NO-Donor Iron Nitrosyl Complex with N-Ethylthiourea Ligand Exhibits Selective Toxicity to Glioma A172 Cells

Abstract
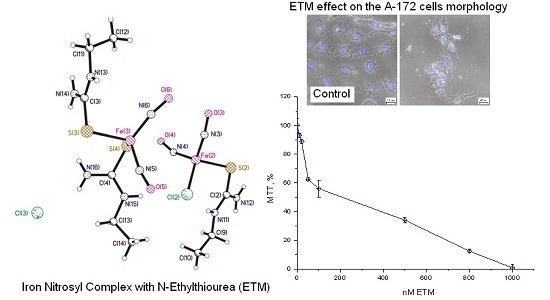
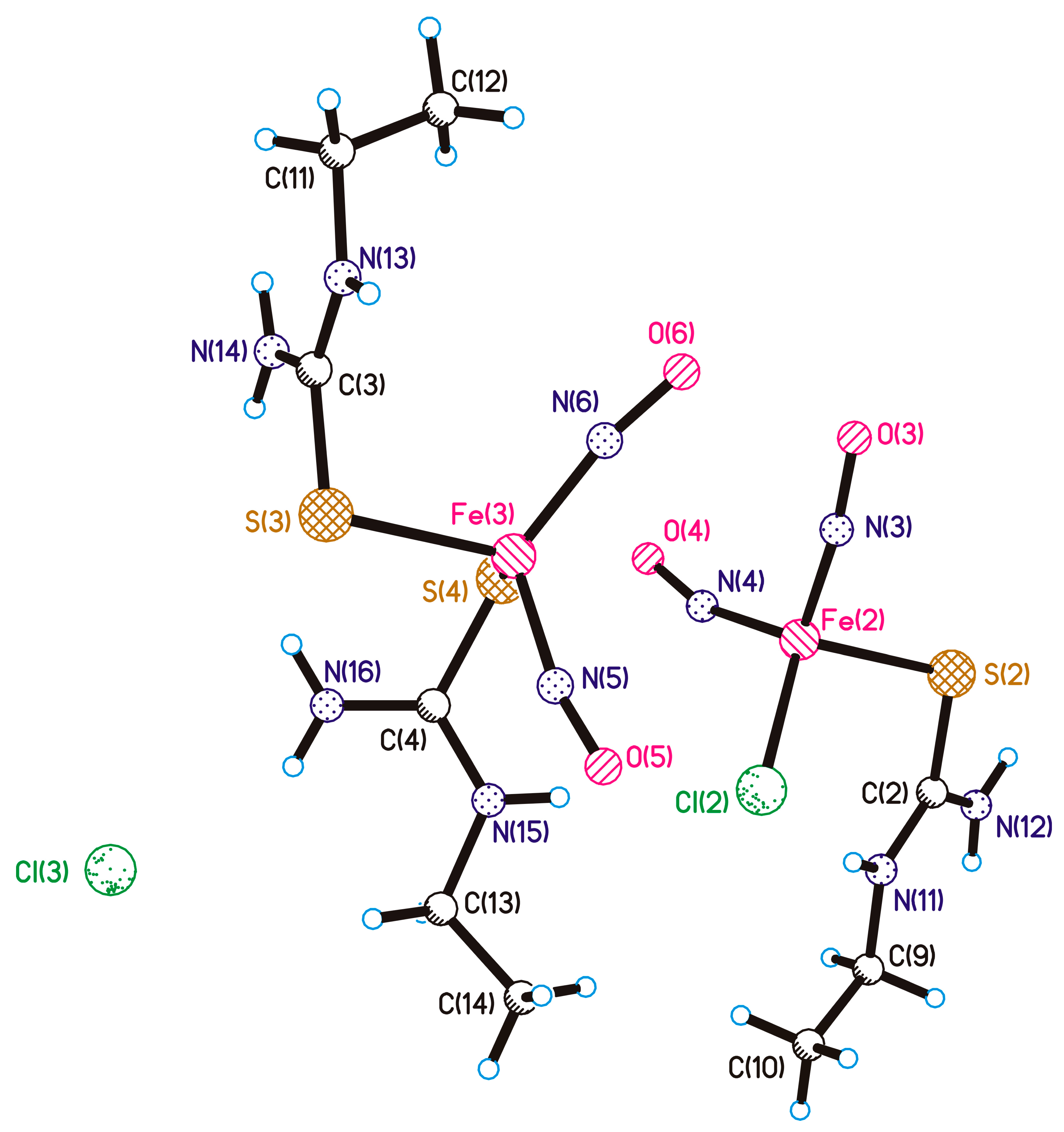
:
1. Introduction
2. Results
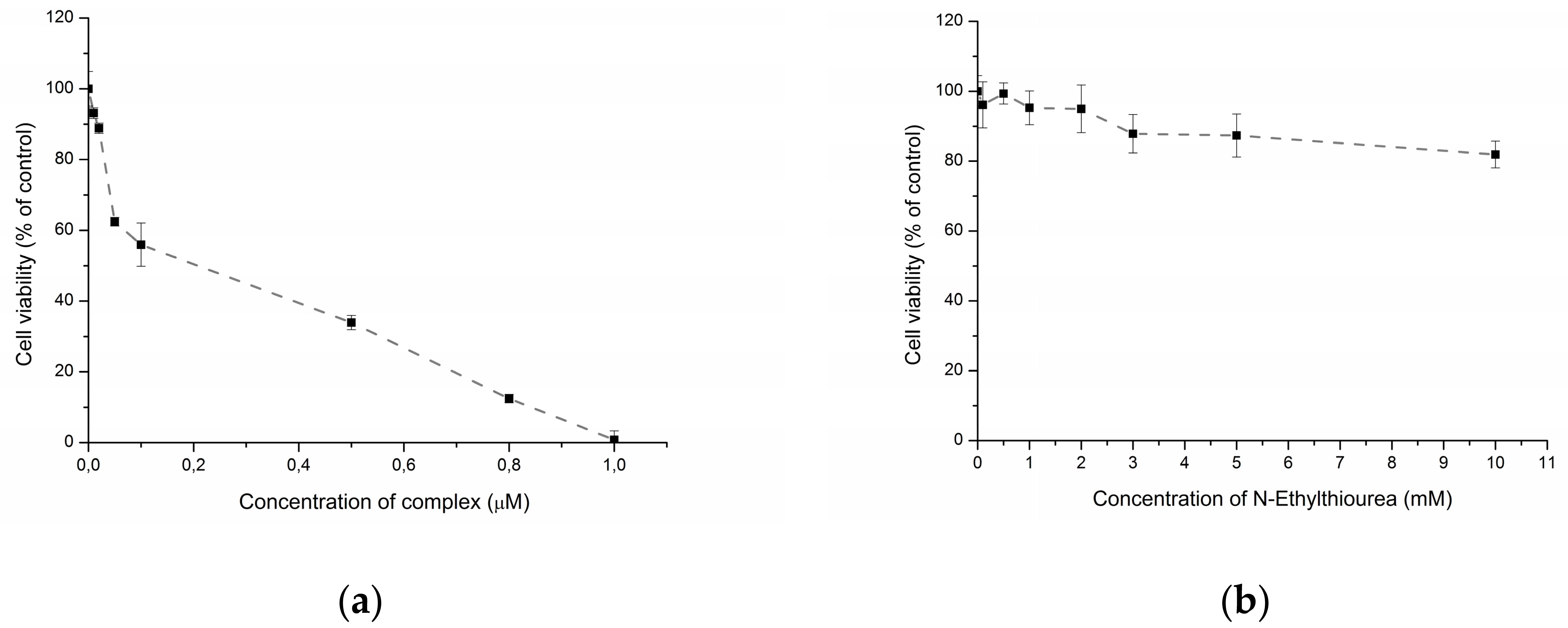
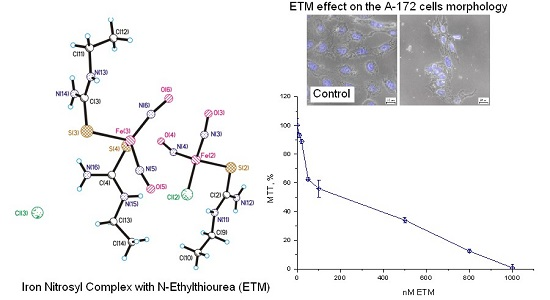
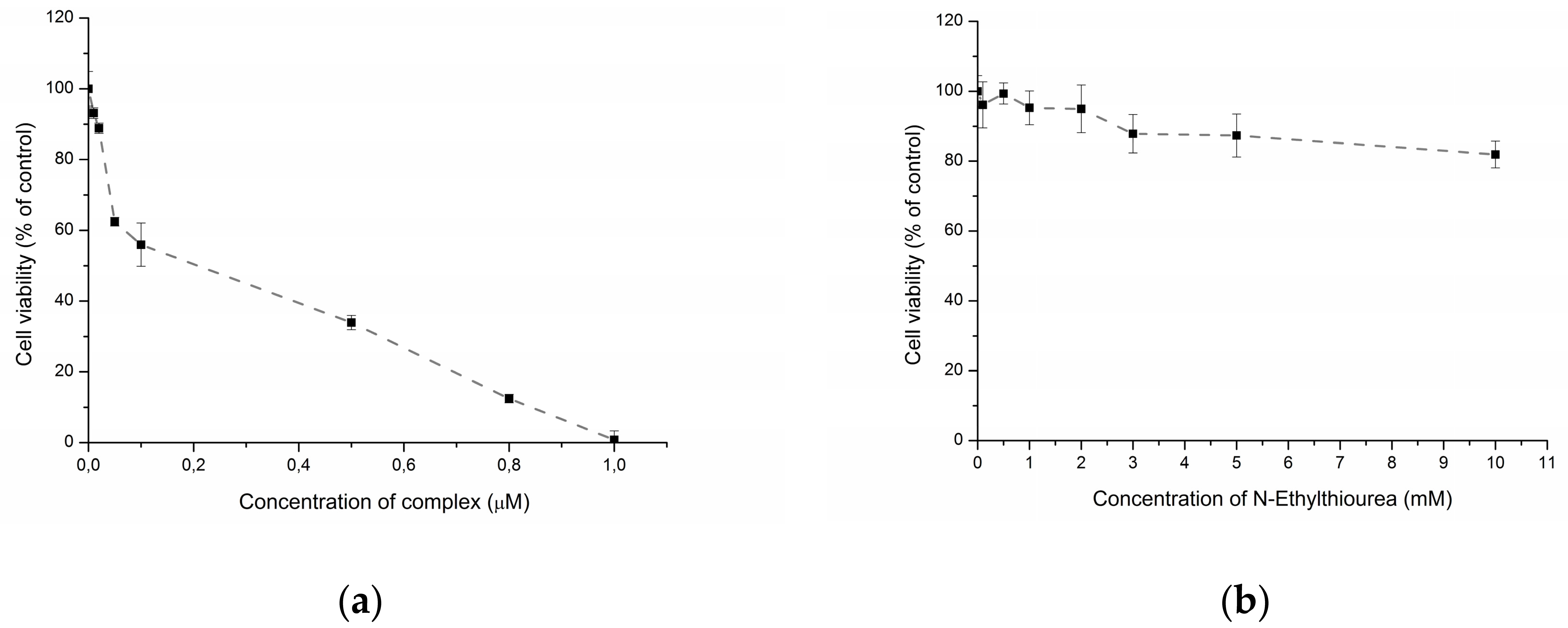
Effects of ETM on Еру Viability of Cells of Different Origin
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. Cell Culture
4.3. Cytotoxicity Studies and Determination of IC50 Doses
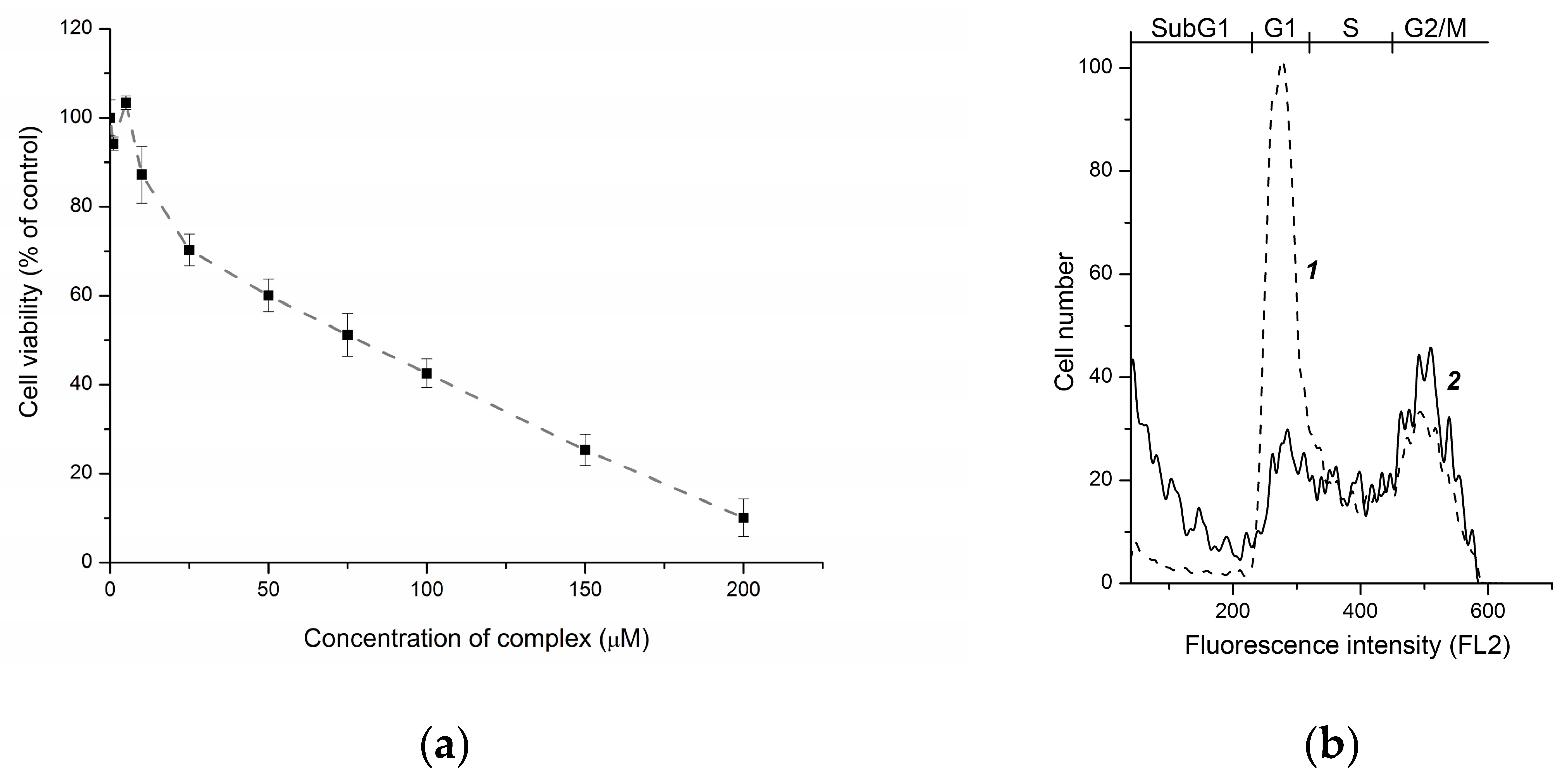
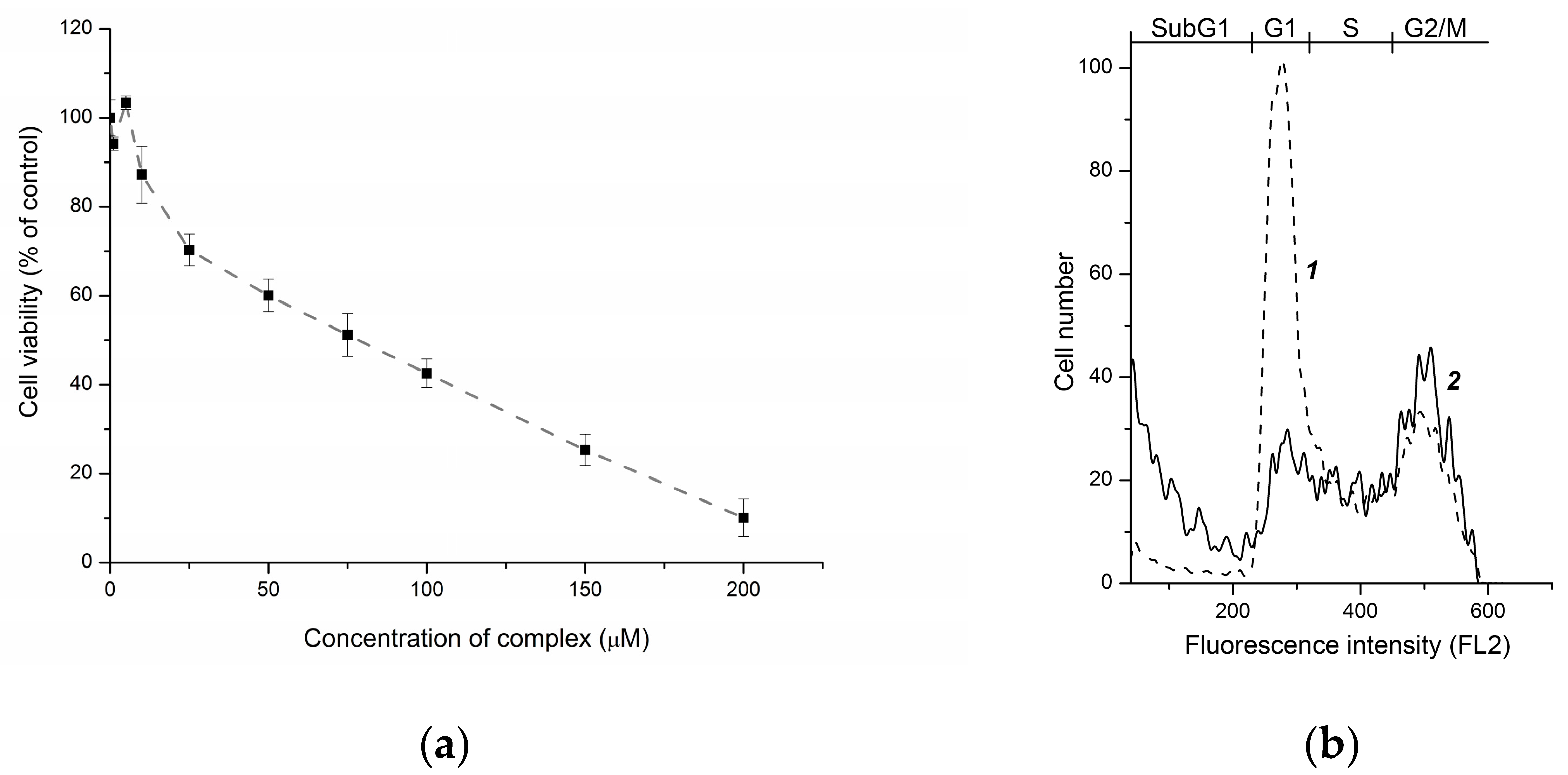
4.4. Flow Cytometry
4.5. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ghimire, K.; Altmann, H.M.; Straub, A.C.; Isenberg, J.S. Nitric oxide: What’s new to NO? Am. J. Physiol. Cell Physiol. 2017, 312, C254–C262. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, L.; Mollica, M.; Re, A.T.; Wu, S.; Zuo, L. Nitric oxide in cancer metastasis. Cancer Lett. 2014, 353, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Hirst, D.; Robson, T. Nitric oxide in cancer therapeutics: Interaction with cytotoxic chemotherapy. Curr. Pharm. Des. 2010, 16, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S.; Chilka, S.; Bonavida, B. Nitric oxide donors: Novel cancer therapeutics (review). Int. J. Oncol. 2008, 33, 909–927. [Google Scholar] [CrossRef] [PubMed]
