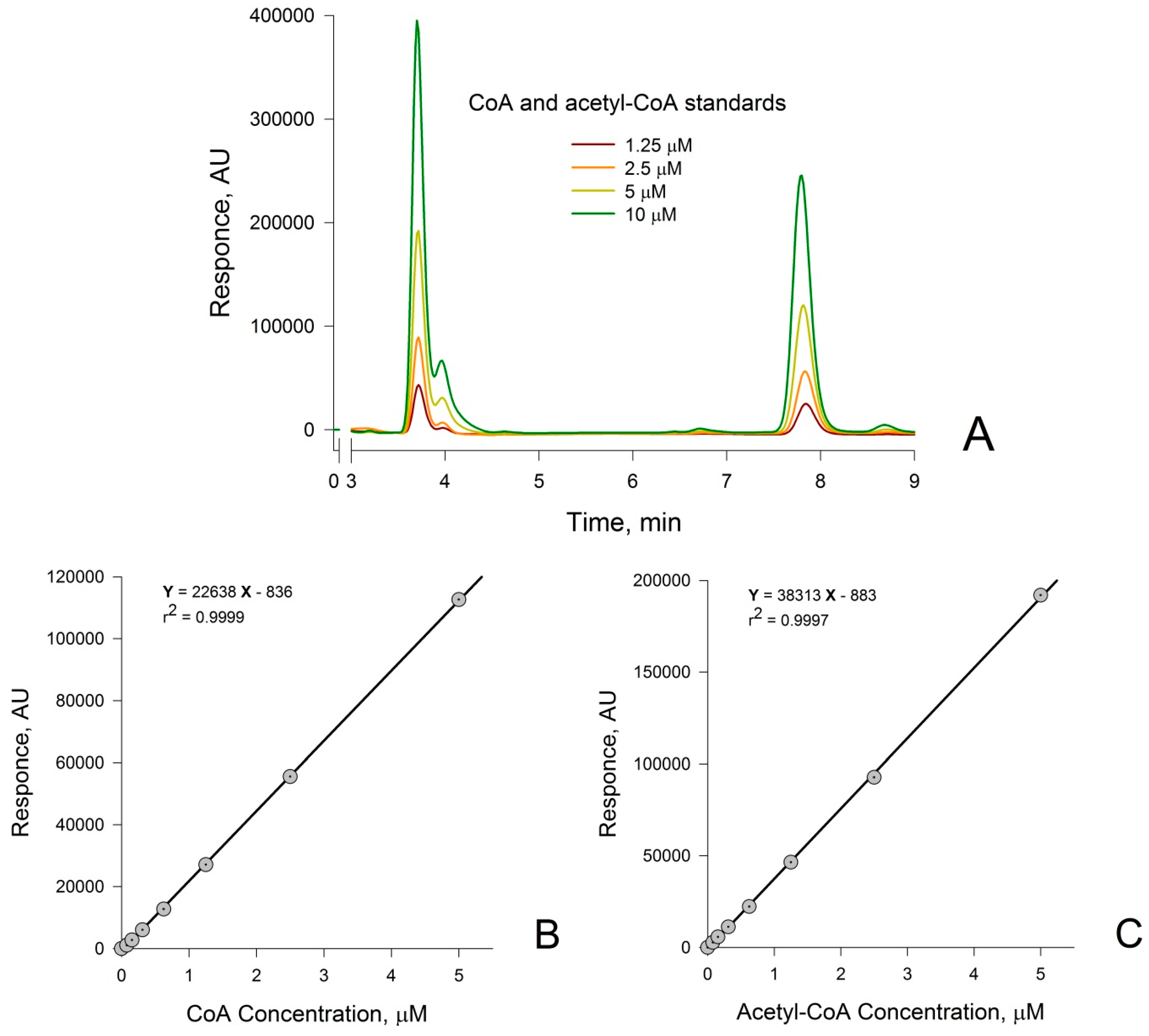
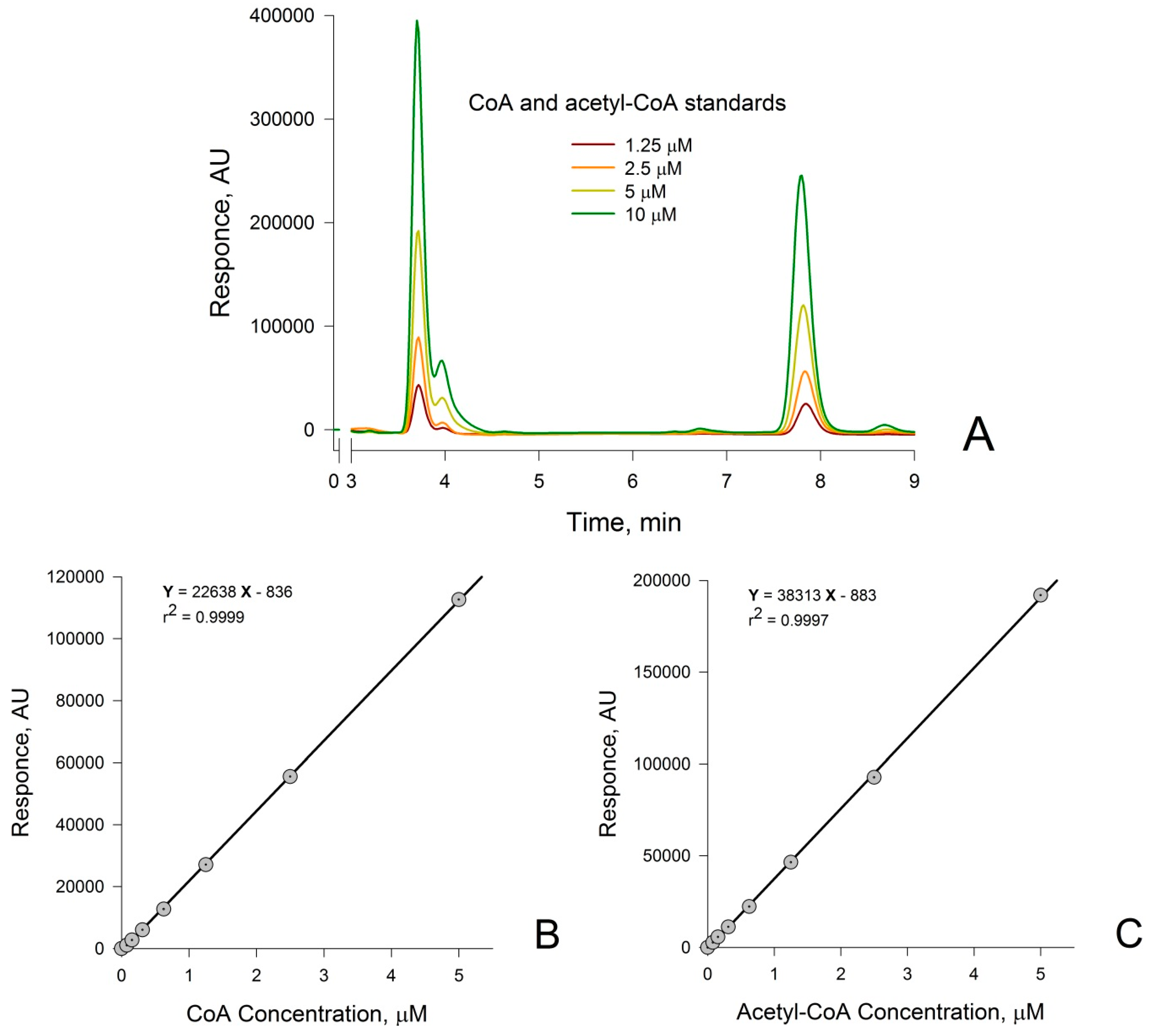
3.1. Measurement of CoA and Acetyl-CoA in Combined Standard Solutions by HPLC
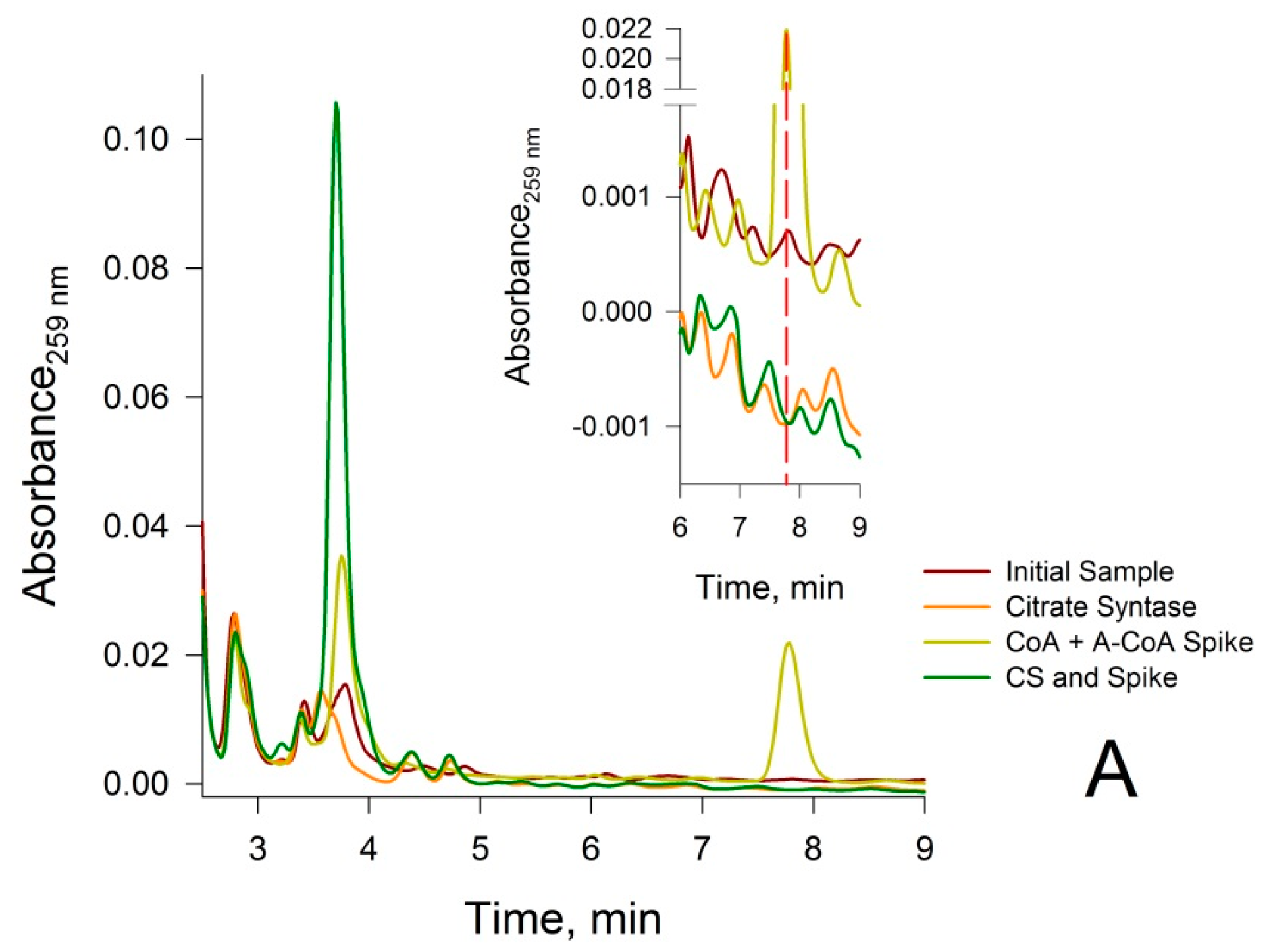
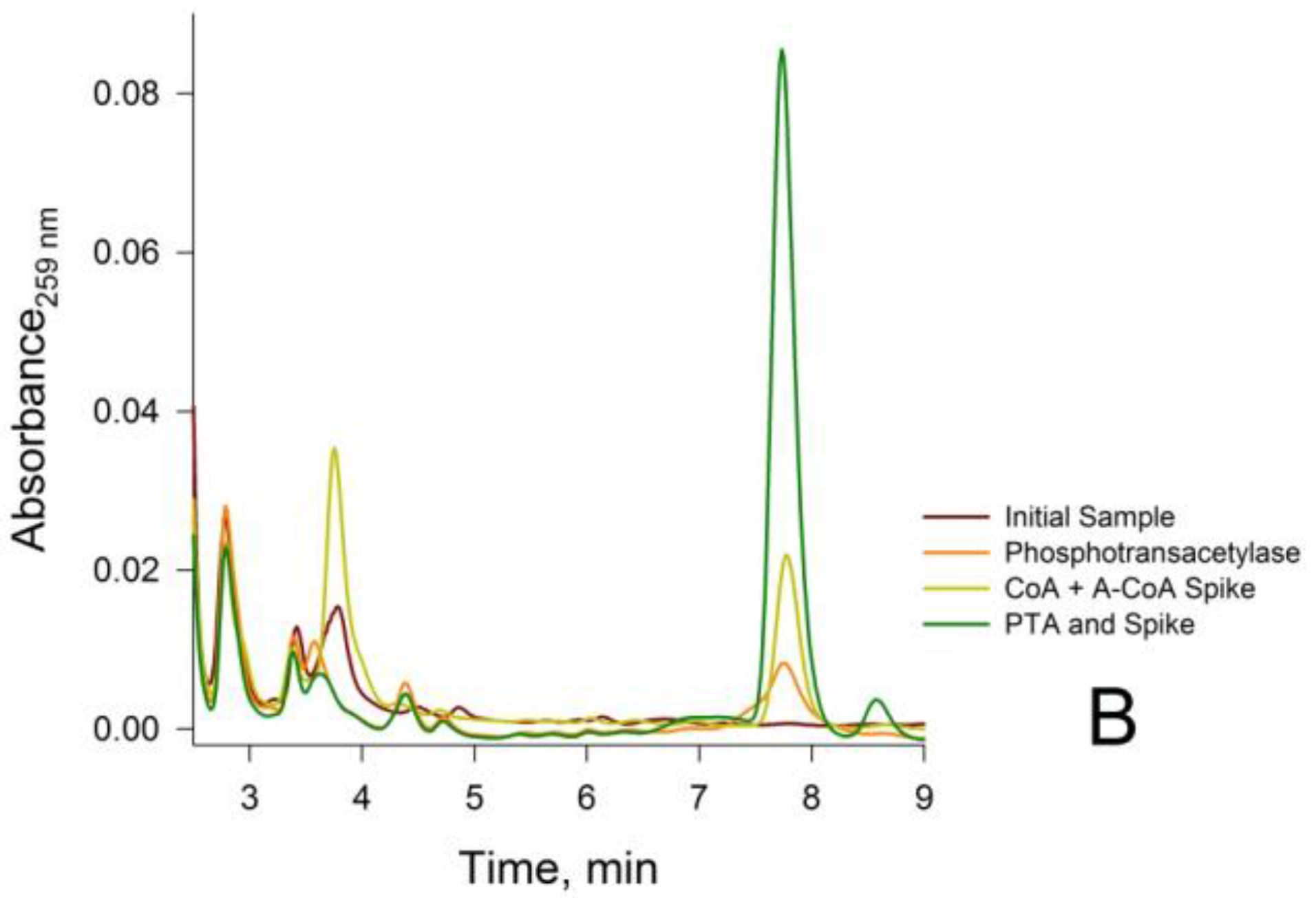
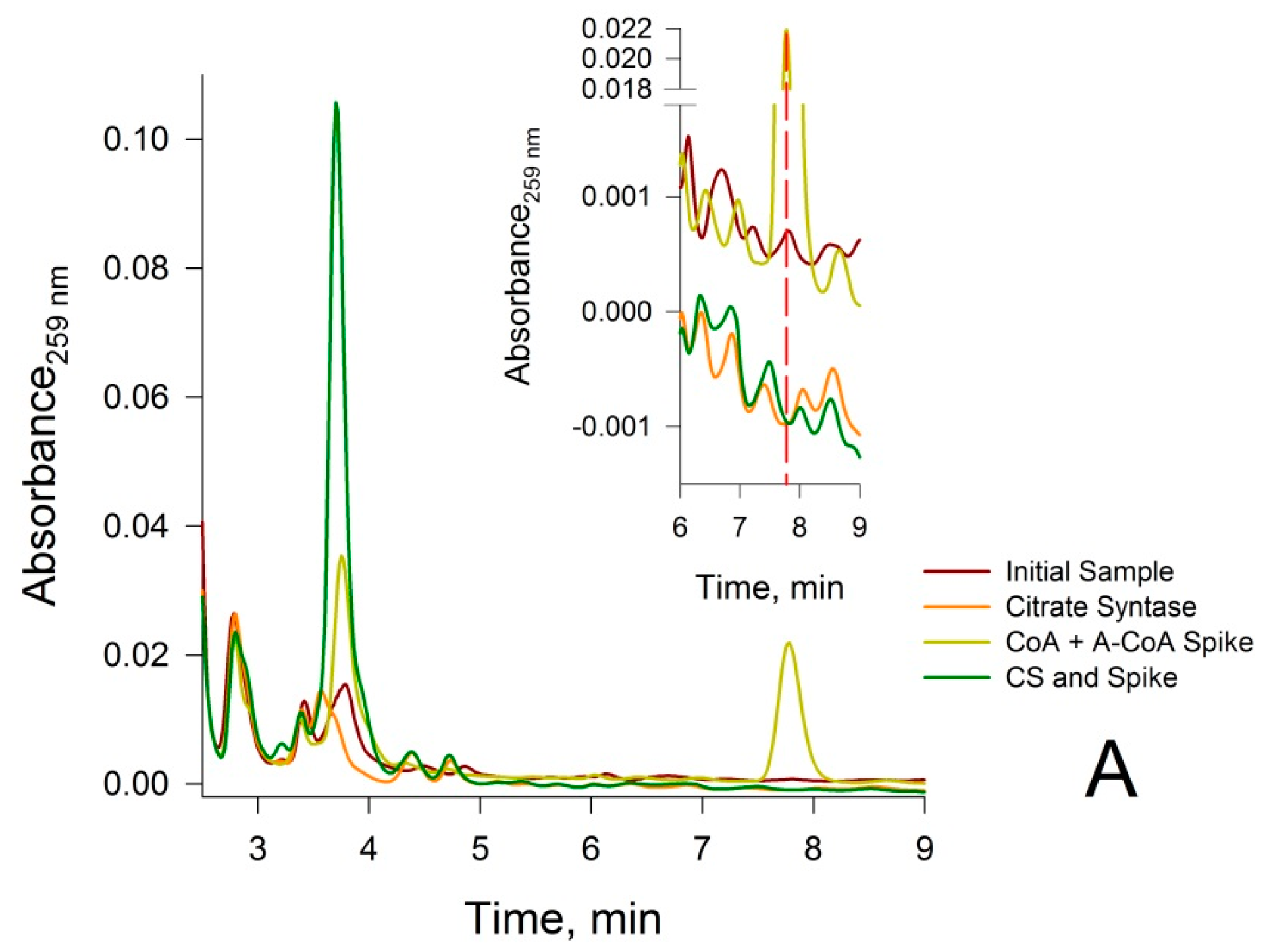
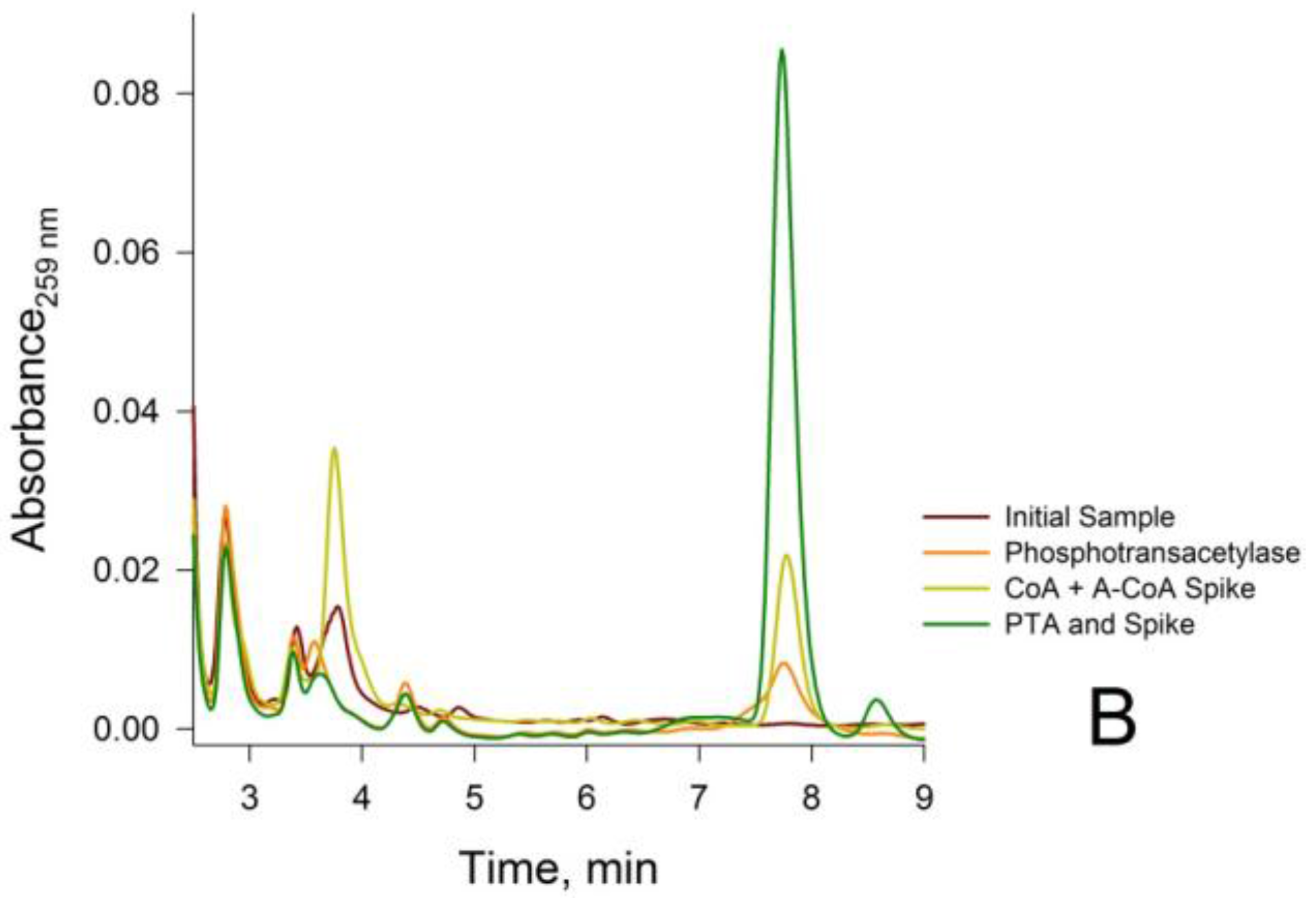
3.2. Confirmation that the HPLC Peaks at 3.8 min and 7.8 min Obtained from a Rat Liver Homogenate Are Due Solely to CoA and Acetyl-CoA, Respectively
3.3. Determination of CoA and Acetyl-CoA Concentrations in Biological Samples
4. Discussion
4.1. WT and mtDNA Mutant Cells
4.2. Rat Liver and Kidney
4.3. Rat Plasma
4.4. Rodent Brain
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leonardi, R.; Jackowski, S. Coenzyme A: When small is mighty. ASBMB Today 2013, 12, 56–57. [Google Scholar]
- Davaapil, H.; Tsuchiya, Y.; Gout, I. Signalling functions of coenzyme A and its derivatives in mammalian cells. Biochem. Soc. Trans. 2014, 42, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Abo Alrob, O.; Lopaschuk, G.D. Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem. Soc. Trans. 