3.2. Chemistry
3.2.1. General Method for Synthesis of Acrylic Acids 1a–1r
Method A: A mixture of the appropriate the appropriate phenylacetic acid (0.50 g, 1 equivalent), benzaldehyde (1 equivalent), acetic anhydride (2 mL) and triethylamine (1 mL) were heated under reflux for 3–5 h. After acidification with concentrated hydrochloric acid (5 mL), the resulting solid was filtered off and recrystallised to yield the required acrylic acid. Method B: A mixture of the appropriate benzaldehyde (1 eq.), the appropriate phenylacetic acid (700 mg, 1 eq.), acetic anhydride (2 mL) and triethylamine (1 mL) were reacted in the microwave reactor at a 120 °C and for 30 min. After acidification with concentrated hydrochloric acid (5 mL), the resulting solid was filtered and recrystallised to afford the required acrylic acid.
(E)-2,3-bis(3,4,5-Trimethoxyphenyl)acrylic acid (1c). 3,4,5-Trimethoxybenzaldehyde (700 mg, 3.59 mmol) and 3,4,5-trimethoxyphenylacetic acid (744 mg, 3.59 mmol) were reacted following the general method A. Recrystallisation from methanol afforded the acrylic acid as fine yellow solid (484 mg, 33%). 3,4,5-Trimethoxybenzaldehyde (606 mg, 3.09 mmol) and 3,4,5-trimethoxyphenylacetic acid (700 mg, 3.09 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as fine yellow solid (1.2 g, 96%), m.p. 169–172 °C. 1H-NMR (CDCl3): δ 7.85 (s, 1H, C=CH), 6.53 (s, 2H, Ar-H), 6.42 (s, 2H, Ar-H), 3.88 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.83 (s, 6H, 2 × OCH3), 3.63 (s, 6H, 2 × OCH3). 13C-NMR (DMSO-d6): δ 171.4, 160.9, 153.1, 152.2, 141.7, 139.0, 137.1, 129.8, 128.7, 107.9, 106.9, 99.9, 60.5 (OCH3), 60.4 (OCH3), 55.2 (OCH3), 55.0 (OCH3). IR: νmax (KBr) cm−1: 3440.90, 2938.31, 1668.56, 1580.98, 1505.11, 1413.46, 1272.44, 1240.40. HRMS (EI): Found 405.1452 [M + H]+, C21H25O8 requires 405.1471. 405.1549.
(E)-3-(3-Hydroxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (1f). 3-Hydroxybenzaldehyde (377 mg, 3.09 mmol), 3,4,5-trimethoxyphenylacetic acid (700 mg, 3.09 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as fine yellow soild (711 mg, 60%), m.p. 230–231 °C. 1H-NMR (DMSO-d6): δ 7.61 (s, 1H, C=CH), 7.05 (t, 1H, 8 Hz, Ar-H), 6.67 (dd, 1H, 8 Hz, 1 Hz, Ar-H), 6.54–6.52 (m, 2H, Ar-H), 6.44 (s, 2H, Ar-H), 3.70 (s, 3H, O CH3), 3.67 (s, 6H, OCH3). 13C-NMR (DMSO-d6): δ 168.40 (COOH), 157.02, 152.93, 139.05, 136.96, 135.59, 132.96, 131.72, 129.19, 121.28, 116.85, 116.27, 106.74, 60.12 (OCH3), 55.91 (OCH3). IR: νmax (KBr) cm−1: 3362.7, 2941.0, 2836.0, 2627.4, 1681.6, 1584.8, 1411.8, 1239.2, 1125.0, 997.3, 965.6, 688.2. HRMS (EI): Found 353.1003 [M + Na]+, C18H18O6Na requires 353.1001. Hydrolysis of (E)-3-(3-acetoxyphenyl)-2-(3,4,5-trimethoxy-phenyl)acrylic acid 1k with NAOH/MeOH for 12 h followed by acidification to pH 4 afforded 1f in 88% yield.
(E)-3-(2,3,4-Trimethoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (1h). 2,3,4-Trimethoxy-benzaldehyde (607 mg, 3.09 mmol) and 3,4,5-trimethoxyphenylacetic acid (700 mg, 3.09 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as fine yellow solid (374 mg, 30%), m.p. 222–225 °C. 1H-NMR (DMSO-d6): δ 7.91 (s, 1H, C=CH), 6.61 (d, 1H, 9 Hz, Ar-H), 6.46 (m, 3H, Ar-H), 3.86 (s, 3H, OCH3), 3.74 (s, 6H, OCH3), 3.70 (s, 3H, OCH3), 3.68 (s, 6H, OCH3). 13C-NMR (DMSO-d6): δ 168.96, 154.79, 153.48, 153.36, 141.88, 137.35, 133.34, 132.05, 124.97, 121.23, 108.20, 107.30, 61.95 (OCH3), 60.88 (OCH3), 60.58 (OCH3), 56.40 (OCH3), 56.31 (OCH3) IR: νmax (KBr) cm−1: 3440.90, 2938.31, 1668.56, 1580.98, 1505.11, 1413.46, 1272.44, 1240.40. HRMS (EI): Found 427.1370 [M + Na]+, C21H24O8Na requires 427.1369.
(E)-3-(4-Hydroxy-3,5-dimethoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (1j). 3,5-Dimethoxy-4-hydroxybenzaldehyde (350 mg, 1.91 mmol) and 3,4,5-trimethoxyphenylacetic acid (433 mg, 1.91 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as pale yellow solid (400 mg, 48%), m.p. 212–214 °C. 1H-NMR (CDCl3): δ 7.86 (s, 1H, C=CH), 6.52 (s, 2H, Ar-H), 6.44 (s, 2H, Ar-H), 3.86 (s, 3H, OCH3), 3.83 (s, 6H, OCH3), 3.59 (s, 6H, OCH3). 13C-NMR (CDCl3): δ 171.94, 168.05, 153.40, 151.32, 141.59, 137.23, 131.51, 130.88, 130.35, 139.41, 107.34, 105.99, 60.34 (OCH3), 55.77 (OCH3), 55.37 (OCH3), 19.97 (CH3). IR: νmax (KBr) cm−1: 3438.9, 2942.4, 1765.4, 1676.7, 1594.8, 1506.7, 1420.4, 1272.3, 1236.7, 1127.5, 999.8, 906.9. HRMS (EI): Found 455.1321 [M + Na]+, C22H24O9Na requires 455.1318.
(E)-3-(3-Acetoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (1k). 3-Hydroxybenzaldehyde (350 mg, 2.3 mmol) and 3,4,5-trimethoxyphenylacetic acid (520 mg, 2.3 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as fine yellow solid (515 mg, 56%), m.p. 176–180 °C. 1H-NMR (CDCl3): δ 7.88 (s, 1H, C=CH), 6.95 (d, 1H, 8Hz, Ar-H), 6.88 (dd, 1H, 18 Hz, 2 Hz, Ar-H), 6.65 (s, 2H, Ar-H), 6.50 (s, 2H, Ar-H), 3.88 (s, 3H, OCH3), 3.81 (s, 6H, O CH3), 2.30 (s, 3H, COCH3). 13C-NMR (CDCl3): δ 171.72, 153.36, 150.13, 141.07, 140.40, 137.21, 132.37, 130.80, 130.36, 124.46, 122.28, 113.19, 105.96, 60.38 (OCH3), 55.72 (OCH3), 54.88 (OCH3), 20.18 (CH3). IR: νmax (KBr) cm−1: 3362.7, 2941.0, 2836.0, 2627.4, 1681.6, 1584.8, 1411.8, 1239.2, 1125.0, 997.3, 965.6, 688.2. HRMS (EI): Found 395.1110 [M + H]+, C20H20O7Na requires 395.1107.
(E)-2-(3,4,5-Trimethoxyphenyl)-3-(5-methylthiophen-2-yl)but-2-enoic acid (1p). 5-Methyl-2-thiophene-carboxaldehyde (389 mg, 0.337 mL, 3.09 mmol) and 3,4,5-trimethoxyphenylacetic acid (700 mg, 2.3 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as a dark yellow solid (375 mg, 36%), m.p. 212–217 °C. 1H-NMR (DMSO-d6): δ 7.87 (s, 1H, C=CH), 7.24 (d, 1H, 3.6 Hz, thiophene-H), 6.74 (d, 1H, 3.6 Hz, thiophene-H), 6.47 (s, 2H, Ar-H), 3.72 (s, 9H, OCH3), 2.31 (s, 3H, CH3). 13C-NMR (DMSO-d6): δ 15.23 (CH3), 55.93 (OCH3), 60.20 (OCH3), 106.94, 125.42, 128.27, 130.99, 133.25, 134.85, 136.05, 137.44, 153.36, 168.10. IR: νmax (KBr) cm−1: 3426.13, 2998.75, 2537.61, 1662.03, 1581.35, 1412.40, 1283.72, 1128.23. HRMS (EI): Found 357.0763 [M + Na]+, C17H18O5NaS requires 357.0773.
(E)-3-(Furan-3-yl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (1q). 3-Furaldehyde (296 mg, 0.26 mL, 3.09 mmol) and 3,4,5-trimethoxyphenylacetic acid (700 mg, 2.3 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as a dark brown solid (460 mg, 49%), m.p. 188–191 °C. 1H-NMR (DMSO-d6): 7.90 (s, 1H, C=CH), 7.74 (s, 1H, C=CH), 7.29–7.25 (m, 1H, C=CH), 6.86 (m, 1H, C=CH), 6.38 (s, 2H, Ar-H), 3.92 (s, 3H, OCH3), 3.81 (s, 6H, OCH3). 13C-NMR (DMSO-d6): 168.3, 153.0, 143.7, 143.6, 139.0, 138.4, 138.0, 124.4, 108.2, 105.1, 60.8 (OCH3), 56.1 (OCH3). IR: νmax (KBr) cm−1: 3447.5, 2938.9, 2627.6, 1674.9, 1582.2, 1504.8, 1909.5, 1504.8, 1909.5, 1236.5, 1127.5, 1007.9, 803.2. HRMS (EI): Found 327.0839 [M + Na]+, C16H16O6Na requires 327.0845.
(E)-3-(3-Methoxy-4-hydroxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (1r). 4-Hydroxy-3-methoxy-benzaldehyde (470 mg, 3.09 mmol) and 3,4,5-trimethoxyphenylacetic acid (700 mg, 3.09 mmol) were reacted following the general method B. Recrystallisation from methanol afforded the acrylic acid as fine yellow needles (923 mg, 83%), m.p. 237–239 °C. 1H-NMR (DMSO-d6): 12.45 (br s, 1H, COOH), 8.98 (br s, 1H, OH), 7.57 (s, 1H, C=CH), 6.81 (d, J = 7 Hz, 1H, Ar-H), 6.60 (d, J = 7 Hz, 1H, Ar-H), 6.53 (s, 1H, Ar-H), 6.44 (s, 2H, Ar-H), 3.73 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 3.69 (s, 6H, OCH3). 13C-NMR (DMSO-d6): 168.61 (COOH), 153.09, 148.89, 145.84, 139.13, 136.90, 132.18, 130.31, 127.02, 122.99, 117.19, 111.49, 106.65, 60.12 (OCH3), 55.91 (OCH3), 55.45 (OCH3). IR: νmax (KBr) cm−1: 3423.9 (br), 2939.8, 1671.3, 1585.2, 1509.6, 1455.3, 1411.1, 1268.3, 1239.3, 1126.2. HRMS (EI): Found 383.1103 [M + Na]+, C19H20O7Na requires 383.1107.
(4-((E/Z)-1-(4-(2-Methylaminoethoxy)phenyl)-2-phenylbut-1-enyl)phenoxy)(tert-butyl)dimethylsilane (2a). (4-((E/Z)-1-(4-(2-Bromoethoxy)phenyl)-2-phenylbut-1-enyl)phenoxy)(tert-butyl) dimethylsilane (1.457 g, 2.69 mmol) was dissolved in a solution (2 M) of methylamine in THF (1.678 g, 26.99 mL, 54 mmol). The reaction mixture was stirred in a sealed pressure tube for 48 h at 60 °C, followed by the addition of dichloromethane (100 mL). The organic phase was washed with a sodium bicarbonate/sodium carbonate (pH 10), (50 mL). The aqueous phase was then extracted with 3 × 50 mL dichloromethane. The organic layers were combined, dried over sodium sulfate and concentrated in vacuo to yield crude product. The material was purified via flash chromatography on silica gel (DCM:EtOAc 3:1) to afford the product as a brown resin. (1.24 g, 94%). 1H-NMR (CDCl3): δ 7.20–6.50 (m, 26H, Ar-H), 4.11 (t, 0.43 × 4H, J = 5.0 Hz, CH2), 3.95 (t, 0.43 × 4H, J = 5.0 Hz, CH2), 3.36 (s, 2H, NH), 2.99 (s, 0.57 × 4H, CH2), 2.89 (s, 0.43 × 4H, CH2) 2.53 (m, 10H, NCH3, CH2), 1.01–0.92 (m, 24H, CH3, (CH3)3), 0.26 (s, 0.43 × 12H, Si(CH3)2), 0.13 (s, 0.57 × 12H, Si(CH3)2). 13C-NMR (CDCl3): δ 156.94, 156.10, 153.84, 153.06, 142.19, 142.11, 140.65, 140.59, 137.53, 137.44, 136.35, 136.06, 136.02, 135.68, 131.57, 131.43, 130.20, 130.12, 129.25, 127.41, 127.33, 125.48, 125.44, 119.12, 118.54, 113.54, 112.80, 65.99, 65.73, 50.12, 50.03, 49.47, 35.39, 35.26, 28.60, 28.47, 25.26, 25.24, 17.75, 13.24, 13.21, −4.80, −4.91. IR: νmax (KBr) cm−1: 3431.2, 1606.0, 1508.0, 1241.3, 1173.2, 1099.5, 1078.8, 1036.9, 938.9, 846.8. HRMS (EI): Found 488.3088 [M + H]+, C31H41NO2Si requires 488.2907.
{2-[4-(1,2-Diphenylbut-1-enyl)phenoxy]ethyl}methylamine (2b) {2-[4-(1,2-Diphenylbut-1-enyl)phenoxy]-ethyl}bromide (0.41 g, 1.00 mmol) was dissolved in anhydrous tetrahydrofuran (20 mL) together with methylamine (in a 20 molar equivalent excess), and sealed in a high pressure tube. The reaction was heated to 60 °C while stirring for 48–72 h. Following addition of sodium carbonate/sodium hydrogen carbonate pH 10 buffer solution (50 mL), the mixture were extracted with DCM (3 × 50 mL). The organic phases were combined, dried over sodium sulfate and the solvent evaporated in vacuo to afford a crude product which was then purified via flash chromatography (DCM:MeOH) to afford the product as a brown oil (0.28 g, 77%, E/Z = 1:1.1). 1H-NMR (CDCl3): δ 0.96–1.02 (m, 6H, CH3), 2.49–2.58 (m, 10H, CH2, CH3), 2.98–3.08 (m, 4H, CH2N), 3.99–4.23 (m, 6H, NH, CH2O), 6.59–7.40 (m, 28H, ArH). 13C-NMR (CDCl3): 13.25, 28.62, 28.64, 35.16, 35.28, 49.76, 49.89, 65.45, 65.78, 112.96, 113.70, 125.28, 125.67, 126.17, 126.92, 127.39, 127.50, 127.72, 129.03, 129.28, 130.23, 130.40, 131.53, 135.44, 135.94, 137.74, 137.89, 141.03, 141.56, 141.89, 142.88, 143.33, 156.06, 156.92. IR: νmax (KBr) cm−1: 3431.2, 1606.0, 1508.0, 1241.3, 1173.2, 1099.5, 1078.8, 1036.9, 938.9, 846.8. HRMS (EI): Found 358.2173 [M + H]+, C25H28NO requires 358.2171.