- Mülsch, A.; Mordvintcev, P.; Vanin, A.F.; Busse, R. The potent vasodilating and guanylyl cyclase activating dinitrosyl-iron (II) complex is stored in a protein-bound form in vascular tissue and is released by thiols. FEBS Lett. 1991, 294, 252–256. [Google Scholar] [CrossRef]
- Richardson, D.R.; Lok, H.C. The nitric oxide-iron interplay in mammalian cells: Transport and storage of dinitrosyl iron complexes. Biochim. Biophys. Acta 2008, 1780, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, H. Coordination Chemistry of Nitrosyls and Its Biochemical Implications. Struct. Bond. 2013, 153, 45–114. [Google Scholar]
- Borodulin, R.R.; Kubrina, L.N.; Mikoyan, V.D.; Poltorakov, A.P.; Shvydkiy, V.O.; Burbaev, D.S.; Serezhenkov, V.A.; Yakhontova, E.R.; Vanin, A.F. Dinitrosyl iron complexes with glutathione as NO and NO(+) donors. Nitric Oxide 2013, 29, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Keszler, A.; Diers, A.R.; Ding, Z.; Hogg, N. Thiolate-based dinitrosyl iron complexes: Decomposition and detection and differentiation from S-nitrosothiols. Nitric Oxide 2017, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Feelisch, M.; te Poel, M.; Zamora, R.; Deussen, A.; Moncada, S. Understanding the controversy over the identity of EDRF. Nature 1994, 368, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Vanin, A.F.; Stukan, R.A.; Manukhina, E.B. Physical properties of dinitrosyl iron complexes with thiol-containing ligands in relation with their vasodilator activity. Biochim. Biophys. Acta 1996, 1295, 5–12. [Google Scholar] [CrossRef]
- Lobysheva, I.I.; Stupakova, M.V.; Mikoyan, V.D.; Vasilieva, S.V.; Vanin, A.F. Induction of the SOS DNA repair response in Escherichia coli by nitric oxide donating agents: Dinitrosyl iron complexes with thiol-containing ligands and S-nitrosothiols. FEBS Lett. 1999, 454, 177–180. [Google Scholar] [PubMed]
- Vasilieva, S.V.; Moshkovskaya, E.Y.; Sanina, N.A.; Aldoshin, S.M.; Vanin, A.F. Genetic signal transduction by nitrosyl-iron complexes in Escherichia coli. Biochemistry 2004, 69, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Kleschyov, A.L.; Strand, S.; Schmitt, S.; Gottfried, D.; Skatchkov, M.; Sjakste, N.; Daiber, A.; Umansky, V.; Munzel, T. Dinitrosyl-iron triggers apoptosis in Jurkat cells despite overexpression of Bcl-2. Free Radic. Biol. Med. 2006, 40, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.N.; Lo, F.C.; Tsai, M.L.; Shih, K.N.; Chiang, M.H.; Lee, G.H.; Liaw, W.F. Dinitrosyl iron complexes [E5Fe(NO)2]− (E = S, Se): A precursor of Roussin’s black salt [Fe4E3(NO)7]−. Inorg. Chim. Acta 2006, 359, 2525–2533. [Google Scholar] [CrossRef]