2014, 42, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San, P.J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, P.K.; Garland, P.B. Variations in tissue contents of coenzyme A thio esters and possible metabolic implications. Biochem. J. 1964, 93, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Jenniskens, F.A.; Jopperi-Davis, K.S.; Walters, L.C.; Schorr, E.N.; Rogers, L.K.; Welty, S.E.; Smith, C.V. Effects of fasting on tissue contents of coenzyme A and related intermediates in rats. Pediatr. Res. 2002, 52, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell. 2011, 42, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Leonardi, R.; Lin, W.; Boyd, V.A.; Goktug, A.; Shelat, A.A.; Chen, T.; Jackowski, S.; Rock, C.O. A high-throughput screen reveals new small-molecule activators and inhibitors of pantothenate kinases. J. Med. Chem. 2015, 58, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Jackowski, S.; Leonardi, R. Deregulated coenzyme A, loss of metabolic flexibility and diabetes. Biochem. Soc. Trans. 2014, 42, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Říčný, J.; Tuček, S. Acetyl coenzyme A in the brain: Radioenzymatic determination, use of microwaves, and postmortem changes. Anal. Biochem. 1980, 103, 369–376. [Google Scholar] [CrossRef]
- Liu, G.; Chen, J.; Che, P.; Ma, Y. Separation and quantitation of short-chain coenzyme A’s in biological samples by capillary electrophoresis. Anal. Chem. 2003, 75, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Allred, J.B.; Guy, D.G. Determination of coenzyme A and acetyl CoA in tissue extracts. Anal. Biochem. 1969, 29, 293–299. [Google Scholar] [CrossRef]
- McDougal, D.B.; Dargar, R.V. A spectrophotometric cycling assay for reduced coenzyme a and its esters in small amounts of tissue. Anal. Biochem. 1979, 97, 103–115. [Google Scholar] [CrossRef]
- Coulier, L.; Bas, R.; Jespersen, S.; Verheij, E.; Van der Werf, M.J.; Hankemeier, T. Simultaneous quantitative analysis of metabolites using ion-pair liquid chromatography electrospray ionization mass spectrometry. Anal. Chem. 2006, 78, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Chu, D.B.; Marx, H.; Sauer, M.; Hann, S.; Koellensperger, G. LC-MS/MS-based analysis of coenzyme A and short-chain acyl-coenzyme A thioesters. Anal. Bioanal. Chem. 2015, 407, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Giljanović, J.; Prkić, A. Determination of coenzyme A (CoASH) in the presence of different thiols by using flow-injection with a UV/Vis spectrophotometric detector and potentiometric determination