3.2.2. General Method for the Synthesis of Endoxifen-Acrylic Acid Conjugates 3a–3m
Step (i): A mixture of the required acrylic acid (1 eq., 0.154 mmol), DCC (1 eq., 0.154 mmol, 32 mg), and HOBt (1 eq., 0.154 mmol, 21 mg) was suspended in 3 mL of anhydrous DCM and stirred for 10 min under a nitrogen atmosphere. The endoxifen derivative 2a (75 mg, 1 eq., 0.154 mmol) was dissolved in 3 mL of anhydrous DCM and slowly added to the mixture via syringe. Reaction was allowed stir for 24–48 h. Reaction was monitored via TLC. The reaction mixture was diluted to 15 mL with anhydrous DCM and filtered to remove DCU. The filtrate was evaporated to dryness under reduced pressure. Purification was not required at this step. Silyl deprotection: The residue was dissolved in THF (3 mL) and stirred under nitrogen. A solution of 0.1 M TBAF (2 eq.) was added to the mixture and allowed to stir for 24 h. The mixture was evaporated to dryness under reduced pressure. The residue was dissolved in DCM and was washed with 10% HCl solution. The resulting organic phase was dried over sodium sulphate and evaporated to dryness under vacuum. The material was purified via flash chromatography on silica gel.
Step (ii): A mixture of the required acrylic acid (1.2 eq.), EDC (1.4 eq.), and HOBt (1.4 eq.) was suspended in anhydrous dichloromethane (3 mL) and stirred for 10 min under a nitrogen atmosphere. The endoxifen derivative 2a (1 eq.) was dissolved in anhydrous dichloromethane (3 mL) and slowly added to the mixture via syringe. Reaction was allowed stir for 16 h. Reaction was monitored via TLC. The reaction mixture was diluted to 15 mL with anhydrous dichloromethane. To this mixture, water (20 mL) was added. The aqueous phase was extracted with DCM (20 mL × 3), brine (50 mL), dried over Na2SO4 and evaporated to dryness in vacuo to yield the crude product. The material was purified via flash chromatography on silica gel. (DCM:EtOAc). The silyl deprotection was performed as above.
(E/Z)-3-(Benzo[d][1,3]dioxol-5-yl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-2-(4-methoxyphenyl)-N-methylacrylamide (3k). Following the general method above, the acrylic acid 1m (1.2 eq., 176 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded the silyl ether as a white solid (261 mg, 83%). HRMS: Found 790.3566 [M + Na]+, C48H53NO6NaSi requires 790.3540. The protected compound (142 mg, 0.185 mmol) was dissolved in THF (3 mL) and stirred under nitrogen. A solution of TBAF (1M in THF, 1 eq., 0.185 mL, 0.185 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure and purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid (109 mg, 91%), m.p. 84–86 °C. 1H-NMR (CDCl3) δ 6.99–7.24 (m, 17H, Ar-H, C=CH), 6.34–6.88 (m, 25H, Ar-H), 5.87 (s, 4H, CH2), 4.00–4.26 (m, 4H, CH2), 3.59–3.86 (m, 10H, OCH3, CH2), 2.96–3.13 (m, 6H, NCH3), 2.38–2.54 (m, 4H, CH2), 0.87–0.96 (m, 6H, CH3). 13C-NMR (CDCl3) δ 172.8 (C=O), 160.7, 159.3, 158.3, 154.9, 154.0, 148.0, 147.3, 147.3, 147.2, 147.1, 142.6, 140.9, 137.8, 135.7, 135.4, 131.98, 131.96, 130.6, 130.3, 130.2, 130.1, 129.7, 129.7, 129.5, 127.81, 127.8, 125.9, 124.0, 115.1, 114.4, 114.2, 113.9, 113.2, 109.1, 108.1, 108.1, 101.0, 100.2 (OCH2O), 65.9 (CH2), 60.4 (CH2), 55.2 (OCH3), 47.6 (CH2), 38.7 (NCH3), 25.6 (CH2), 13.6 (CH3). IR: νmax (KBr) cm−1: 3239.3, 2929.9, 1735.6, 1605.7, 1508.4, 1244.2, 1036.7, 835.5 HRMS (EI): Found 676.2685 [M + Na]+, C42H39NO6Na requires 676.2675.
(E/Z)-N-(2-(4-((E)-1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-3-(4-methoxy-3-nitro-phenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (3a). Following the general method above, the acrylic acid 1a (1.2 eq., 191 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol), and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). The crude product was afforded as a brown resin. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) to afford the product as a white solid. (309 mg, 88%), HRMS: Found 881.3841 [M + Na]+, C50H58N2O9NaSi requires 881.3809. The product was then deprotected. The protected compound (182 mg, 0.212 mmol) was dissolved in 3 mL THF and stirred under nitrogen environment. A solution of TBAF (1M in THF, 1 eq., 0.212 mL, 0.212 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (146 mg, 93%), m.p. 77–78 °C. 1H-NMR (CDCl3) δ 7.58–7.73 (m, 2H, Ar-H), 6.99–7.17 (m, 12H, Ar-H, C=CH), 6.09–6.88 (m, 24H, Ar-H), 4.01–4.28 (m, 4H, CH2), 3.53–3.93 (m, 28H, CH2, OCH3), 2.99–3.20 (m, 6H, NCH3), 2.36–2.50 (m, 4H, CH2), 0.84–0.96 (m, 6H, CH3). 13C-NMR (CDCl3): δ 172.8 (C=O), 172.7 (C=O), 157.2, 156.3, 155.9, 155.1, 154.1, 153.2, 142.7, 142.6, 141.2, 141.1, 140.9, 137.8, 137.7, 137.1, 136.7, 136.3, 135.7, 135.3, 132.1, 132.0, 130.8, 130.7, 129.7, 127.8, 125.9, 115.1, 114.4, 113.9, 113.1, 113.1, 105.3, 105.3, 66.7, 66.4, 65.1, 64.8, 60.9, 60.5, 56.1, 49.2, 48.3, 48.2, 37.7, 37.6, 35.7, 35.7, 35.2, 35.1, 34.2, 34.1, 32.1, 32.0, 31.6, 31.6, 29.1, 29.0 (CH2), 13.7 (CH3). IR: νmax (KBr) cm−1: 3240.6, 2933.8, 1733.9, 1606.5, 1505.9, 1238.4, 1126.7, 834.2. HRMS (EI): Found 743.2974 [M − H]−, C44H43N2O9 requires 743.2969.
(E/Z)-2-(3,5-Dimethoxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-3-(3,4,5-trimethoxyphenyl)acrylamide (3b). Following the general method above, the acrylic acid analogue 1b was reacted with endoxifen derivative 2a. The final product was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a brown resin (33%). 1H-NMR (CDCl3): δ 7.17–6.37 (m, 38H, Ar-H, C=CH), 4.25–4.09 (m, 38H, OCH3, NCH2, OCH2), 3.23–3.05 (m, 6H, NCH3), 2.52–2.48 (m, 4H, CH2), 0.94–0.91 (m, 6H, CH3), 13C-NMR (CDCl3): δ 160.56, 154.86, 153.90, 152.22, 152.20, 140.38, 137.34, 137.30, 131.56, 131.52, 130.20, 130.13, 129.97, 129.23, 127.38, 125.44, 114.67, 113.95, 113.47, 112.71, 106.28, 106.25, 100.06, 60.45 (OCH3), 55.34 (OCH3), 54.94 (OCH3), 53.01, 48.70 (NCH3), 33.41 (CH2), 28.63 (CH2), 28.54 (CH2), 25.10 (CH2), 24.45 (CH2), 13.22 (CH3), 13.17 (CH3) IR: νmax (KBr) cm−1: 3167.5, 2935.3, 1737.4, 1581.6, 1505.6, 1233.1, 1125.0, 1003.7 834.9 HRMS (EI): Found 752.3223 [M + Na]+, C45H47NO8Na requires 752.3199.
(E/Z)-N-(2-(4-((Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-2,3-bis(3,4,5-trimethoxyphenyl)acrylamide (3c). Following the general method above, the acrylic acid 1c (1.2 eq., 198 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol), and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol) to afford the silyl ether which was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) to afford a white solid. (315 mg, 88%). HRMS: Found 898.4193 [M + H]+, C52H63NO9NaSi requires 896.4170. The protected compound (158 mg, 0.18 mmol) was dissolved in 3 mL THF and treated with a solution of TBAF (1 M in THF, 1 eq., 0.18 mL, 0.18 mmol) was added to the mixture and allowed to stir for 1 h. Purifiication by flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) afforded the product as a white solid. (128 mg, 94%), m.p. 77–78 °C. 1H-NMR (CDCl3) δ 6.97–7.19 (m, 14H, Ar-H, C=CH), 6.30–6.89 (m, 24H, Ar-H), 4.02–4.27 (m, 4H, CH2), 3.52–3.95 (m, 40H, OCH3), 2.98–3.29 (m, 6H, NCH3), 2.37–2.53 (m, 4H, CH2), 0.83–0.96 (m, 6H, CH3), 13C-NMR (CDCl3), δ 171.3 (C=O), 155.0, 154.1, 153.4, 152.7, 152.7, 142.6, 142.5, 141.0, 137.8, 137.8, 137.6, 136.6, 135.6, 135.2, 132.0, 131.9, 130.6, 130.6, 130.5, 129.6, 129.6, 127.8, 125.9, 115.0, 114.3, 113.9, 113.1, 106.7, 106.6, 106.1, 106.1, 60.9, 60.82 (OCH3), 60.81 (OCH3), 60.4 (OCH3), 56.1 (OCH3), 55.8 (OCH3), 53.4, 47.7, 39.0 (NCH3) 29.1 (CH2), 29.0 (CH2), 13.6 (CH3), 13.56 (CH3) IR: νmax (KBr) cm−1: 3167.5, 2935.3, 1737.4, 1581.6, 1505.6, 1233.1, 1125.0, 1003.7 834.9. HRMS (EI): Found 782.3301 [M + Na]+, C46H49NO9Na requires 782.3305.
(E/Z)-2-(3,5-Dimethoxyphenyl)-3-(3-hydroxy-4-methoxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methylacrylamide (3d). Following the general method above, the acrylic acid analogue 1d was reacted with endoxifen derivative 2a. Following deprotection, the final product was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a brown resin (54%). 1H-NMR (CDCl3): δ 7.19–6.37 (m, 40H, Ar-H, C=CH), 4.24–3.60 (m, 26H, OCH3, O-CH2, N-CH2), 3.18–3.04 (m, 6H, NCH3), 2.53–2.47 (m, 4H, CH2), 0.95–0.91 (m, 6H, CH3). 13C-NMR (CDCl3): δ 160.44 (C=O), 156.55, 154.65, 153.69, 146.00, 145.98, 144.60, 144.58, 142.17, 140.49, 140.38, 137.38, 135.30, 135.15, 131.56, 130.18, 129.25, 127.98, 127.38, 127.35, 125.44, 121.62, 115.01, 114.63, 113.90, 113.48, 112.74, 109.71, 106.29, 100.13, 55.40 (OCH3), 54.89 (OCH3), 53.00 (CH2), 48.75 (NCH3), 33.41 (OCH2), 28.61 (CH2CH3), 28.55 (CH2CH3), 25.11 (CH2), 24.45 (CH2), 13.21 (CH3), 13.18 (CH3). IR: νmax (KBr) cm−1: 3379.5, 2933.2, 1735.1, 1606.3, 1581.7, 1505.9, 1238.9, 1124.6, 1026.5, 833.4. HRMS (EI): Found 708.2925 [M + Na]+, C43H43NO7Na requires 708.2937.
(E/Z)-3-(3,4-Dihydroxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (3e). Following the general method above, the acrylic acid 1e (1.2 eq., 170 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol), and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol) to afford the silyl ether as a which was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) to afford a white solid. (237 mg, 71%). HRMS: Found 838.3751 [M + Na]+, C49H57NO8NaSi requires 838.3751. The silyl ether (157 mg, 0.192 mmol) was dissolved in 3 mL THF and stirred under nitrogen environment. A solution of TBAF (1 M in THF, 1 eq., 0.192 mL, 0.192 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified via flash chromatography over silica gel to afford the product as a white solid. (113 mg, 84%), m.p. 63–69 °C. 1H-NMR (CDCl3): δ 7.13–6.49 (m, 38H, Ar-H, C=CH), 4.22–3.52 (m, 26H, OCH3, O-CH2, N-CH2), 3.04–2.94 (m, 6H, NCH3), 2.47 (m, 4H, CH2), 1.32–1.28 (m, 6H, CH3). 13C-NMR (CDCl3): δ 156.62, 154.66, 153.66, 152.88, 145.98, 145.96, 144.60, 142.12, 137.33, 135.20, 132.01, 131.54, 130.22, 130.10, 129.58, 129.25, 128.00, 127.32, 127.30, 125.46, 121.50, 115.10, 114.64, 114.05, 113.40, 112.65, 109.76, 105.58, 65.71, 55.57, 55.44, 48.72, 33.44, 28.62, 25.31, 24.54, 13.22, 13.17. IR: νmax (KBr) cm−1: 3250.6, 2930.1, 1735.4, 1605.6, 1605.1, 1244.1, 1037.1, 835.6 HRMS (EI): Found 724.2881 [M + Na]+, C43H43NO8Na requires 724.2886.
(E/Z)-3-(3-Hydroxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (3f). Following the general method above, the acrylic acid analogue 1f was reacted with endoxifen derivative 2a. The product was purified by flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a brown resin (45%). 1H-NMR (CDCl3): δ 7.15–6.38 (m, 40H, Ar-H, C=CH), 4.27–3.45 (m, 26H, OCH3, N-CH2, O-CH2), 3.26–3.07 (m, 6H, NCH3), 2.49–2.47 (m, 4H, CH2), 0.94–0.91 (m, 6H, CH3) 13C-NMR (CDCl3): δ 171.96, 171.90, 156.64, 155.82, 154.44, 153.53, 152.67, 142.16, 142.65, 140.66, 140.52, 137.24, 137.15, 136.42, 136.28, 135.92, 135.28, 134.91, 131.59, 131.55, 130.25, 130.18, 129.82, 129.50, 129.45, 129.23, 128.87, 127.40, 125.56, 120.82, 120.68, 115.64, 114.94, 114.62, 113.91, 113.40, 112.66, 105.56, 105.47, 105.49, 65.83, 66.46, 55.60, 55.41, 53.01, 47.46, 38.71, 38.63, 28.63, 28.55, 13.20, 13.17. IR: νmax (KBr) cm−1: 3250.6, 2930.1, 1735.4, 1605.6, 1605.1, 1244.1, 1037.1, 835.6. HRMS (EI): Found 708.2937 [M + Na]+, C43H43NO7Na requires 708.2937.
(E/Z)-3-(3,4-Dimethoxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (3g). Following the general method above, the acrylic acid 1g (1.2 eq., 184 mg, 0.492 mmol) ), EDC (1.4 eq., 110 mg, 0.572 mmol), and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded a white solid, (300 mg, 87%). HRMS: Found 866.4046 [M + Na]+, C51H61NO8NaSi requires 866.4046. The silyl ether (176 mg, 0.21 mmol) was dissolved in THF (3 mL) and stirred under nitrogen. A solution of TBAF (1 M in THF, 1 eq., 0.21 mL, 0.21 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (139 mg, 91%), m.p. 63–66 °C. 1H-NMR (CDCl3) δ 6.99–7.19 (m, 13H, Ar-H, C=CH), 6.29–6.87 (m, 25H, Ar-H), 5.89 (br. s., 1H, OH), 4.02–4.26 (m, 4H, CH2), 3.51–3.87 (m, 34H, OCH3, CH2), 2.98–3.24 (m, 6H, NCH3), 2.38–2.51 (m, 4H, CH2), 0.85–0.95 (m, 6H, CH3). 13C-NMR (CDCl3) δ 171.6 (C=O), 161.0, 154.9, 154.0, 152.7, 152.6, 142.6, 141.0, 137.9, 137.7, 136.7, 135.7, 132.0, 130.6, 130.4, 129.7, 129.6, 127.8, 127.8, 125.9, 115.0, 114.3, 113.9, 113.2, 106.8, 106.7, 100.5, 60.8 (CH2), 60.4 (OCH3), 55.8 (OCH3), 55.4 (OCH3), 38.9 (NCH3), 25.6 (CH2), 13.6 (CH3), 13.6 (CH3). IR: νmax (KBr) cm−1: 3242.0, 2934.9, 2837.9, 2837.6, 1736.1, 1600.2, 1505.9, 1421.4, 1239.7, 1155.4, 833.2 HRMS (EI): Found 752.3210 [M + Na]+, C45H47NO8Na requires 752.3199.