- Wen, Y.D.; Ho, Y.L.; Shiau, R.J.; Yeh, J.K.; Wu, J.Y.; Wang, W.L.; Chiou, S.J. Synergistic antitumor effect of curcumin and dinitrosyl iron complexes for against melanoma cells. J. Organomet. Chem. 2010, 695, 352–359. [Google Scholar] [CrossRef]
- Shiau, R.J.; Wu, J.Y.; Chiou, S.J.; Wen, Y.D. Effects of curcumin on nitrosyl-iron complex-mediated DNA cleavage and cytotoxicity. Planta Med. 2012, 78, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Giliano, N.Y.; Konevega, L.V.; Noskin, L.A.; Serezhenkov, V.A.; Poltorakov, A.P.; Vanin, A.F. Dinitrosyl iron complexes with thiol-containing ligands and apoptosis: Studies with HeLa cell cultures. Nitric Oxide 2011, 24, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Lu, C.Y.; Chen, Y.L.; Lo, F.C.; Wang, T.Y.; Chen, Y.J.; Liaw, W.F.; Wang, Y.M. Water-Soluble Dinitrosyl Iron Complex (DNIC): A Nitric Oxide Vehicle Triggering Cancer Cell Death via Apoptosis. Inorg. Chem. 2016, 55, 9383–9392. [Google Scholar] [CrossRef] [PubMed]
- Stupina, T.S.; Parkhomenko, I.I.; Balalaeva, I.V.; Kostyuk, G.V.; Sanina, N.A.; Terent´ev, A.A. Cytotoxic properties of the nitrosyl iron complex with phenylthiyl. Russ. Chem. Bull. Int. Ed. 2011, 60, 1488–1494. [Google Scholar] [CrossRef]