of CoASH using an iodide ISE. Molecules 2009, 15, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Vadali, R.V.; Bennett, G.N.; San, K.Y. Cofactor engineering of intracellular CoA/acetyl-CoA and its effect on metabolic flux redistribution in Escherichia coli. Metab. Eng. 2004, 6, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Pham, U.; Gout, I. Methods for measuring CoA and CoA derivatives in biological samples. Biochem. Soc. Trans. 2014, 42, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Pham, U.; Hu, W.; Ohnuma, S.; Gout, I. Changes in Acetyl CoA levels during the early embryonic development of Xenopus laevis. PLoS ONE 2014, 9, e97693. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Nakai, T.; Fukuwatari, T. Simultaneous high-performance liquid chromatography determination of coenzyme A, dephospho-coenzyme A, and acetyl-coenzyme A in normal and pantothenic acid-deficient rats. Anal. Biochem. 2012, 430, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Shurubor, Y.I.; Cooper, A.J.L.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Krasnikov, B.F. Simultaneous determination of tricarboxylic acid cycle metabolites by high-performance liquid chromatography with ultraviolet detection. Anal. Biochem. 2016, 503, 8–10. [Google Scholar] [CrossRef] [PubMed]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Human Mol. Genet. 2010, 19, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Clark-Matott, J.; Saleem, A.; Dai, Y.; Shurubor, Y.; Ma, X.; Safdar, A.; Beal, M.F.; Tarnopolsky, M.; Simon, D.K. Metabolomic analysis of exercise effects in the POLG mitochondrial DNA mutator mouse brain. Neurobiol. Aging 2015, 36, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Dunn, D.A.; Cannon, M.V.; Irwin, M.H.; Pinkert, C.A. Animal models of human mitochondrial DNA mutations. Biochim. Biophys. Acta 2012, 1820, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, S.H.; Ferry, J.G. Steady-state kinetic analysis of phosphotransacetylase from Methanosarcina thermophile. J. Bacteriol. 2006, 188, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Bergmeyer, H.U. Methods of Enzymatic Analysis, 2nd ed.; Verlag Chemie Weinheim Academic Press, Inc.: New York, NY, USA, 1974; pp. 2287–2288. [Google Scholar]
- Snoswell, A.M.; Koundakjian, P.P. Relationships between carnitine and coenzyme A esters in tissues of normal and alloxan-diabetic sheep. Biochem. J. 1972, 127, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Guo, T.; Gross, V.; Lazarev, A.; Koh, C.C.; Gillessen, S.; Joerger, M.; Jochum, W.; Aebersold, R. Reproducible tissue homogenization and protein extraction for quantitative proteomics using micropestle-assisted pressure-cycling technology. J. Proteome Res. 2016, 15, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.; Wu, J.W.; Wang, S.P.; Allard, P.; Mamer, O.A.; Sweetman, L.; Moser, A.B.; Kratz, L.; Alvarez, F.; Robitaille, Y.; et al. A liver-specific defect of Acyl-CoA degradation produces hyperammonemia, hypoglycemia and a distinct hepatic Acyl-CoA pattern. PLoS ONE 2013, 8, e60581. [Google Scholar] [CrossRef] [PubMed]
- Piantoni, P.; Ylioja, C.M.; Allen, M.S. Feed intake is related to changes in plasma nonesterified fatty acid concentration and hepatic acetyl CoA content following feeding in lactating dairy cows. J. Dairy Sci. 2015, 98, 6839–6847. [Google Scholar] [CrossRef] [PubMed]
- Brass, E.P.; Hoppel, C.L. Relationship between acid-soluble carnitine and coenzyme A pools in vivo. Biochem. J. 1980, 190, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Passonneau, J.V.; Hasselberger, F.X.; Schulz, D.W. Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J. Biol. Chem. 1964, 239, 18–30. [Google Scholar] [PubMed]