(E/Z)-N-(2-(4-((Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-3-(2,3,4-tri-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylamide (3h). Following the general method above, the acrylic acid analogue 1h was reacted with endoxifen derivative 2a. The final product was purified by flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a brown resin (26%). 1H-NMR (CDCl3): δ 7.16–6.43 (m, 36H, Ar-H, C=CH), 4.12–3.55 (m, 40H, OCH3, N-CH2, O-CH2), 3.12–3.10 (m, 6H, NCH3), 2.50–2.47 (m, 4H, CH2), 0.94–0.91 (t, 6H, J = 7.5Hz CH3) 13C-NMR (CDCl3): δ 170.7 (C=O), 155.0 (Cq), 154.2 (Cq), 153.4 (Cq), 152.7 (Cq), 152.7 (Cq), 142.6 (Cq), 142.5 (Cq), 141.0 (Cq), 137.8 (Cq), 137.8 (Cq), 137.6 (Cq), 136.6 (Cq), 135.6 (Cq), 135.2 (Cq), 132.0 (ArC), 131.9 (ArC), 130.6 (ArC), 130.6 (ArC), 130.5 (ArC), 129.6 (ArC), 129.6 (ArC), 127.8 (ArC), 125.9 (ArC), 115.0 (ArC), 114.3 (ArC), 113.9 (ArC), 113.1 (ArC), 106.7 (ArC), 106.6 (ArC), 106.1 (ArC), 106.1 (ArC), 60.9 (CH2), 60.82 (OCH3), 60.81 (OCH3), 60.4 (OCH3), 56.1 (OCH3), 55.8 (OCH3), 53.4, 47.7, 39.0 (NCH3) 29.1 (CH2), 29.0 (CH2), 13.6 (CH3), 13.56 (CH3). IR: νmax (KBr) cm−1: 3167.5, 2935.3, 1737.4, 1581.6, 1505.6, 1233.1, 1125.0, 1003.7, 834.9. HRMS (EI): Found 782.3310 [M + Na]+, C46H49NO9Na requires 782.3305.
(E/Z)-2-(3-Hydroxy-4-methoxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)-ethyl)-N-methyl-3-(3,4,5-trimethoxyphenyl)acrylamide (3i). Following the general method above, the acrylic acid 1i (1.2 eq., 177 mg, 0.492 mmol)), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded the product as a white solid. (289 mg, 85%), HRMS: Found 852.3920 [M + Na]+, C50H59NO8NaSi requires 852.3908. The silyl ether (137 mg, 0.16 mmol) was dissolved in THF (3 mL) and stirred under nitrogen. A solution of TBAF (1 M in THF, 1 eq., 0.16 mL, 0.16 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified by flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (103 mg, 90%), m.p: 81–83 °C. (DCM:EtOAc). 1H-NMR (CDCl3) δ 6.99–7.19 (m, 15H, Ar-H, C=CH), 5.85–6.88 (m, 23H, Ar-H), 4.05–4.29 (m, 4H, CH2), 3.49–3.89 (m, 28H, OCH3, CH2), 2.91–3.25 (m, 6H, NCH3), 2.39–2.50 (m, 4H, CH2), 0.85–0.94 (m, 6H, CH3). 13C-NMR (CDCl3) δ 171.2 (C=O), 168.7, 154.9, 154.0, 153.4, 151.7, 151.7, 135.7, 133.3, 132.0, 131.9, 130.7, 130.6, 129.6, 129.6, 128.3, 127.9, 127.8, 125.9, 115.0, 114.3, 113.9, 113.1, 106.1, 106.0, 60.8 (OCH3), 60.4 (CH2), 56.3 (OCH3), 56.2 (OCH3), 56.1 (OCH3), 55.9 (OCH3), 29.04 (CH2), 28.97 (CH2), 13.6 (CH3), 13.57 (CH3). IR: νmax (KBr) cm−1: 3428.1, 2935.6, 2346.1, 1766.2, 1596.7, 1508.0, 1239.8, 1127.5, 835.0. HRMS (EI): Found 716.3226 [M + H]+, C44H46NO8 requires 716.3223.
(E/Z)-3-(4-Hydroxy-3,5-dimethoxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)-phenoxy)ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (3j). Following the general method above, the acrylic acid 1j (1.2 eq., 194 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded the product as a white solid. (315 mg, 76%), HRMS: Found 882.4022 [M + Na]+, C51H61NO9NaSi requires 882.4013. The silyl ether (158 mg, 0.18 mmol) was dissolved in THF (3 mL) and stirred under nitrogen environment. A solution of TBAF (1 M in THF, 1 eq., 0.18 mL, 0.18 mmol) was added to the mixture and allowed to stir for 1 h. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid (128 mg, 94%), m.p: 76–79 °C. 1H-NMR (CDCl3) δ 6.97–7.19 (m, 14H, Ar-H, C=CH), 6.30–6.89 (m, 24H, Ar-H), 4.02–4.27 (m, 4H, CH2), 3.52–3.95 (m, 34H, CH2, OCH3), 2.98–3.29 (m, 6H, NCH3), 2.37–2.53 (m, 4H, CH2), 0.83–0.96 (m, 6H, CH3). 13C-NMR (CDCl3) δ 171.3 (C=O), 155.0, 154.1, 153.4, 152.7, 152.7, 142.6, 142.5, 141.0, 137.8, 137.8, 137.6, 136.6, 135.6, 135.2, 132.0, 131.9, 130.6, 130.6, 130.5, 129.6, 129.6, 127.8, 125.9, 115.0, 114.3, 113.9, 113.1, 106.7, 106.6, 106.1, 106.1, 60.9 (CH2), 60.82 (OCH3), 60.81 (OCH3), 60.4 (OCH3), 56.1 (OCH3), 55.8 (OCH3), 53.4, 47.7, 39.0 (NCH3) 29.1 (CH2), 29.0 (CH2), 13.6 (CH3), 13.56 (CH3). IR: νmax (KBr) cm−1: 3167.5, 2935.3, 1737.4, 1581.6, 1505.6, 1233.1, 1125.0, 1003.7 834.9. HRMS (EI): Found 768.3133 [M + Na]+, C45H47NO9Na requires 768.3149.
(E/Z)-N-(2-(4-((E)-1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-3-(5-methyl-thiophen-2-yl)-2-(3,4,5-trimethoxyphenyl)acrylamide (3l). Following the general method above, the acrylic acid 1p (1.2 eq., 165 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). The product was purified by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) to afford a white solid (184 mg, 56%). HRMS: Found 826.3604 [M + Na]+, C48H57NO6NaSiS requires 826.3574. The silyl ether (101 mg, 0.126 mmol) was dissolved in (THF) 3 mL and stirred under nitrogen environment. A solution of TBAF (1 M in THF, 1 eq., 0.126 mL, 0.126 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (70 mg, 81%), m.p: 83–84 °C. 1H-NMR (CDCl3): δ 5.82–7.24 (m, 34H, Ar-H, C=CH), 3.99–4.33 (m, 4H, CH2), 3.72–3.96 (m, 22H, CH2, OCH3), 2.97–3.37 (m, 6H, NCH3), 2.43–2.56 (m, 4H, CH2), 2.37 (d, J = 3.26 Hz, 6H, CH3), 0.94 (s, 6H, CH3). 13C-NMR (CDCl3): δ 171.3 (C=O), 157.1, 156.3, 154.9, 154.0, 153.6, 153.6, 142.8, 142.7, 142.6, 142.5, 141.1, 141.0, 138.3, 138.2, 137.7, 136.9, 136.0, 135.8, 135.4, 132.3, 132.3, 132.0, 131.4, 131.4, 130.7, 129.7, 127.9, 125.9, 125.7, 125.6, 124.7, 124.7, 115.1, 114.4, 113.9, 113.2, 106.4, 106.3, 61.0 (OCH3), 60.5 (CH2), 56.3 (OCH3), 56.2 (OCH3), 38.7 (NCH3) (CH2), 29.1 (CH2), 29.0 (CH2), 15.5 (CH3), 13.7 (CH3), 13.6 (CH3). IR: νmax (KBr) cm−1: 3255.2, 2961.7, 2932.4, 1735.6, 1605.5, 1580.2, 1505.9, 1238.4, 1120.2, 834.0. HRMS (EI): Found 712.2720 [M + Na]+, C42H43NNaO6S requires 712.2709.
(E/Z)-3-(Furan-3-yl)-N-(2-(4-((E)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (3m). Following the general method above, the acrylic acid 1q (1.2 eq., 149 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded the product as a white solid. (89 mg, 32%), HRMS: Found 796.3656 [M + Na]+, C47H55NO7NaSi requires 796.3646. The silyl ether (70 mg, 0.09 mmol) was dissolved in THF (3 mL) and stirred under nitrogen. A solution of TBAF (1M in THF, 1 eq., 0.09 mL, 0.09 mmol) was added to the mixture and allowed to stir for 1 h. The solvent was evaporated under reduced pressure. Purification by flash chromatography over silica gel afforded the product as a white solid. (29 mg, 50%), m.p: 81–84 °C. 1H-NMR (CDCl3) δ 7.55–7.66 (m, 2H, Ar-H, CCH2), 5.90–7.47 (m, 40H, Ar-H, C=CH, C=C2), 4.00–4.32 (m, 4H, CH2), 3.66–3.99 (m, 22H, OCH3, CH2), 3.02–3.30 (m, 6H, NCH3), 2.43–2.56 (m, 4H, CH2), 0.90–1.00 (m, 6H, CH3). 13C-NMR (CDCl3): δ 171.7 (C=O), 154.8, 153.9, 153.4, 153.3, 143.6, 142.9, 142.9, 142.6, 142.5, 141.1, 137.9, 137.7, 135.8, 135.6, 135.5, 135.5, 132.0, 130.7, 130.7, 129.7, 127.9, 126.0, 121.3, 121.3, 115.1, 114.4, 113.9, 113.1, 110.0, 106.0, 105.9, 66.2 (OCH3), 61.0 (OCH3), 60.5 (CH2), 56.1 (OCH3), 39.1 (NCH3), 29.1 (CH2), 29.0 (CH2), 13.7 (CH3), 13.6 (CH3). HRMS (EI): Found 682.2791 [M + Na]+, C41H41NNaO7 requires 682.2781.
(E)-N,N-Diethyl-3-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylamide (3q). The acrylic acid 1n (1 eq., 0.87 mmol, 0.3 g) was reacted with Mukaiyama’s reagent (3 eq., 2.61 mmol, 0.67 g) in dry DCM (20 mL). After stirring for 5 min, diethylamine (1 eq., 0.87 mmol, 0.06 g) and trimethylamine (5 eq., 4.35 mmol, 0.44 g, 0.60 mL) were added. The reaction was stirred at room temperature for 3 h. After completion the mixture was diluted with DCM and washed with HCl 10% (10 mL), NaHCO3 sat (10 mL), water (10 mL), brine (10 mL) and dried over sodium sulphate. The crude material was then purified via flash column chromatography (eluent, DCM: ethylacetate 1:1) to afford the product as a pale yellow oil, (63%, 0.215 g). 1H-NMR (CDCl3) δ 7.08 (d, J = 8.8 Hz, 2H, Ar-H), 6.70 (d, J = 8.7 Hz, 2H, Ar-H), 6.55 (s, 1H, CH), 6.54 (s, 2H, Ar-H), 3.82 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.66 (s, 6H, OCH3), 3.39 (s, 4H, CH2), 1.06 (d, J = 62.4 Hz, 6H, CH3). 13C-NMR (CDCl3) δ 171.10 (C=O), 159.08 (C), 153.22 (2 × C), 137.70 (CH), 136.21 (C), 130.96 (C), 130.73 (2 × CH), 128.46 (C), 127.90 (C), 113.48 (2 × CH), 105.94 (2 × CH), 60.90 (OCH3), 56.03 (2 × OCH3), 55.17 (OCH3), 43.02 (2 × CH2), 14.15 (2 × CH3). IR: νmax (KBr) cm−1: 3368, 2935, 2836, 1668, 1603, 1578, 1507, 1454, 1379, 1275, 1237, 1122, 1028, 1002, 908, 811, 729, 637, 569. HRMS (APCI): Found 400.2113 (M + H) C23H30NO5 requires 400.2118.
(E)-2-(4-Methoxyphenyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (3n). The acrylic acid 1p (1 eq., 0.87 mmol, 0.3 g) was reacted with oxalyl chloride (4 eq., 3.48 mmol, 0.44 g, 0.3 mL) and DMF in catalytic amount and stirred in dry DCM at room temperature overnight. The product was concentrated and used for the following step without purification. The residue of the chlorination was redissolved in DCM and reacted with 4 mL of 28% aqueous ammonium hydroxide to afford the product as a yellow oil (50%). 1H-NMR (DMSO-d6), δ 7.37 (s, 1H, CH), 7.20 (s, 1H, H), 7.09 (d, J = 8.7 Hz, 2H, Ar-H), 6.99 (d, J = 8.8 Hz, 2H, Ar-H), 6.62 (s, 1H, H), 6.30 (s, 2H, Ar-H), 3.74 (s, 3H, OCH3), 3.57 (s, 3H, OCH3), 3.45 (s, 6H, OCH3). 13C-NMR (DMSO-d6) δ 169.25, 159.38 (C), 152.65, 137.96, 135.68, 134.79, 131.20, 130.83, 129.11, 115.00, 107.90, 60.41 (OCH3), 55.73 (2 × OCH3), 55.65 (OCH3). IR: νmax (KBr) cm−1: 3375, 2935, 2839, 1672 1593, 1579, 1451, 1424, 1301, 1210, 1234, 1121, 904, 833, 807, 775, 730, 688, 610, 565. HRMS (APCI): Found 344.1485 (M + H) C19H22NO5 requires 344.1492.
(E)-N,N-diethyl-3-(3-hydroxy-4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl) acrylamide (3o). Following the general method given above for compound 3q, the acrylic acid 1l (1 eq., 1.38 mmol, 0.5 g) was reacted with Mukaiyama’s reagent (3 eq., 4.16 mmol, 1.06 g) in dry DCM (20 mL). After stirring for 5 min, diethylamine (1 eq., 1.38 mmol, 0.1 g) and trimethylamine (5 eq., 6.9 mmol, 0.69 g, 0.96 mL) were added. The reaction was stirred at room temperature for 3 h. After completion, the mixture was diluted with DCM and washed with HCl 10% (10 mL), NaHCO3 sat (10 mL), water (10 mL), brine (10 mL) and dried over sodium sulphate., to afford the product as an oil 1H-NMR (CDCl3) δ 7.26 (s, 1H, Ar-H), 7.21 (d, J = 8.7 Hz, 1H, Ar-H), 6.93 (d, J = 8.7 Hz, 1H, Ar-H), 6.68 (s, 1H, Ar-H), 6.52 (s, 2H, Ar-H), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.71 (s, 6H, OCH3), 3.59 (s, 4H, CH2), 1.16 (s, 3H, CH3), 0.97 (s, 3H, CH3). 13C-NMR (CDCl3), δ 159.20, 153.43, 149.35, 148.41, 138.93, 138.79, 130.11, 129.46, 126.99, 123.63, 120.79, 113.07, 106.02, 60.97 (OCH3), 56.48 (2 × OCH3), 56.41 (OCH3), 46.58 (2 × CH2), 13.89 (CH3), 12.68 (CH3). IR: νmax (KBr) cm−1: 3399, 2935, 1651, 1637, 1578, 1506, 1448, 1413, 1381, 1288, 1238, 1157, 1119, 1017, 971, 808, 773, 569. HRMS (APCI): Found 416.2049 (M + H) C23H30NO6 requires 416.2067.