- Sanina, N.A.; Kozub, G.I.; Zhukova, O.S.; Emel’yanova, N.S.; Kondrat’eva, T.A.; Korchagin, D.V.; Shilov, G.V.; Ovanesyan, N.S.; Aldoshin, S.M. Synthesis, structure, NO donor activity of iron–sulfur nitrosyl complex with 2-aminophenol-2-yl and its antiproliferative activity against human cancer cells. J. Coord. Chem. 2013, 66, 3602–3618. [Google Scholar] [CrossRef]
- Stupina, N.S.; Terent’ev, A.A.; Antonova, N.O.; Balalaeva, I.V.; Sanina, N.A.; Aldoshin, S.M. Influence of sulfur-nitrosyl iron complexes of “µ-S” structural type on NF-κB nuclear factor. Int. Sci. J. Med. Biol. Sci. 2014, 1, 23–29. [Google Scholar]
- Sanina, N.A.; Shmatko, N.Y.; Korchagin, D.V.; Shilov, G.V.; Terent’ev, A.A.; Stupina, T.S.; Balakina, A.A.; Komleva, N.V.; Ovanesyan, N.S.; Kulikov, A.V.; et al. A New member of the cationic dinitrosyl iron complexes family incorporating N-ethylthiourea is effective against human HeLa and MCF-7 tumor cell lines. J. Coord. Chem. 2016, 5, 812–825. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014, 16, iv1–iv63. [Google Scholar] [CrossRef] [PubMed]
- Vakilian, A.; Khorramdelazad, H.; Heidari, P.; Rezaei, Z.S.; Hassanshahi, G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 2017, 103, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, M.; Endo, S.; Hirashima, Y.; Hamada, H.; Ogiichi, T.; Takaku, A. Growth inhibition and radiosensitization of cultured glioma cells by nitric oxide generating agents. J. Neurooncol. 