- Kato, T. Ischemic effect on CoASH and acetyl-CoA concentration levels in cerebrum, cerebellum and liver of mice. J. Neurochem. 1978, 31, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, C.; Nistér, M. Protein extraction from solid tissue. Methods Mol. Biol. 2011, 675, 307–312. [Google Scholar] [PubMed]
- Song, W.J.; Jackowski, S. Cloning, sequencing, and expression of the pantothenate kinase (coaA) gene of Escherichia coli. J. Bacteriol. 1992, 174, 6411–6417. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Rehg, J.E.; Rock, C.O.; Jackowski, S. Pantothenate Kinase 1 is required to support the metabolic transition from the fed to the fasted state. PLoS ONE 2010, 5, e11107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Sample | N | CoA | Acetyl-CoA |
|---|---|---|---|
| WT cells b | 6 | 0.467 ± 0.015 | 0.162 ± 0.004 |
| Mut cells b | 6 | 0.040 ± 0.001 e | 0.140 ± 0.009 f |
| WT mouse cortex c | 6 | 0.017 ± 0.006 | 0.008 ± 0.003 |
| Mut mouse cortex c | 6 | 0.012 ± 0.004 | 0.005 ± 0.001 |
| Rat liver b | 16 | 0.872 ± 0.122 | 0.194 ± 0.038 |
| Rat kidney b | 16 | 0.191 ± 0.062 | 0.013 ± 0.006 |
| Rat brain b | 16 | 0.079 ± 0.024 | 0.028 ± 0.011 |