(E)-3-(4-Methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylamide (
3p). To a solution of acrylic acid
1l (2 mmol, 0.68 g) in dichloromethane (7 mL), triethylamine (1.2 mL, 8.6 mmol) and thionyl chloride (0.27 mL, 3.72 mmol) were added dropwise. The reaction mixture was stirred at room temperature for 2 h whereupon it was concentrated to dryness under reduce pressure. To 30 mL of a 28% aqueous NH
4OH solution a solution of the crude residue in dichloromethane (30 mL) was added. The reaction mixture was vigorously stirred at room temperature overnight, then diluted with water and extracted with dichloromethane. The combined organic layers were dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduce pressure. The residue was purified by flash chromatography (eluent, DCM/ethyl acetate,
v/v 1:1) to afford the product as white solid (15%, 0.1 g) [
71].
1H-NMR (DMSO-
d6) δ 7.43 (s, 1H, Ar-H), 7.20 (s, 1H, NH), 6.99 (d,
J = 8.8 Hz, 2H, Ar-H), 6.79 (d,
J = 8.9 Hz, 2H, Ar-H), 6.59 (s, 1H, NH), 6.44 (s, 2H, Ar-H), 3.72 (s, 3H, OCH
3), 3.70 (s, 3H, OCH
3), 3.69 (s, 6H, OCH
3).
13C-NMR (CDCl
3) δ 169.02, 160.20, 154.28, 137.69, 132.23, 130.83, 127.67, 127.10, 113.77, 106.28, 61.04 (OCH
3), 56.22 (2 × OCH
3), 55.20 (OCH
3). IR: ν
max (KBr) cm
−1: 3421, 3298, 3145, 2941, 1673, 1589, 1503, 1452, 1421, 1409, 1368, 1295, 1250, 1234, 1182, 1026, 994, 822, 805, 772, 725, 691, 652. HRMS (EI): Found 344.2194 (M + H) C
19H
21NO
5 requires 344.1498.
3.2.3. General Method for the Synthesis of Endoxifen-Cinnamic Acid/3-Phenylpropanoic Acid Conjugates 5a–5d
Step (A): A mixture of the appropriate cinnamic acid/3-phenylpropanoic acid (1 eq., 0.154 mmol), DCC (1 eq., 0.154 mmol, 32 mg), and HOBt (1 eq., 0.154 mmol, 21 mg) were suspended in anhydrous dichloromethane (3 mL) and stirred for 10 min under a nitrogen. The silyl-protected endoxifen derivative 2a (75 mg, 1 eq., 0.154 mmol) was dissolved in anhydrous DCM (3 mL) and slowly added to the mixture via syringe. The reaction was allowed stir for 24–48 h and monitored via TLC. The reaction mixture was diluted to 15 mL with anhydrous DCM and filtered to remove DCU. The filtrate was evaporated to dryness under reduced pressure and the residue was dissolved in THF (3 mL) and stirred under nitrogen atmosphere. A solution of 0.1 M TBAF (2 eq.) was added to the mixture and allowed to stir for 24 h. The solvent was evaporated to dryness under reduced pressure. The residue was dissolved in DCM and was washed with 10% HCl solution. The resulting organic phase was dried over sodium sulphate and evaporated to dryness. The material was purified via flash chromatography on silica gel, (eluent: DCM:EtOAc).
Step (B): A mixture of the appropriate cinnamic acid/3-phenylpropanoic acid (1.2 eq), EDC (1.4 eq.), and HOBt (1.4 eq.) were suspended in anhydrous dichloromethane (3 mL) and stirred for 10 min under a nitrogen atmosphere. The protected endoxifen (1 eq.) 2a was dissolved in anhydrous dichloromethane (3 mL) and slowly added to the mixture via syringe and the reaction was stirred for 16 h and monitored via TLC. The reaction mixture was diluted to 15 mL with anhydrous dichloromethane. To this mixture, water (20 mL) was added. The aqueous phase was extracted with DCM (20 mL × 3), brine (50 mL), dried over Na2SO4 and evaporated to dryness in vacuo to yield the crude product. The material was purified via flash chromatography on silica gel. (eluent: DCM:EtOAc). The above residue was dissolved in THF (3 mL) and stirred under nitrogen atmosphere. A solution of TBAF (1 M in THF, 1 eq.) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified via flash chromatography over silica gel (eluent: DCM:EtOAc) to afford the product.
(E/Z)-3-(3-Hydroxy-4-methoxyphenyl)-N-(2-(4-((Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)-ethyl)-N-methylacrylamide (5a). Following the general method above, the cinnamic acid 4a (1.2 eq., 118 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 10:1 to 10:1) affordrd the product as a white solid, (236 mg, 87%). HRMS: Found 686.3317 [M + Na]+, C46H49NO5NaSi requires 686.3278. The silyl ether (126 mg, 0.189 mmol) was dissolved in THF (3 mL). A solution of TBAF (1 M in THF, 1 eq., 0.189 mL, 0.189 mmol) was added to the mixture and stirred under nitrogen for 1 h. The solvent was evaporated under reduced pressure. Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) afforded the product as a white solid. (94 mg, 94%), m.p: 77–78 °C. 1H-NMR (CDCl3) δ 7.51–7.65 (m, 2H, C=CH), 6.91–7.18 (m, 18H, Ar-H), 6.62–6.87 (m, 13H, Ar-H), 5.79–6.52 (m, 7H, Ar-H, C=CH), 3.94–4.21 (m, 4H, CH2), 3.80–3.92 (m, 8H, OCH3, 0.5 × CH2), 3.75 (s, 2H, CH2), 3.03–3.34 (m, 6H, NCH3), 2.45 (m, 4H, CH2), 0.90 (t, J = 7.04 Hz, 6H, CH3). 13C-NMR (CDCl3) δ 171.3 (C=O), 167.2, 167.2, 157.2, 156.3, 155.0, 154.1, 148.2, 145.8, 143.0, 142.7, 142.6, 140.9, 140.8, 137.8, 136.6, 136.2, 135.7, 135.3, 132.0, 132.0, 130.7, 130.6, 129.7, 128.7, 127.8, 125.8, 121.9, 121.6, 115.4, 115.1, 114.3, 113.9, 113.1, 112.6, 112.6, 110.5, 66.8, 66.5, 60.4 (CH2), 56.0 (OCH3), 48.6, 37.7 (NCH3), 35.1 (NCH3), 29.01 (CH2), 28.99 (CH2), 13.6 (CH3). IR: νmax (KBr) cm−1: 3385.2, 2930.3, 1733.5, 1643.5, 1584.1, 1504.9, 1265.0, 1238.0, 1025.7, 834.0. HRMS (EI): Found 572.2413 [M + Na]+, C35H35NO5Na requires 572.2413.
(E/Z)-3-(3-hydroxy-4-methoxyphenyl)-N-(2-(4-(1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methylpropanamide (5b). Following the general method above, the 3-phenylpropanoic acid 4b (1.2 eq., 96.5 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded the product as a white solid. (234 mg, 86%). HRMS: Found 688.3438 [M + Na]+, C41H51NO5NaSi requires 688.3434. The silyl ether (144 mg, 0.219 mmol) was dissolved in THF (3 mL) and stirred under nitrogen. A solution of TBAF (1M in THF, 1 eq., 0.219 mL, 0.219 mmol) was added to the mixture and stirred for 1 h. The solvent was evaporated and the material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (108 mg, 90%), m.p. 78–80 °C. (DCM:EtOAc). 1H-NMR (CDCl3) δ 7.01–7.20 (m, 12H, Ar-H), 5.57–6.86 (m, 20H, Ar-H), 3.50–4.17 (m, 13H, OCH3, CH2), 2.40–3.09 (m, 18H, NCH3, CH2), 0.75–0.98 (m, 6H, CH3). 13C-NMR (CDCl3) δ 172.91 (C=O), 172.85 (C=O), 157.2, 156.3, 156.0, 154.9, 154.0, 145.5, 145.5, 145.5, 145.0, 145.0, 142.7, 142.6, 141.0, 140.9, 137.8, 137.8, 136.6, 136.1, 135.8, 135.7, 135.4, 134.4, 134.4, 132.0, 132.0, 131.9, 130.7, 130.6, 130.6, 129.7, 127.8, 127.8, 125.9, 119.8, 119.8, 115.0, 114.5, 114.5, 114.3, 113.9, 113.1, 113.1, 110.7, 110.7, 66.6 (CH2), 66.3 (CH2), 64.9 (CH2), 60.4 (CH2), 55.9 (OCH3), 49.1 (CH2), 48.2 (CH2), 48.1 (CH2), 37.6 (NCH3), 37.5 (NCH3), 35.6 (CH2), 35.5 (CH2), 35.1 (CH2), 35.0 (CH2), 34.2, 34.1, 31.0 (CH2), 30.7 (CH2), 30.6 (CH2), 29.0 (CH2), 28.98 (CH2), 13.6 (CH3), 13.6 (CH3). IR: νmax (KBr) cm−1: 3441.1, 3232.4, 2931.4, 1733.5, 1607.4, 1508.4, 1272.0, 1239.5, 1029.1, 834.8. HRMS (EI): Found 574.2565 [M + Na]+, C35H37NO5Na requires 574.2569.
(E/Z)-N-(2-(4-((Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)-N-methyl-3-(3,4,5-tri-methoxyphenyl)acrylamide (5c). Following the general method above, the cinnamic acid 4c (1.2 eq., 117 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol) and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen derivative 2a (1 eq., 200 mg, 0.41 mmol). Purification by flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) afforded the product as a white solid, (258 mg, 89%). HRMS: Found 730.3569 [M + Na]+, C43H53NO6NaSi requires 730.3540. The silyl ether (160 mg, 0.226 mmol) was dissolved in THF (3 mL) and stirred under nitrogen. A solution of TBAF (1 M in THF, 1 eq., 0.226 mL, 0.226 mmol) was added to the mixture stirred for 1 h. The solvent was evaporated under reduced pressure. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (127 mg, 95%), m.p: 66–68 °C. 1H-NMR (CDCl3) δ 7.53–7.65 (m, 2H, C=CH), 7.01–7.20 (m, 14H, Ar-H), 6.43–6.89 (m, 18H, Ar-H, C=CH), 3.96–4.25 (m, 4H, CH2), 3.74–3.95 (m, 22H, OCH3, CH2), 3.04–3.39 (m, 6H, CH3), 2.46 (q, J = 7.04 Hz, 4H, CH2), 0.84–0.98 (m, 6H, CH3), OH not observed. 13C-NMR (CDCl3), δ 169.6 (C=O), 153.4, 153.3, 132.0, 130.7, 129.7, 127.8, 125.9, 116.4, 115.0, 114.3, 113.9, 113.1, 105.4, 105.1, 60.9 (CH2), 60.4 (OCH3), 56.2 (OCH3), 56.2 (OCH3), 37.9 (NCH3), 29.0 (CH2), 13.6 (CH3) IR: νmax (KBr) cm−1: 3236.4, 2934.8, 1735.4, 1647.2, 1583.3, 1506.3, 1240.5, 1089.3, 825.9. HRMS (EI): Found 616.2691 [M + Na]+, C37H39NO6Na requires 616.2675.
N-(2-(4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenoxy)ethyl)-3-(3,4,5-trimethoxyphenyl)-N-methylpropanamide (5d). (i) Following the general method above, the cinnamic acid/3-phenylpropanoic acid analogue 4d was reacted with endoxifen derivative 2a. The crude mixture was then taken to the next step without further purification. The final product was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a brown resin (29%). (ii) As per general method, the 3-phenylpropanoic acid 4d (1.2 eq., 118 mg, 0.492 mmol), EDC (1.4 eq., 110 mg, 0.572 mmol), and HOBt (1.4 eq., 77.6 mg, 0.572 mmol) was reacted with endoxifen 2a (1 eq., 200 mg, 0.41 mmol). The crude product was afforded as a brown resin. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 20:1 to 10:1) to afford the product as a white solid. (264 mg, 92%). HRMS: Found 732.3723 [M + Na]+, C43H55NO6NaSi requires 732.3696. The product was then deprotected. The protected compound (156 mg, 0.22 mmol) was dissolved in THF (3 mL) and stirred under nitrogen environment. A solution of TBAF (1 M in THF, 1 eq., 0.22 mL, 0.22 mmol) was added to the mixture and allowed to stir for 1 h. The mixture was evaporated to dryness under reduced pressure. The material was purified via flash chromatography over silica gel (DCM:EtOAc, gradient 6:1 to 3:1) to afford the product as a white solid. (121 mg, 93%), m.p. 73–75 °C. 1H-NMR (CDCl3): δ 7.18–6.40 (m, 15 H, Ar-H), 4.20–4.13 (m, 1H, CH2-O-), 4.05–3.97 (m, 1H, -OCH2O-), 3.85–3.83 (m, 9H, OCH3), 3.79–3.77 (m, 1H, CH2-NCH3), 3.70–3.66 (m, 1H, CH2-NCH3), 3.11 (s, 1H, NCH3), 3.08 (s, 1H, NCH3), 3.00 (s, 0.5H, NCH3), 2.99 (s, 0.5H, NCH3), 2.92 (m, 2H, Ar-CH2), 2.80–2.58 (m, 2H, -O-CH2), 2.52–2.48 (m, 2H, CH2-CH3), 0.95 (t, 3H, 7.5 Hz, CH3), OH not observed. 13C-NMR (CDCl3): δ 170.27 (C=O), 156.75, 154.48, 152.11, 143.74, 142.31, 140.93, 137.36, 137.54, 136.15, 134.91, 133.88, 131.60, 131.40, 130.23, 130.19, 129.26, 127.38, 125.45, 119.39, 114.59, 144.10, 113.90, 113.42, 112.07, 110.25, 66.18 (OCH2), 65.88 (OCH2), 56.88 (OCH3), 55.64 (OCH3), 55.50 (OCH3), 53.01, 47.74 (CH2), 37.24 (CH3N), 37.18 (CH3N), 35.17 (CH2O), 35.42 (OCH2), 33.86 (CH3N), 30.73 (CH2), 30.52 (CH2), 28.46 (CH2CH3), 13.60 (CH3), 13.17 (CH3). IR: νmax (KBr) cm−1: 3251.9, 2933.8, 1735.3, 1607.2, 1589.8, 1508.4, 1239.2, 1127.4, 833.7. HRMS (EI): Found 618.2691 [M + Na]+, C37H41NO6Na requires 618.2691.