1999, 42, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Viani, P.; Giussani, P.; Brioschi, L.; Bassi, R.; Anelli, V.; Tettamanti, G.; Riboni, L. Ceramide in nitric oxide inhibition of glioma cell growth. Evidence for the involvement of ceramide traffic. J. Biol. Chem. 2003, 278, 9592–9601. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shi, Y.; Li, S.; Qi, Q.; Gu, L.; Song, J.; Wang, P.G. A glycosylated nitric oxide donor, beta-Gal-NONOate, and its site-specific antitumor activity. Arch. Pharm. (Weinheim) 2006, 339, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Janjetovic, K.; Misirkic, M.; Vucicevic, L.; Harhaji, L.; Trajkovic, V. Synergistic antiglioma action of hyperthermia and nitric oxide. Eur. J. Pharmacol. 2008, 583, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, P.R.; Wang, P.G.; Lampidis, T.J.; Ardalan, B.; Braunschweiger, P. Differential expression of Glut 1 mRNA and protein levels correlates with increased sensitivity to the glyco-conjugated nitric oxide donor (2-glu-SNAP) in different tumor cell types. J. Chemother. 2008, 20, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Weyerbrock, A.; Baumer, B.; Papazoglou, A. Growth inhibition and chemosensitization of exogenous nitric oxide released from NONOates in glioma cells in vitro. J. Neurosurg. 