| Rat plasma d | 16 | 0.009 ± 0.001 | 0.0003 ± 0.0001 |
| Bergmeyer [27] a | Shibata et al. [21] N = 5 | Present Findings b N = 16 | ||||
|---|---|---|---|---|---|---|
| Liver | Kidney | Liver | Kidney | Liver | Kidney | |
| CoA | 135–158 | 68 | 161 | 68 | 87.2 ± 12.2 (63–124) | 19.1 ± 6.2 (8–31) |
| Acetyl-CoA | 22–47 | 18–43 | 9 | 5 | 19.4 ± 3.8 (10–35) | 1.30 ± 0.60 (0.2–3.6) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shurubor, Y.I.; D’Aurelio, M.; Clark-Matott, J.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Cooper, A.J.L.; Krasnikov, B.F. Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection. Molecules 2017, 22, 1388. https://doi.org/10.3390/molecules22091388
Shurubor YI, D’Aurelio M, Clark-Matott J, Isakova EP, Deryabina YI, Beal MF, Cooper AJL, Krasnikov BF. Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection. Molecules. 2017; 22(9):1388. https://doi.org/10.3390/molecules22091388
Chicago/Turabian StyleShurubor, Yevgeniya I., Marilena D’Aurelio, Joanne Clark-Matott, Elena P. Isakova, Yulia I. Deryabina, M. Flint Beal, Arthur J. L. Cooper, and Boris F. Krasnikov. 2017. "Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection" Molecules 22, no. 9: 1388. https://doi.org/10.3390/molecules22091388