[4-(tert-Butyldimethylsilanyloxy)phenyl]-(4-hydroxyphenyl)methanone (7a). 4,4′-Dihydroxybenzo-phenone (6, 6.00 g, 28 mmol) and imidazole (2.094 g, 31 mmol) were dissolved in DMF (20 mL) with stirring. A solution of tert-butyldimethylsilyl chloride (4.22 g, 28 mmol) in DMF (20 mL) was added to the mixture over 1 h. The reaction mixture was allowed to stir at room temperature for 16 h followed by the addition of ethyl acetate (100 mL) and 10% hydrochloric acid (50 mL). The organic layer was washed with water (100 mL), brine (100 mL), dried over sodium sulphate and evaporated to dryness in vacuo to yield crude product. The material was purified via flash chromatography on silica gel (n-hexane:DCM, 5:1) to afford the product 7a as a light yellow oil. (3.34 g, 27%). 1H-NMR (CDCl3): δ 7.76 (dd, 4H, J = 6.5 Hz, Ar-H), 6.94 (dd, 4H, J = 7.5 Hz, Ar-H), 1.02 (s, 9H, CH3), 0.27 (s, 6H, CH3). 13C-NMR (CDCl3): δ 195.66, 160.25, 159.47, 132.41, 132.02, 130.50, 129.37, 119.30, 114.90, 25.16 (CH3), 17.80, −4.78 (SiC). IR: νmax (KBr) cm−1: 3362.6, 295.9, 2930.0, 2856.9, 1641.6, 1602.3, 1580.3, 1508.1, 1470.5, 1313.0, 1278.1, 1161.1, 913.4, 839.1, 771.9. HRMS (EI): Found 329.1579 [M + H]+, C19H25O3Si requires 329.1573. The diprotected benzophenone bis(4-((tert-butyldimethylsilyl)oxy)phenyl)methanone was also obtained as a colourless oil (34%). 1H-NMR (CDCl3): δ 7.75 (d, 4 H, J = 8.5 Hz, Ar-H), 6.91 (d, 4H, J = 8.5 Hz, Ar-H), 1.02 (s, 18 H, CH3), 0.26 (s, 12 H, CH3). 13C-NMR (CDCl3): δ 194.1 (C=O), 159.0, 131.7, 130.8, 119.2, 119.1, 25.2 (CH2), 17.8, −4.79 (SiC) IR: νmax (Film) cm−1: 2956.0, 2930.8, 2886.7, 2858.8, 1653.1, 1599.1, 1508.0, 1267.5, 1161.2, 1006.4, 909.2. HRMS (EI): Found 437.1937 [M + Na]+, C23H34O3Si2Na requires 437.1944.
3.2.4. General Method for Cyclofenil Derivatives 8a–8e via McMurry Coupling
Titanium tetrachloride (4.5 eq., 5.2 g, 37.4 mmol, 3 mL) was added via a syringe dropwise to zinc dust (9 eq., 3.58 g, 54.81 mmol) in dry THF (50 mL) and the mixture was refluxed for 2 h in darkness and under nitrogen. The appropriate phenolic ketone 7a (1 eq.) and cyclic ketone (3 eq.) were dissolved in dry THF (40 mL). This solution was added to the titanium tetrachloride/zinc mixture carefully via a syringe. The reaction mixture was then refluxed for a further 3 h. and then cooled and then diluted with EtOAc (75 mL) and 10% K2CO3 solution. The mixture was filtered under vacuum and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with 10% K2CO3 solution (20 mL), water (50 mL) and brine (50 mL) then dried over Na2SO4, filtered and the solvent was evaporated under reduced pressure to afford the crude product. The material was purified via flash chromatography over silica gel to afford the product.
4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclopentylidene)methyl)phenol (8a). Following the general method above, using, TiCl4 (4.5 eq., 5.2 g, 27.4 mmol, 3 mL), Zn dust (9 eq., 3.58 g, 54.81 mmol), 7a (1 eq., 2 g, 6.09 mmol) and cyclopentanone (3 eq., 1.54 g, 18.26 mmol, 1.88 mL). The crude material was purified via flash chromatography over silica gel (n-hexane:DCM, 3:1) to afford the product as an orange resin (2.25 g, 97%). 1H-NMR (CDCl3) δ 7.01–7.11 (m, 4H, (Ar-H)), 6.74–6.82 (m, 4H, (Ar-H)), 2.40 (t, J = 7.00 Hz, 4H, (2 × CH2)), 1.69 (quin, J = 3.50 Hz, 4H, (2 × CH2)), 1.01 (s, 9H, (3 × CH3)), 0.22 (s, 6H, (2 × CH3)). 13C-NMR (CDCl3) δ 153.4, 153.1, 141.6, 136.4, 135.7, 131.5, 130.0, 129.7, 118.9, 114.3, 32.8 (CH2), 26.5 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3400.5 (br.), 2955.5, 2930.6, 2358.3, 1635.33, 1598.3, 1507.4, 1270.0 (br.), 1163.4, 911.7. HRMS (EI): Found 379.2101 [M − H]−, C24H31O2Si requires 379.2093.
4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclohexaneylidene) methyl) phenol (8b). Following the general method above, TiCl4 (4.5 eq., 5.2 g, 27.4 mmol, 3 mL), Zn dust (9 eq., 3.58 g, 54.81 mmol), 7a (1 eq., 2 g, 6.09 mmol) and cyclohexaneanone (3 eq., 1.79 g, 18.26 mmol, 1.9 mL) were reacted. The crude material was purified via flash chromatography over silica gel (n-Hexane:DCM, 3:1) to afford the product as an orange resin (2.1 g, 87%). 1H-NMR (CDCl3) δ 6.95–7.04 (m, 4H, Ar-H), 6.74–6.79 (m, 4H, Ar-H), 2.23–2.31 (m, 4H, 2 × CH2), 1.55–1.69 (m, 6H, 3 × CH2), 1.02 (s, 9H, 3 × CH3), 0.24 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 153.3, 153.2, 137.8, 136.0, 135.6, 133.1, 130.7, 130.4, 118.8, 114.2, 32.1 (CH2), 28.2 (CH2), 26.4 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3390.8, 2928.4, 2855.8, 1698.7, 1604.9, 1505.7, 1471, 1463.2, 1447.1, 1258.7, 1167.1, 1099.0, 915.8, 802.1. HRMS (EI): Found 395.2405 [M + H]+, C25H35O2Si requires 395.2406.
4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cycloheptylidene)methyl)phenol (8c). Following the general method above, TiCl4 (4.5 eq., 5.2 g, 27.4 mmol, 3 mL), Zn dust (9 eq., 3.58 g, 54.81 mmol), 7a (1 eq., 2 g, 6.09 mmol) and cycloheptanone (3 eq., 2.04 g, 18.26 mmol, 2.15 mL) were reacted. The crude material was purified via flash chromatography over silica gel (n-hexane:DCM, 3:1) to afford the product as an orange resin (2.1 g, 84%). 1H-NMR (CDCl3) δ 6.99–7.06 (m, 4H, Ar-H), 6.73–6.79 (m, 4H, Ar-H), 2.33 (s, 4H, 2 × CH2), 1.59 (s, 8H, 4 × CH2), 1.00 (s, 9H, 3 × CH3), 0.21 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 153.1, 139.1, 136.4, 136.1, 135.8, 130.1, 129.9, 118.9, 114.3, 33.0 (CH2), 32.96 (CH2), 29.0 (CH2), 27.7 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3401.2, 2929.5, 2857.5, 1647.7, 1599.7, 1507.2, 1462.9, 1268.2, 1163.6, 1013.5, 912.9, 840.0, 805.4, 781.4. HRMS (EI): Found 409.2574 [M + H]+, C26H37O2Si requires 409.2563.
4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclooctylidene)methyl)phenol (8d). Following the general method above, TiCl4 (4.5 eq., 5.2 g, 27.4 mmol, 3 mL), Zn dust (9 eq., 3.58 g, 54.81 mmol), 7a (1 eq., 2 g, 6.09 mmol) and cyclooctanone (3 eq., 2.30 g, 18.26 mmol, 2.4 mL) were reacted. The crude material was purified via flash chromatography over silica gel (n-Hexane:DCM, 3:1) to afford the product as an orange resign (2.3 g, 89%). 1H-NMR (CDCl3). δ 6.99–7.11 (m, 4H, Ar-H), 6.73–6.80 (m, 4H, Ar-H), 2.28 (t, J = 6.50 Hz, 4H, 2 × CH2), 1.63–1.71 (m, 2H, CH2), 1.47–1.62 (m, 8H, 4 × CH2), 0.99 (s, 9H, 3 × CH3), 0.21 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 153.1, 139.4, 136.7, 136.3, 135.8, 129.8, 129.5, 119.1, 114.4, 32.0 (CH2), 26.1 (CH2), 25.9 (CH2), 25.8 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.9 (CH3). IR: νmax (KBr) cm−1: 3345.2 (br.), 2928.7, 2856.5, 1603.4, 1503.5, 1253.8 (br.), 1166.7, 917.0. HRMS (EI): Found 445.2519 [M + Na]+, C27H38NaO2Si requires 455.2539.
4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(4-methylcyclohexaneylidene) methyl) phenol (8e). Following the general method above, TiCl4 (4.5 eq., 5.2 g, 27.4 mmol, 3 mL), Zn dust (9 eq., 3.58 g, 54.81 mmol), 7a (1 eq., 2 g, 6.09 mmol) and 4-methylcyclohexaneanone (3 eq., 2.05 g, 18.27 mmol, 2.24 mL) were reacted. The crude material was purified via flash chromatography over silica gel (n-Hexane:DCM, 3:1) to afford the product as an orange resin (2.15 g, 86%). 1H-NMR (CDCl3) δ 7.04–7.08 (m, 4H, Ar-H), 6.80–6.86 (m, 4H, Ar-H), 2.69 (d, J = 13.1 Hz, 4H, 2 × CH2), 2.05 (m, 5H, CH, 2 × CH2), 1.09 (s, 9H, 3 × CH3), 1.02 (d, 3H, J = 6.5 Hz, CH3), 0.30 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 153.4, 153.3, 137.4, 136.2, 135.6, 133.3, 130.7, 130.5, 118.9, 114.4, 32.4 (CH), 29.0 (CH2), 26.6 (CH2), 25.2 (CH3), 17.8 (C(CH3)3), −4.7 (CH3). IR: νmax (KBr) cm−1: 3369.1, 3031.2, 2919.0, 2855.1, 1704.1, 1603.2. 1505.4, 1471.7, 1362.1, 1253.0, 1167.5, 1099.6, 1007.4, 916.0839, 780.7, 734.2. HRMS (EI): Found 409.2570 [M + H]+, C26H37O2Si requires 409.2563.
3.2.5. General Method for the Preparation of Bromoethyl Ethers 9a–9e
The appropriate phenol 8a–8e (1 eq.) was dissolved in 1,2-dibromoethane (~50 eq.) with stirring. Tetrabutylammonium hydrogen sulfate (5 mmol) was added followed by 1 M NaOH solution (50 mL). The biphasic mixture was stirred vigorously at room temperature for 16 h. followed by addition of DCM (100 mL) and NaHCO3 solution (100 mL). The aqueous layer was extracted with DCM (2 × 100 mL) and the organic extracts were combined and washed with water (50 mL), brine (50 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified via flash chromatography over silica gel (eluent: DCM:n-hexane) to afford the product.
(4-((4-(2-Bromoethoxy)phenyl)(cyclopentylidene)methyl)phenoxy)(tert-butyl) dimethylsilane (9a). Following the general method above, phenol 8a (1 eq., 2.1 g, 5.52 mmol), 1,2-dibromoethane (~50 eq. 52.29 g, 275.8 mmol, 24 mL) and tetrabutylammonium hydrogen sulfate (1.7 g, 5 mmol) were reacted. The crude product was purified via flash chromatography over silica gel (n-hexane:DCM, 4:1) to afford the product as a clear resin (2.5 g, 93%). 1H-NMR (CDCl3) δ 6.67–7.46 (m, 8H, Ar-H), 4.23–4.39 (m, 2H, CH2), 3.59–3.75 (m, 2H, CH2), 1.46–1.78 (m, 4H, 2 × CH2), 1.01 (s, 9H, 3 × CH3), 0.23 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 156.9, 154.6, 130.0, 129.7, 128.9, 128.8, 128.6, 120.1, 118.9, 113.6, 113.6, 113.5, 67.3 (CH2), 32.8 (CH2), 28.7 (CH2), 26.6 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR:νmax (KBr) cm−1: 2955.0, 2858.7, 1651.1, 1597.8, 1508.3, 1255.3 (br.), 1169.7, 913.6. HRMS (EI): Found 487.1666 [M + H]+, C26H36BrO2Si requires 487.1668.
(4-((4-(2-Bromoethoxy)phenyl)(cyclohexaneylidene)methyl)phenoxy)(tert-butyl)dimethylsilane (9b). Using the general method above, phenol 8b (1 eq., 2.1 g, 5.32 mmol), 1,2-dibromoethane (50 eq., 52.29 g, 275.8 mmol, 24 mL) and tetrabutylammonium hydrogen sulfate (1.7 g, 5 mmol) were reacted together. The crude product was purified via flash chromatography over silica gel (n-hexane:DCM, 4:1) to afford the product as a clear resin (2.44 g, 91%). 1H-NMR (CDCl3) δ 7.06 (d, J = 8.53 Hz, 2H, Ar-H), 6.98 (d, J = 8.28 Hz, 2H, Ar-H), 6.85 (d, J = 8.78 Hz, 2H, Ar-H), 6.76 (d, J = 8.78 Hz, 2H, Ar-H), 4.30 (t, J = 6.27 Hz, 2H, CH2), 3.66 (t, J = 6.27 Hz, 2H, CH2), 2.23–2.31 (m, 4H, 2 × CH2), 1.59–1.68 (m, 6H, 3 × CH2), 1.01 (s, 9H, 3 × CH3), 0.23 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 156.3, 153.8, 138.4, 136.8, 136.3, 133.5, 131.0, 130.8, 119.3, 114.1, 67.8 (CH2), 32.5 (CH2), 29.2 (CH2), 28.7 (CH2), 26.9 (CH2), 25.7 (CH3), 18.2 (C(CH3)3), −4.4 (CH3). IR: νmax (KBr) cm−1: 2930.1, 2857.5, 1654.4, 1600.6, 1508.2, 1471.9, 1254.5, 1168.0, 913.8, 838.9, 804.6, 781.1. HRMS (EI): Found 501.1827 [M + H]+, C27H38BrO2Si requires 501.1824.
(4-((4-(2-Bromoethoxy)phenyl)(cycloheptylidene)methyl)phenoxy)(tert-butyl)dimethylsilane (9c). Following the general method above, phenol 8c (1 eq., 2.35 g, 5.70 mmol), 1,2-dibromoethane (50 eq. 56.64 g, 298.8 mmol, 26 mL) and tetrabutylammonium hydrogen sulfate (1.7 g, 5 mmol) were reacted together. The crude product was purified via flash chromatography over silica gel (n-hexane:DCM, 4:1) to afford the product as a clear resin (2.76 g, 94%). 1H-NMR (CDCl3) δ 7.09 (d, J = 8.53 Hz, 2H, Ar-H), 7.00 (d, J = 8.53 Hz, 2H, Ar-H), 6.84 (d, J = 8.53 Hz, 2H, Ar-H), 6.75 (d, J = 8.53 Hz, 2H, Ar-H), 4.29 (t, J = 6.27 Hz, 2H, CH2), 3.65 (t, J = 6.27 Hz, 2H, CH2), 2.32 (s, 4H, 2 × CH2), 1.59 (s, 8H, 4 × CH2), 0.99 (s, 9H, 3 × CH3), 0.20 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 155.7, 153.2, 139.2, 136.7, 136.3, 135.8, 130.1, 129.8, 118.9, 113.6, 67.3 (CH2), 32.9 (CH2), 29.0 (CH2), 28.8 (CH2), 27.7 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.9 (CH3). IR: νmax (KBr) cm−1: 2929.0, 2856.8, 1602.8, 1507.6, 1471.7, 1254.1, 1168.0, 1016.7, 913.9, 839.4, 781.0. HRMS (TOF-MS): Found 514.1895 [M + H]+, C28H39O2BrSi requires 514.1903.