2009, 110, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Safdar, S.; Taite, L.J. Targeted diazeniumdiolates: Localized nitric oxide release from glioma-specific peptides and proteins. Int. J. Pharm. 2012, 422, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-κB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.P.; Nozell, S.E.; Benveniste, E.T. NF-κB and STAT3 signaling in glioma: Targets for future therapies. Expert Rev. Neurother. 2010, 10, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Puliyappadamba, V.T.; Hatanpaa, K.J.; Chakraborty, S.; Habib, A.A. The role of NF-κB in the pathogenesis of glioma. Mol. Cell. Oncol. 2014, 1, e963478. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.E.; Hess, D.T.; Stamler, J.S. S-nitrosylation: Physiological regulation of NF-κB. Proc. Natl. Acad. Sci. USA 2004, 101, 8841–8842. [Google Scholar] [CrossRef] [PubMed]
- Colasanti, M.; Persichini, T. Nitric oxide: An inhibitor of NF-μB/Rel system in glial cells. Brain Res. Bull. 2000, 52, 155–161. [Google Scholar] [CrossRef]
- Rieger, J.; Ständer, M.; Löschmann, P.A.; Heneka, M.; Dichgans, J.; Klockgether, T.; Weller, M. Synthesis and biological effects of NO in malignant glioma cells: Modulation by cytokines including CD95L and TGF-β, dexamethasone, and p53 gene transfer. Oncogene 1998, 17, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 2004, 281, 301–311. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Mean IC50 ± SD (µM) |