(4-((4-(2-Bromoethoxy)phenyl)(cyclooctylidene)methyl)phenoxy)(tert-butyl)dimethylsilane (9d). Following the general method above, phenol 8d (1 eq., 2.33 g, 5.70 mmol), 1,2-dibromoethane (50 eq. 56.64 g, 298.8 mmol, 26 mL) and tetrabutylammonium hydrogen sulfate (1.7 g, 5 mmol) were reacted together. The crude product was purified via flash chromatography over silica gel (n-hexane:DCM, 4:1) to afford the product as a clear resin (2.83 g, 94%). 1H-NMR (CDCl3) δ 7.12 (d, J = 8.53 Hz, 2H, Ar-H), 7.03 (d, J = 8.03 Hz, 2H, Ar-H), 6.85 (d, J = 8.53 Hz, 2H, Ar-H), 6.76 (d, J = 8.53 Hz, 2H, Ar-H), 4.28 (t, J = 6.27 Hz, 2H, CH2), 3.65 (t, J = 6.27 Hz, 2H, CH2), 2.24–2.32 (m, 4H, 2 × CH2), 1.63–1.71 (m, 2H, CH2), 1.48–1.63 (m, 8H, 4 × CH2), 0.99 (s, 9H, 3 × CH3), 0.20 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 155.6, 153.1, 139.6, 137.0, 136.5, 135.7, 129.7, 129.5, 119.1, 113.8, 67.3 (CH2), 32.0 (CH2), 28.8 (CH2), 26.1 (CH2), 26.0 (CH2), 25.8 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 2929.6, 2857.7, 1651.2, 1598.9, 1508.5, 1255.3 (br.), 1168.2, 912.8. HRMS (EI): Found 529.2139 [M + H]+, C29H42BrO2Si requires. 529.2137.
(4-((4-(2-Bromoethoxy)phenyl)(4-methylcyclohexaneylidene) methyl)phenoxy) (tert-butyl)dimethylsilane (9e). Following the general method above, phenol 8e (1 eq., 2.3 g, 5.63 mmol), 1,2-dibromoethane (50 eq. 56.64 g, 298.8 mmol, 26 mL) and tetrabutylammonium hydrogen sulfate (1.7 g, 5 mmol) were reacted together. The crude product was purified via flash chromatography over silica gel (n-hexane:DCM, 4:1) to afford the product as a clear resin (2.61 g, 90%). 1H-NMR (CDCl3) δ 7.09 (d, J = 8.5 Hz, 2H, Ar-H), 7.02 (d, J = 8.0 Hz, 2H, Ar-H), 6.87 (d, J = 8.5 Hz, 2H, Ar-H), 6.80 (d, J = 8.0 Hz, 2H, Ar-H), 4.31 (t, J = 6.3 Hz, 2H, CH2), 3.67 (t, J = 6.3 Hz, 2H, CH2), 2.64 (t, J = 11.5 Hz, 2H, CH2), 1.96–2.03 (m, 2H, CH2), 1.82 (d, J = 12.0 Hz, 2H, CH2), 1.61–1.72 (m, 1H, CH), 1.08–1.18 (m, 2H, CH2), 1.04 (s, 9H, CH3), 0.98 (d, J = 6.5 Hz, 3H, CH3), 0.21 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 155.9, 153.4, 137.6, 136.3, 135.9, 133.2, 130.6, 130.4, 118.8, 118.8, 113.6, 69.3 (CH2), 36.5 (CH2), 32.4 (CH), 31.4 (CH2), 28.8 (CH2), 25.3 (CH3), 17.77 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 2951.7, 2927.8, 2857.0, 1604.5, 1507.3, 1472.0, 1457.4, 1254.1, 1168.2, 1017.15, 914.7, 839.7, 804.7, 780.5. HRMS (EI): Found 515.2006 [M + H]+, C28H40O2SiBr requires 515.1981.
3.2.6. General Method for Preparation of Amines 10a–10e
Methylamine (2 M in THF) (20 eq.) was added to the alkyl bromide compound 9a–9e and sealed in a high pressure tube. The reaction mixture was heated to 60 °C while stirring for 48 h. The reaction mixture was allowed sufficient time to cool, allowing the internal pressure to decrease prior to opening the pressure tube. The solvent was evaporated under reduced pressure. The oil was dissolved in DCM (30 mL) and was washed with a pH10 aq. solution (30 mL) which was then extracted with DCM (2 × 30 mL). The organic layers were combined and washed with water (50 mL), brine (50 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure to obtain a brown oil. The crude material was purified via flash chromatography on silica gel. (DCM:EtOAc).
2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclopentylidene)methyl)phenoxy)-N-methylethanamine (10a). Following the general method above, 9a (1 g, 2.05 mmol) and methylamine (20 eq., 41.02 mmol, 20 mL) were reacted together. The crude material was purified via flash chromatography on silica gel (DCM:EtOAc, 2:1) to afford a brown resin (737 mg, 82%). 1H-NMR (CDCl3) δ 7.10 (d, J = 8.53 Hz, 2H, Ar-H), 7.03 (d, J = 8.53 Hz, 2H, Ar-H), 6.86 (d, J = 8.53 Hz, 2H, Ar-H), 6.76 (d, J = 8.03 Hz, 2H, Ar-H), 4.12–4.19 (m, 2H, CH2), 3.03–3.16 (m, 2H, CH2), 2.58 (s, 3H, CH3), 2.34–2.44 (m, 4H, 2 × CH2), 1.63–1.73 (m, 4H, 2 × CH2), 1.00 (s, 9H, 3 × CH3), 0.20 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 156.0 (C-O-CH2), 153.2, 141.8, 136.4, 136.3, 131.4, 129.9, 129.7, 118.9, 113.4, 65.1 (CH2), 49.5 (CH2), 32.8 (CH3), 26.49 (CH2), 26.47 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3343.1 (br.), 2955.1, 2930.6, 2857.7, 1660.9, 1604.1, 1508.4, 1254.1 (br.), 1168.9, 915.4. HRMS (EI): Found 438.2838 [M + H]+, C27H40NO2Si requires 438.2828.
2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclohexaneylidene)methyl)phenoxy)-N-methylethanamine (10b). Following the general method above, 9b (1 g, 2.0 mmol) and methylamine (20 eq., 41.02 mmol, 20 mL) were reacted together. The crude material was purified via flash chromatography on silica gel (DCM:EtOAc, 2:1) to afford a brown resin (781 mg, 86%). 1H-NMR (CDCl3) δ 7.03 (d, J = 8.5 Hz, 2H, Ar-H), 6.96 (d, J = 8.5 Hz, 2H, Ar-H), 6.84 (d, J = 8.5 Hz, 2H, Ar-H), 6.74 (d, J = 8.5 Hz, 2H, Ar-H), 4.13–4.17 (m, 2H, CH2), 3.08 (d, J = 6.5 Hz, 2H, CH2), 2.58 (s, 3H, CH3), 2.24 (d, J = 5.0 Hz, 4H, 2 × CH2), 1.56–1.63 (m, 6H, 3 × CH2), 0.99 (s, 9H, CH3), 0.21 (s, 6H, CH3). 13C-NMR (CDCl3) δ 156.1, 153.3, 137.9, 136.0, 135.8, 133.0, 130.5, 130.3, 118.8, 113.4, 65.1 (CH2), 49.5 (CH2), 34.7 (NCH3), 32.1 (CH2), 32.0 (CH2), 28.2 (CH2), 26.4 (CH3), 17.71 (C(CH3)3), −4.83 (CH3). IR: νmax (KBr) cm−1: 3435.1, 2928.5, 2854.3, 1606.2, 1507.9, 1471.3, 1255.0, 1168.7, 915.1, 835.8, 778.9. HRMS (EI): Found 452.2982 [M + H]+, C28H42NO2Si requires 452.2985.
2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cycloheptylidene)methyl)phenoxy)-N-methylethanamine (10c). Following the general method above, 9c (1 g, 1.94 mmol) and methylamine (~20 eq., 41.02 mmol, 20 mL) were reacted together. The crude material was purified via flash chromatography over silica gel (DCM:EtOAc, 2:1) to afford a brown resin (813 mg, 90%). 1H-NMR (CDCl3) δ 7.07 (d, J = 8.8 Hz, 2H, Ar-H), 7.00 (d, J = 8.3 Hz, 2H, Ar-H), 6.83 (d, J = 8.3 Hz, 2H, Ar-H), 6.74 (d, J = 8.3 Hz, 2H, Ar-H), 4.08 (d, J = 6.5 Hz, 2H, CH2), 3.61 (d, J = 6.5 Hz, 2H, CH2), 2.99 (s, 1H, NH), 2.52 (s, 3H, CH3), 2.29–2.33 (m, 4H, 2 × CH2), 1.55–1.64 (m, 8H, 4 × CH2), 0.99 (s, 9H, CH3), 0.20 (s, 6H, CH3). 13C-NMR (CDCl3) δ 156.7, 153.6, 139.5, 136.8, 136.7, 136.4, 132.0, 130.4, 130.3, 128.5, 120.0, 119.3, 113.8, 113.6, 65.1 (CH2), 49.5 (CH2), 33.5 (NCH3), 33.4 (CH2), 29.4 (CH2), 28.2 (CH2), 28.1 (CH2), 25.7 (CH3), 18.1 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3436.1, 2928.1, 2855.9, 1604.3, 1507.1, 1471.7, 1253.4, 1171.7, 1045.2, 914.6, 839.2, 805.0, 780.6. HRMS (EI): Found 446.3152 [M + H]+, C29H44NO2Si requires 446.3141.
2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclooctylidene)methyl)phenoxy)-N-methylethanamine (10d). Following the general method above, 9d (1 g, 1.89 mmol) and methylamine (20 eq., 41.02 mmol, 20 mL) were reacted together. The crude material was purified via flash chromatography over silica gel (DCM:EtOAc, 2:1) to afford a brown resin (761 mg, 84%). 1H-NMR (CDCl3) δ 7.06–7.15 (m, 2H, Ar-H), 6.98–7.06 (m, 2H, Ar-H), 6.85–6.93 (m, 2H, Ar-H), 6.71–6.79 (m, 2H, Ar-H), 4.16–4.26 (m, 2H, CH2), 3.10–3.24 (m, 2H, CH2), 2.60 (s, 3H, CH3), 2.20–2.32 (m, 4H, 2 × CH2), 1.62–1.73 (m, 2H, CH2), 1.45–1.62 (m, 8H, 4 × CH2), 0.99 (s, 9H, 3 × CH3), 0.19 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 155.4 (C-O-CH2), 153.1, 139.6, 137.1, 136.5, 135.6, 129.6, 129.4, 119.1, 113.8, 63.3 (CH2), 48.5 (CH2), 33.6 (CH3), 32.0 (CH2), 26.0 (CH2), 25.9 (CH2), 25.7 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3413.8 (br.), 2927.7, 2856.0, 1643.2, 1605.9, 1506.7, 1241.1 (br.), 1168.5, 835.8. HRMS (EI): Found 480.3301 [M + H]+, C30H46NO2Si requires 480.3298.
2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(4-methylcyclohexaneylidene)methyl)phenoxy)-N-methyl-ethanamine (10e). Following the general method above, 9e (1 g, 1.94 mmol) and methylamine (~20 eq., 41.02 mmol, 20 mL) were reacted together. The crude material was purified via flash chromatography on silica gel (DCM:EtOAc, 2:1) to afford a brown resin (722 mg, 80%). 1H-NMR (CDCl3) δ 7.04 (d, J = 7.0 Hz, 2H, Ar-H), 6.97 (d, J = 8.5 Hz, 2H, Ar-H), 6.84 (d, J = 8.5 Hz, 2H, Ar-H), 6.75 (d, J = 8.5 Hz, 2H, Ar-H), 4.09 (t, J = 4.5 Hz, 2H, CH2), 3.01 (s, 2H, CH2), 2.75 (s, 1H, NH), 2.54–2.62 (m, 5H, CH3, CH2), 1.95 (t, J = 12.5 Hz, 2H, CH2), 1.78 (d, 2H, CH2), 1.60–1.64 (m, 1H, CH), 1.09 (q, J = 12.0 Hz, 2H, CH2), 1.00 (s, 9H, CH3), 0.94 (d, J = 6.6 Hz, 3H, CH3), 0.22 (s, 6H, CH3). 13C-NMR (CDCl3) δ 156.5, 153.3, 137.4, 135.9, 135.8, 133.2, 130.5, 130.4, 130.4, 118.8, 113.3, 66.1 (CH2), 50.2 (CH2), 36.4 (CH2), 35.6 (NCH3), 32.4 (CH), 31.3 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3435.6, 2946.9, 2927.7, 2856.3, 1605.4, 1507.9, 1457.6, 1471.9, 1253.3, 1168.5, 1045.1, 915.2, 838.1, 805.0, 780.0. HRMS (EI): Found 466.3156 [M + H]+, C29H44NO2Si requires 466.3141.
3.2.7. General Method for Preparation of Cyclofenil Derivatives 11a–11e
The appropriate silyl ether 10a–10e was dissolved in THF (3 mL) while stirring under N2. An equimolar quantity of TBAF (1 M in THF) was added and the mixture was allowed to stir for 30 min. The reaction was monitored via TLC. The solvent was evaporated to dryness. The residue was redissolved in DCM (20 mL) and washed with a 10% HCl solution (10 mL). The organic phase was dried over Na2SO4 and evaporated to dryness in vacuo. The residue is purified via flash chromatography on silica gel to afford the products.
4-(Cyclopentylidene(4-(2-(methylamino)ethoxy)phenyl)methyl)phenol (11a). Following the general deprotection method, the cyclofenil derivative 10a (200 mg, 0.457 mmol) and TBAF (1 M in THF) (0.457 mmol, 0.457 ml) were reacted together in THF (3 mL). The residue was purified via flash chromatography on silica gel (DCM:MeOH, gradient 50:1 to 40:1) to afford the product as a pale pink solid (107 mg, 73%), m.p. 160–163 °C. 1H-NMR (CDCl3) δ 9.23–9.42 (m, 1H, OH), 7.03 (d, J = 8.53 Hz, 2H, Ar-H), 6.92 (d, J = 8.53 Hz, 2H, Ar-H), 6.86 (d, J = 8.53 Hz, 2H, Ar-H), 6.68 (d, J = 8.53 Hz, 2H, Ar-H), 4.00 (t, J = 5.52 Hz, 2H, CH2), 2.84 (t, J = 5.27 Hz, 2H, CH2), 2.35 (s, 3H, NCH3), 2.26–2.34 (m, 4H, 2 × CH2), 1.54–1.69 (m, 4H, 2 × CH2). 13C-NMR (CDCl3) δ 157.1, 155.9, 140.9, 136.3, 134.5, 132.4, 130.3, 115.2, 114.3, 67.2 (CH2), 50.6 (CH2), 36.4 (NCH3), 33.2 (CH2), 26.9 (CH2). IR:νmax (KBr) cm−1: 3438.3 (br.), 2952.55, 2864.3, 2591.1, 1608.2, 1507.1, 1268.9, 1235.6, 1053.8, 834.8. HRMS (EI): Found 324.1950 [M + H]+, C21H26NO2 requires 324.1964.
4-(Cyclohexaneylidene(4-(2-(methylamino)ethoxy)phenyl)methyl)phenol (11b). Following the general deprotection method above, the cyclofenil derivative 10b (200 mg, 0.442 mmol) and TBAF (1 M in THF) (0.442 mmol, 0.442 ml) were reacted together in THF (3 mL). The residue was purified via flash chromatography on silica gel (DCM:MeOH, gradient 50:1 to 40:1) to afford the product as a white solid (116 mg, 78%), m.p. 169–170 °C. 1H-NMR (DMSO-d6) δ 6.95 (d, J = 8.53 Hz, 2H, Ar-H), 6.77–6.88 (m, 4H, Ar-H), 6.67 (d, J = 8.53 Hz, 2H, Ar-H), 3.98 (t, J = 5.52 Hz, 2H, CH2), 2.80 (t, J = 5.65 Hz, 2H, CH2), 2.33 (s, 3H, NCH3), 2.09–2.23 (m, 4H, 2 × CH2), 1.46–1.64 (m, 6H, 3 × CH2) (OH not observed). 13C-NMR (DMSO-d6) δ 157.3, 157.0, 137.0, 135.9, 134.2, 133.2, 130.9, 130.8, 115.3, 114.3, 67.5 (CH2), 50.8 (CH2), 36.6 (NCH3), 32.4 (CH2), 28.6 (CH2), 23.5 (CH2). IR: νmax (KBr) cm−1: 3438.2 (br.), 2930.0, 2847.3, 2660.1, 2591.11, 1607.8, 1508.4, 1465.7, 1302.6, 1273.0, 1168.4, 1041.8, 833.7. HRMS (EI): Found 338.2104 [M + H]+, C22H28NO2 requires 338.2120.
4-(Cycloheptylidene(4-(2-(methylamino)ethoxy)phenyl)methyl)phenol (11c). Following the general deprotection method above, the cyclofenil derivative 10c (200 mg, 0.429 mmol) and TBAF (1 M in THF) (0.429 mmol, 0.429 mL) were reacted together in THF (3 mL). The residue was purified via flash chromatography on silica gel (DCM:MeOH, gradient 50:1 to 40:1) to afford the product as a pale pink solid (122 mg, 81%), m.p. 169–171 °C. 1H-NMR (DMSO-d6) δ 7.00 (d, J = 8.78 Hz, 2H, Ar-H), 6.80–6.95 (m, 4H, Ar-H), 6.67 (d, J = 8.53 Hz, 2H, Ar-H), 3.97 (t, J = 5.65 Hz, 2H, CH2), 2.80 (t, J = 5.52 Hz, 2H, CH2), 2.33 (s, 3H, NCH3), 2.18–2.27 (m, 4H, 2 × CH2), 1.46–1.63 (m, 8H, 4 × CH2). 13C-NMR (DMSO-d6) δ 157.2, 156.0, 138.4, 137.1, 136.4, 134.5, 130.3, 130.3, 115.2, 114.3, 67.5 (CH2), 50.8 (CH2), 36.6 (NCH3), 33.3 (CH2), 29.2 (CH2), 28.0 (CH2), 23.5 (CH2). IR: νmax (KBr) cm−1: 3440.6 (br.), 2929.7, 2849.4, 2575.8, 1607.4, 1505.9, 1463.3, 1274.6, 1240.8, 1168.6, 1045.2, 828.8. HRMS (EI): Found 352.2261 [M + H]+, C23H30NO2 requires 352.2277.
4-(Cyclooctylidene(4-(2-(methylamino)ethoxy)phenyl)methyl)phenol (11d). Following the general deprotection method above, the cyclofenil 10d (200 mg, 0.417 mmol) and TBAF (1 M in THF) (0.417 mmol, 0.417 mL) were reacted together in THF (3 mL). The residue was purified via flash chromatography on silica gel (DCM:MeOH, gradient 50:1 to 40:1) to afford the product as a clear oil (119 mg, 78%). 1H-NMR (DMSO-d6) δ 7.02 (d, J = 8.53 Hz, 2H, Ar-H), 6.80–6.93 (m, 4H, Ar-H), 6.68 (d, J = 8.53 Hz, 2H, Ar-H), 3.96 (t, J = 5.65 Hz, 2H, CH2), 2.80 (t, J = 5.77 Hz, 2H, CH2), 2.32 (s, 3H, NCH3), 2.13–2.26 (m, 4H, 2 × CH2), 1.40–1.68 (m, 10H, 5 × CH2). 13C-NMR (DMSO-d6) δ 157.1, 156.8, 138.5, 137.2, 136.8, 134.1, 130.0, 129.8, 115.5, 114.5, 67.5 (CH2), 50.8 (CH2), 36.6 (NCH3), 32.4 (CH2), 26.4 (CH2), 26.2 (CH2), 23.5 (CH2). IR: νmax (KBr) cm−1: 3438.1 (br.), 2932.3, 2871.9, 2852.2, 2640.4, 2566.5, 1607.9, 1587.1, 1505.9, 1455.8, 1274.9, 1239.5, 1168.1, 1046.6, 838.9. HRMS (EI): Found 366.2414 [M + H]+, C24H32NO2 requires 366.2433.
4-((4-(2-(Methylamino)ethoxy)phenyl)(4-methylcyclohexaneylidene)methyl)phenol (11e). Following the general deprotection method above, the cyclofenil derivative 10e (200 mg, 0.429 mmol) and TBAF (1 M in THF) (0.429 mmol, 0.429 mL) were reacted together in THF (3 mL). The residue was purified via flash chromatography on silica gel (DCM:MeOH, gradient 50:1 to 40:1) to afford the product as a pale pink solid (124 mg, 82%), m.p: 74–76 °C. 1H-NMR (DMSO-d6) δ 6.94 (d, J = 8.50 Hz, 2H, Ar-H), 6.78–6.88 (m, 4H, Ar-H), 6.68 (d, J = 8.53 Hz, 2H, Ar-H), 3.98 (t, J = 5.52 Hz, 2H, CH2), 2.80 (t, J = 5.40 Hz, 2H, CH2), 2.48 (t, J = 16.10 Hz, 2H, CH2), 2.33 (s, 3H, NCH3), 1.90 (dt, J = 3.14, 12.86 Hz, 2H, CH2), 1.72 (d, J = 11.80 Hz, 2H, CH2), 1.51–1.63 (m, 3H, CH2, CH), 0.90 (d, J = 6.27 Hz, 3H, CH3). 13C-NMR (DMSO-d6) δ 157.3, 156.6, 136.7, 135.9, 134.3, 133.6, 130.9, 130.8, 115.2, 114.3, 67.5 (CH2), 50.8 (CH2), 36.6 (NCH3), 32.7 (CH), 31.7 (CH2), 23.5 (CH2), 22.4 (CH3). IR:νmax (KBr) cm−1: 3439.7 (br.), 2945.8, 2913.1, 2887.4, 2866.9, 1607.06, 1505.9, 1456.2, 1270.3, 1234.5, 1168.2, 1043.6, 830.4. HRMS (EI): Found 352.2264 [M + H]+, C23H30NO2 requires 352.2277.
3.2.8. General Method for the Synthesis of OTBDMS Cyclofenil-Acrylic Acid Conjugates 12a–12e
A mixture of the required acrylic acid 1l (1.2 eq), EDC (1.4 eq.) and HOBt (1.4 eq.) was suspended in anhydrous dichloromethane (3 mL) and stirred for 10 min under a nitrogen atmosphere. The appropriate cyclofenil derivative 10a–10e (1 eq.) was dissolved in anhydrous dichloromethane (3 mL) and slowly added to the mixture via syringe. The reaction was allowed stir for 16 h. The reaction was monitored via TLC. The reaction mixture was diluted to 15 mL with anhydrous dichloromethane. To this mixture, water (20 mL) was added. The aqueous phase was extracted with DCM (20 mL × 3), brine (50 mL), dried over Na2SO4 and evaporated to dryness in vacuo to yield the crude product. The material was purified via flash chromatography on silica gel using DCM:EtOAc as eluent.
(E)-N-(2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclopentylidene)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxy phenyl) acrylamide (12a). The acrylic acid 1l (1.2 eq., 196 mg, 0.54 mmol), EDC (1.4 eq., 120 mg, 0.63 mmol), and HOBt (1.4 eq., 85 mg, 0.63 mmol) were reacted with protected cyclofenil derivative 10a (1 eq., 200 mg, 0.45 mmol) following the general method above. The product was obtained as a brown resin. The material was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 15:1 to 10:1) to afford the product as a yellow solid (284 mg, 81%), m.p. 87–89 °C, which was used immediately in the following reaction without further purification. 1H-NMR (CDCl3) δ 6.97–7.16 (m, 4H, Ar-H, C=CH), 6.52–6.88 (m, 10H, Ar-H), 4.25 (m, 1H, 0.5 × OCH2), 3.60–3.92 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.08–3.23 (m, 3H, NCH3), 2.40 (s, 4H, 2 × CH2), 1.69 (s, 4H, 2 × CH2), 1.00 (s, 9H, 3 × CH3), 0.22 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 169.0, 153.2, 152.8, 145.8, 144.6, 141.8, 129.9, 129.7, 118.9, 115.0, 113.2, 109.7, 105.5, 66.0 (CH2), 60.5 (OCH3), 55.4 (OCH3), 38.6 (CH2), 32.8 (CH2), 26.5 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3350.9 (br.), 2954.2, 2936.7, 2859.3, 1603.9, 1581.5, 1505.9, 1463.8, 1242.5 (br.), 1127.1, 912.2. HRMS (EI): Found 780.3922 [M + H]+, C46H58NO8Si requires 780.3932.
(E)-N-(2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclohexaneylidene)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (12b). The acrylic acid 1l (1.2 eq., 189 mg, 0.526 mmol)), EDC (1.4 eq., 118 mg, 0.616 mmol) and HOBt (1.4 eq., 83 mg, 0.616 mmol) were reacted with protected cyclofenil derivative 10b (1 eq., 200 mg, 0.44 mmol) following the general method above. The product was obtained as a brown resin. The material was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 15:1 to 10:1) to afford the product as a yellow solid (272 mg, 78%), m.p. 88–90 °C. which was used immediately in the following reaction without further purification. 1H-NMR (CDCl3) δ 6.90–7.09 (m, 4H, Ar-H, C=CH), 6.52–6.85 (m, 10H, Ar-H), 4.17–4.29 (m, 1H, 0.5 × OCH2), 3.57–3.91 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.07–3.26 (m, 3H, NCH3), 2.18–2.30 (m, 4H, 2 × CH2), 1.52–1.69 (m, 6H, 3 × CH2), 1.00 (s, 9H, 3 × CH3), 0.21 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 170.0, 153.3, 152.8, 145.9, 144.6, 130.5, 130.3, 121.5, 118.8, 115.0, 113.1, 109.7, 105.5, 66.1 (CH2), 60.5 (OCH3), 55.4 (OCH3), 32.0 (CH2), 28.2 (CH2), 26.4 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). HRMS (EI): Found 794.4079 [M + H]+, C47H60NO8Si requires 794.4088.
(E)-N-(2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cycloheptylidene)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (12c). The acrylic acid 1l (1.2 eq., 185 mg, 0.516 mmol), EDC (1.4 eq., 118 mg, 0.616 mmol) and HOBt (1.4 eq., 83 mg, 0.616 mmol) were reacted with protected cyclofenil derivative 10c (1 eq., 200 mg, 0.43 mmol) following the general method above. The product was obtained as a brown resin. The material was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 15:1 to 10:1) to afford the product as a yellow solid (284 mg, 81%), m.p. 84–86 °C which was used immediately in the following reaction without further purification. 1H-NMR (CDCl3) δ 6.93–7.12 (m, 4H, Ar-H, C=CH), 6.50–6.86 (m, 10H, Ar-H), 5.64 (br. s., 1H, OH), 4.17–4.27 (m, 1H, 0.5 × OCH2), 3.55–3.95 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.06–3.24 (m, 3H, NCH3), 2.24–2.37 (m, 4H, 2 × CH2), 1.49–1.64 (m, 8H, 4 × CH2), 0.99 (s, 9H, 3 × CH3), 0.20 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 172.1, 156.6, 153.7, 153.3, 146.4, 145.1, 139.7, 137.9, 136.8, 136.3, 135.9, 130.5, 130.3, 129.9, 128.6, 122.0, 119.4, 115.5, 113.7, 110.2, 105.8, 66.5 (CH2), 60.9 (OCH3), 56.0 (OCH3), 55.9 (OCH3), 47.7 (CH2), 39.0 (NCH3), 33.5 (CH2), 33.4 (CH2), 29.4 (CH2), 28.2 (CH2), 28.1 (CH2), 25.7 (CH3), 18.2 (C(CH3)3), −4.4 (CH3). IR: νmax (KBr) cm−1: 3436.1 (br.). HRMS (EI): Found 830.4050 [M + H]+, C48H61NNaO8Si requires 830.4064.
(E)-N-(2-(4-((4-((tert-Butyldimethylsilyl)oxy)phenyl)(cyclooctylidene)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (12d). The acrylic acid 1l (1.2 eq., 180 mg, 0.5 mmol)), EDC (1.4 eq., 118 mg, 0.616 mmol) and HOBt (1.4 eq., 83 mg, 0.616 mmol) were reacted with protected cyclofenil derivative 10d (1 eq., 200 mg, 0.416 mmol) following the general method above. The product was afforded as a brown resin. The material was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 15:1 to 10:1) to afford the product as a yellow solid (243 mg, 71%), m.p. 89–91 °C which was used immediately in the following reaction without further purification. 1H-NMR (CDCl3) δ 6.95–7.15 (m, 4H, Ar-H, C=CH), 6.51–6.87 (m, 10H, Ar-H), 4.17–4.27 (m, 1H, 0.5 × OCH2), 3.56–3.94 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.05–3.23 (m, 3H, NCH3), 2.20–2.34 (m, 4H, 2 × CH2), 1.62–1.73 (m, 2H, CH2), 1.48–1.61 (m, 8H, 4 × CH2), 0.99 (s, 9H, 3 × CH3), 0.20 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 171.1, 156.4, 153.1, 152.8, 145.9, 144.7, 139.5, 136.6, 135.7, 135.4, 129.7, 129.4, 128.1, 121.5, 119.1, 115.0, 113.4, 109.7, 105.5, 66.1 (CH2), 60.5 (OCH3), 60.0 (OCH3), 55.6 (OCH3), 55.4 (OCH3), 47.2 (CH2), 38.6 (NCH3), 32.1 (CH2), 32.0 (CH2), 26.1 (CH2), 26.0 (CH2), 25.9 (CH2), 25.8 (CH2), 25.2 (CH3), 17.7 (C(CH3)3), −4.9 (CH3). IR: νmax (KBr) cm−1:3425.7 (br.), 2929.0, 2856.1, 1633.8, 1603.9, 1581.6, 1505.9, 1463.7, 1240.2, 1126.2, 838.3. HRMS (EI): Found 844.4200 [M + H]+, C49H63NNaO8Si requires 844.4221.
(E)-N-(2-(4-((4-((tert-butyldimethylsilyl)oxy)phenyl)(4-methylcyclo hexaneylidene)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (12e). The acrylic acid 1l (1.2 eq., 189 mg, 0.526 mmol), EDC (1.4 eq., 118 mg, 0.616 mmol) and HOBt (1.4 eq., 83 mg, 0.616 mmol) were reacted with protected cyclofenil derivative 10e (1 eq., 200 mg, 0.429 mmol) following the general method above The product was afforded as a brown resin. The material was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 15:1 to 10:1) to afford the product as a yellow solid (294 mg, 85%), m.p. 89–93 °C which was used immediately in the following reaction without further purification. 1H-NMR (CDCl3) δ 6.88–7.09 (m, 4H, Ar-H, C=CH), 6.53–6.86 (m, 10H, Ar-H), 5.50–5.89 (br. s., 1H, OH), 4.23 (m, 1H, 0.5 × OCH2), 3.57–3.92 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.07–3.24 (m, 3H, NCH3), 2.50–2.64 (m, 2H, CH2), 1.87–2.00 (m, 2H, CH2), 1.72–1.78 (m, 2H, CH2), 1.05–1.14 (m, 2H, CH2), 1.00 (s, 9H, 3 × CH3), 0.91–0.97 (m, 3H, CH3), 0.21 (s, 6H, 2 × CH3). 13C-NMR (CDCl3) δ 171.7, 156.3, 153.3, 152.8, 145.9, 144.7, 137.5, 135.9, 135.4, 133.1, 130.5, 130.3, 128.1, 121.5, 118.8, 115.0, 113.1, 109.7, 105.5, 66.1 (CH2), 60.5 (OCH3), 60.0 (OCH3), 55.6 (OCH3), 55.4 (OCH3), 47.2 (CH2), 38.5 (NCH3) 36.4 (CH2), 32.3 (CH), 31.3 (CH2), 25.2 (CH3), 21.6 (CH3), 17.7 (C(CH3)3), −4.8 (CH3). IR: νmax (KBr) cm−1: 3436.6 (br.), 2929.1, 2852.2, 1628.9, 1604.4, 1581.8, 1505.9, 1463.4, 1239.6, 1126.1, 839.5. HRMS (EI): Found 808.4211 [M + H]+, C48H62NO8Si requires 808.4239.