|---|---|
| Vero | 359.80 ± 14.94 |
| A431 | 412.84 ± 35.76 |
| BT-20 | 393.62 ± 10.56 |
| BT-474 | 202.71 ± 9.25 ** |
| Hs578T | 275.30 ± 22.88 * |
| HepG2 | 356.07 ± 8.30 |
| Caco2 | 240.89 ± 5.43 ** |
| A172 | 0.41 ± 0.15 ** |
| PANC-1 | 638.95 ± 17.62 ** |
| SubG1 | G1 | S | G2/M | |
|---|---|---|---|---|
| control | 5.72% | 48.54% | 21.90% | 23.84% |
| ETM, 50 μM, 24 h | 30.90% | 15.08% | 21.71% | 32.31% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanina, N.; Shmatko, N.; Stupina, T.; Balakina, A.; Terent’ev, A. NO-Donor Iron Nitrosyl Complex with N-Ethylthiourea Ligand Exhibits Selective Toxicity to Glioma A172 Cells. Molecules 2017, 22, 1426. https://doi.org/10.3390/molecules22091426
Sanina N, Shmatko N, Stupina T, Balakina A, Terent’ev A. NO-Donor Iron Nitrosyl Complex with N-Ethylthiourea Ligand Exhibits Selective Toxicity to Glioma A172 Cells. Molecules. 2017; 22(9):1426. https://doi.org/10.3390/molecules22091426
Chicago/Turabian StyleSanina, Nataliya, Natal’ya Shmatko, Tatiyana Stupina, Anastasiya Balakina, and Alexei Terent’ev. 2017. "NO-Donor Iron Nitrosyl Complex with N-Ethylthiourea Ligand Exhibits Selective Toxicity to Glioma A172 Cells" Molecules 22, no. 9: 1426. https://doi.org/10.3390/molecules22091426