3.2.9. General Method for Deprotection of tert-Butyldimethylsilyl Ethers 12a–12e
The appropriate silyl ether 12a–12e was dissolved in a minimum amount of THF (3 mL) while stirring under N2. An equimolar quantity of TBAF (1 M in THF) was added and the mixture was allowed to stir for 30 min. The reaction was monitored via TLC. The solvent was evaporated to dryness. The residue was redissolved in DCM (20 mL) and washed with a 10% HCl solution (10 mL). The organic phase was dried over Na2SO4 and evaporated to dryness in vacuo. The residue is purified via flash chromatography on silica gel to afford the product.
(E)-N-(2-(4-(Cyclopentylidene(4-hydroxyphenyl)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (13a). The protected cyclofenil-acrylic acid derivative 12a (150 mg, 0.192 mmol) and TBAF (1 M in THF) (0.192 mmol, 0.192 mL) were reacted together in THF (3 mL) following the general method above. The residue was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 5:1 to 2:1) to afford the product as a yellow solid (116 mg, 91%), m.p. 159–162 °C. 1H-NMR (CDCl3) δ 6.96–7.14 (m, 4H, Ar-H, C=CH), 6.47–6.91 (m, 10H, Ar-H), 4.19–4.28 (m, 1H, 0.5 × OCH2), 3.56–3.95 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.10–3.26 (m, 3H, NCH3), 2.31–2.45 (m, 4H, 2 × CH2), 1.62–1.74 (m, 4H, 2 × CH2). 13C-NMR (CDCl3) δ 171.5, 156.1, 153.7, 152.9, 145.9, 144.6, 141.7, 136.3, 135.5, 135.2, 131.3, 129.9, 128.0, 121.6, 115.0, 114.4, 113.2, 109.7, 105.5, 66.0 (CH2), 60.5 (OCH3), 55.6 (OCH3), 55.4 (OCH3), 47.4 (CH2), 32.8 (CH3), 29.3 (CH2), 26.5 (CH2). IR: νmax (KBr) cm−1: 3433.5 (br.), 2933.4, 2862.0, 1603.9, 1508.1, 1408.8, 1238.4, 1125.3, 835.2. HRMS (EI): Found 688.2868 [M + Na]+, C40H43NNaO8 requires 688.2886.
(E)-N-(2-(4-(Cyclohexaneylidene(4-hydroxyphenyl)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (13b). The protected cyclofenil-acrylic acid derivative 12b (150 mg, 0.189 mmol) and TBAF (1 M in THF) (0.189 mmol, 0.189 ml) were reacted together in THF (3 mL) following the general method above. The residue was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 5:1 to 2:1) to afford the product as a yellow solid (119 mg, 93%), m.p. 166–170 °C. 1H-NMR (CDCl3) δ 6.87–7.07 (m, 4H, Ar-H), 6.50–6.84 (m, 10H, Ar-H), 4.17–4.31 (m, 1H, 0.5 × CH2), 3.54–3.95 (m, 15H, 0.5 × CH2, CH2, 3 × OCH3), 3.08–3.24 (m, 3H, NCH3), 2.17–2.30 (m, 4H, 2 × CH2), 1.50–1.67 (m, 6H, 3 × CH2). 13C-NMR (CDCl3) δ 172.1, 154.0, 152.9, 146.0, 144.7, 137.8, 137.3, 135.2, 134.9, 132.9, 130.5, 130.5, 129.6, 128.0, 121.6, 115.0, 114.3, 113.2, 109.7, 105.5, 66.0 (CH2), 60.5 (OCH3), 60.0 (OCH3), 55.6 (OCH3), 55.4 (OCH3), 47.3 (CH2), 38.7 (CH3), 32.0 (CH2), 28.2 (CH2), 26.4 (CH2). IR: νmax (KBr) cm−1: 3425.4 (br.), 2926.5, 2847.3, 1605.2, 1582.5, 1510.9, 1449.1, 1273.8, 1237.5, 1125.7, 1026.9, 835.4. HRMS (EI): Found 702.3026 [M + Na]+, C41H45NNaO8 requires 702.3043.
(E)-N-(2-(4-(Cycloheptylidene(4-hydroxyphenyl)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (13c). The protected cyclofenil-acrylic acid derivative 12c (150 mg, 0.185 mmol) and TBAF (1 M in THF) (0.185 mmol, 0.185 ml) were reacted together in THF (3 mL) following the general method above. The residue was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 5:1 to 2:1) to afford the product as a yellow solid (113 mg, 98%), m.p. 162–163 °C. 1H-NMR (CDCl3) δ 6.92–7.12 (m, 4H, Ar-H, C=CH), 6.49–6.84 (m, 10H, Ar-H), 4.16–4.26 (m, 1H, 0.5 × OCH2), 3.53–3.93 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.0. 13C-NMR (CDCl3) δ 172.2, 156.6, 154.3, 153.3, 146.4, 145.1, 139.6, 137.8, 136.9, 136.2, 135.8, 135.6, 130.5, 130.4, 130.1, 128.5, 122.0, 115.5, 114.9, 113.7, 110.2, 106.1, 66.4 (CH2), 60.9 (OCH3), 60.5 (CH2), 56.0 (OCH3), 55.9 (OCH3), 53.5 (CH2), 50.1 (CH2), 47.8 (CH2), 39.1 (NCH3), 33.4 (CH2), 29.4 (CH2), 28.1 (CH2). IR: νmax (KBr) cm−1: 3437.4 (br.), 2923.4, 2842.2, 160.3, 1582.3, 1506.0, 1410.0, 1273.7, 1238.2, 1125.3, 831.2. HRMS (EI): Found 716.3185 [M + Na]+, C42H47NNaO8 requires 716.3199.
(E)-N-(2-(4-(Cyclooctylidene(4-hydroxyphenyl)methyl)phenoxy)ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide (13d). The protected cyclofenil-acrylic acid derivative 12e (150 mg, 0.182 mmol) and TBAF (1 M in THF) (0.182 mmol, 0.182 ml) were reacted together in THF (3 mL) following the general method above. The residue was purified via flash chromatography on silica gel (DCM:EtOAc, gradient 5:1 to 2:1) to afford the product as a yellow solid (102 mg, 79%), m.p. 158–159 °C. 1H-NMR (CDCl3) δ 6.92–7.13 (m, 4H, Ar-H, C=CH), 6.50–6.86 (m, 10H, Ar-H), 4.16–4.25 (m, 1H, 0.5 × OCH2), 3.51–3.95 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.05–3.24 (m, 3H, NCH3), 2.17–2.36 (m, 4H, 2 × CH2), 1.62–1.74 (m, 2H, CH2), 1.45–1.61 (m, 8H, 4 × CH2). 13C-NMR (CDCl3) δ 172.4, 156.6, 153.8, 152.9, 146.0, 144.7, 139.4, 139.4, 137.3, 135.6, 135.2, 133.9, 129.6, 129.6, 121.6, 115.0, 114.6, 113.4, 109.8, 105.5, 65.9 (CH2), 60.5 (OCH3), 60.1 (CH2), 55.5 (OCH3), 55.4 (OCH3), 39.1 (NCH3), 32.0 (CH2), 26.1 (CH2), 25.9 (CH2), 25.8 (CH2). IR: νmax (KBr) cm−1: 3437.6 (br.), 2927.9, 2847.3, 1604.1, 1582.3, 1505.9, 1274.6, 1238.3, 1125.6, 831.6. HRMS (EI): Found 730.3336 [M + Na]+, C43H49NNaO8 requires 730.3356.
(E)-3-(3-Hydroxy-4-methoxyphenyl)-N-(2-(4-((4-hydroxyphenyl)(4-methylcyclohexaneylidene)methyl)-phenoxy)ethyl)-N-methyl-2-(3,4,5-trimethoxy phenyl) acrylamide (13e). The protected cyclofenil-acrylic acid derivative 12e (150 mg, 0.186 mmol) and TBAF (1 M in THF) (0.186 mmol, 0.186 ml) were reacted together in THF (3 mL) following the general method above. The residue was purified via flash chromatography on silica gel (DCM:EtOAc; gradient 5:1 to 2:1) to afford the product as a yellow solid (110 mg, 86%), m.p. 158–161 °C. 1H-NMR (CDCl3) δ 6.87–7.06 (m, 4H, Ar-H), 6.51–6.84 (m, 10H, Ar-H), 4.17–4.27 (m, 1H, 0.5 × CH2), 3.54–3.94 (m, 15H, 0.5 × OCH2, NCH2, 4 × OCH3), 3.08–3.25 (m, 3H, NCH3), 2.49–2.64 (m, 2H, CH2), 1.93 (t, J = 11.54 Hz, 2H, CH2), 1.76 (d, J = 10.54 Hz, 2H, CH2), 1.55–1.68 (m, 1H, CH), 1.01–1.15 (m, 2H, CH2), 0.87–0.98 (m, 3H, CH3). 13C-NMR (CDCl3) δ 172.2, 154.1, 152.8, 146.0, 144.7, 137.4, 134.8, 133.1, 130.5, 130.5, 128.0, 121.6, 115.0, 114.3, 113.2, 109.8, 105.5, 65.9 (CH2), 60.5 (OCH3), 60.1 (OCH3), 55.5 (OCH3), 55.4 (OCH3), 47.4 (CH3), 38.7 (NCH3), 36.4 (CH2), 32.3 (CH), 31.3 (CH2), 21.6 (CH3). IR: νmax (KBr) cm−1: 3421.5 (br.), 2912.0, 2840.1, 1605.5, 1582.3, 1508.4, 1454.1, 1274.2, 1237.9, 1125.8, 830.8. HRMS (EI): Found 694.3359 [M + H]+, C42H48NO8 requires 694.3380.
N-{2-[4-(1,2-Diphenylbut-1-enyl)phenoxy]ethyl}-N-methylsuccinamic acid (14a). (i) The amine 2b (0.10 g, 0.28 mmol) and succinic anhydride (0.03 g, 0.28 mmol) were dissolved in dry DCM (5 mL). The reaction was allowed stir at room temperature for 16 h. (The reaction was monitored via TLC, DCM:MeOH, 4:1). The reaction mixture was diluted with DCM (10 mL), washed with 1 M NaOH solution (10 mL). The aqueous phase was extracted with DCM (10 mL × 3). The combined organic layers were acidified with dilute HCl dropwise, washed with water (10 mL) and brine (10 mL), dried over sodium sulfate and then evaporated to dryness in vacuo to afford the product 14a (0.13 g, 97%) as a light brown oil. (ii) Succinic acid (0.05 g, 0.42 mmol), dicyclohexylcarbodiimide (DCC) (0.09 g, 0.42 mmol) and 1-hydroxy benzotriazole hydrate (HOBt) were dissolved in dry DCM (5 mL) under a N2 environment. The mixture was allowed to stir for 20 minutes before adding a solution of 1b (0.15 g, 0.42 mmol) in dry DCM (3 mL). The reaction was allowed stir at room temperature for 24 h until no starting material was visible by TLC (DCM:MeOH = 4:1). The reaction mixture was filtered to remove the dicyclohexylurea and then evaporated in vacuo and the residue was purified via flash chromatography on silica gel (DCM:MeOH = 20:1) to afford an isomeric mixture (E:Z = 1:1) of the product as a light brown oil (0.17 g, 90% yield). 1H-NMR (CDCl3): δ 0.95–1.00 (m, 6H, CH3), 2.47–2.57 (m, 4H, CH2), 2.61–2.86 (m, 8H, succinic CH2), 2.99–3.21 (m, 6H, NCH3), 3.65–4.18 (m, 8H, CH2N, CH2O), 6.54–7.40 (m, 28 H, ArH), 8.72 (s, 2H, COOH). 13C-NMR (CDCl3): δ 13.65, 28.08, 28.46, 28.50, 29.06, 29.58, 29.88, 34.19, 34.26, 37.48, 37.55, 48.29, 48.33, 49.17, 49.26, 64.71, 65.04, 66.25, 66.59, 113.22, 113.97, 114.00, 125.71, 125.75, 126.09, 126.14, 126.59, 127.34, 127.81, 127.91, 127.95, 128.15, 129.47, 129.71, 130.68, 130.77, 131.98, 132.06, 135.86, 136.32, 136.37, 136.83, 138.14, 138.29, 141.50, 141.69, 142.02, 142.14, 142.26, 142.32, 142.42, 143.21, 143.29, 143.75, 155.98, 156.36, 156.86, 157.21, 172.32, 172.69, 176.78, 176.85. IR: νmax (KBr) cm−1: 3054.8, 2969.2, 1732.2, 1606.5, 1507.4, 1463.1, 1442.2, 1407.6, 1283.3, 1241.3, 1174.0, 1138.9, 1074.1, 1054.4, 1030.3, 909.8, 732.1. HRMS (EI): Found 480.2162 [M + Na]+, C29H31NO4Na requires 480.2151.
N-[2-(4-{1-[4-(tert-Butyldimethylsilanyloxy)phenyl]-2-phenylbut-1-enyl}phenoxy)ethyl]-N-methyl-succinamic acid (14b). The amine 2b (0.20 g, 0.41 mmol, E/Z = 1:1) and succinic anhydride (0.04 g, 0.41 mmol) were dissolved in dry DCM (5mL). The reaction was allowed stir at room temperature for 16 h and monitored via TLC (DCM:MeOH = 4:1). The reaction mixture was diluted with DCM (10 mL) and washed with 1M NaOH solution (10 mL). The aqueous phase was extracted with DCM (10 mL × 3). The combined organic layers were acidified with dilute HCl dropwise, washed with water (10 mL) and brine (10 mL), dried over sodium sulfate and then evaporated to dryness in vacuo to afford an isomeric mixture (E:Z = 1:1) of the product (222 mg, 92%) as a light brown oil. 1H-NMR (CDCl3): δ 0.12–0.25 (m, 12H, Si(CH3)2), 0.94–1.02 (m, 24H, SiC(CH3)3), 2.48–2.52 (m, 4H, CH2), 2.65–2.87 (m, 8H, 4 × CH2), 3.00–3.22 (m, 6H, NCH3), 3.70–4.17 (m, 8H, CH2N, CH2O), 6.49–7.20 (m, 26H, ArH). 13C-NMR (CDCl3): δ −4.92, −4.80, 13.18, 13.22, 17.75, 25.22, 25.24, 28.46, 28.60, 37.04, 37.10, 47.91, 65.78, 66.12, 112.65, 113.42, 118.53, 119.10, 125.46, 127.31, 127.39, 127.43, 129.26, 130.09, 130.22, 130.30, 131.38, 131.57, 131.65, 135.73, 136.20, 136.27, 137.37, 137.44, 140.65, 142.08, 153.06, 156.68, 172.69, 176.78. IR: νmax (KBr) cm−1: 3435.7, 2927.5, 1696.5, 1624.0, 1603.6, 1509.3, 1417.2, 1248.5, 1202.1, 1167.1, 928.4, 837.0. HRMS (EI): Found 610.2972 [M + Na]+, C35H45NO5SiNa requires 610.2965.
N-(2-{4-[1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-N-methylsuccinamic acid (14c). The protected acid 14b (0.09 g, 0.15 mmol) was treated with TBAF following the general deprotection method above and afforded an isomeric mixture (E:Z = 1:1) of product 13c as a brown oil (0.06 g, 82%). 1H-NMR (CDCl3): δ 0.92–0.96 (m, 6H, CH3), 2.47–2.70 (m, 12H, CH2, succinic CH2), 2.95–3.16 (m, 6H, NCH3), 3.61–3.76 (m, 4H, CH2N), 3.96–3.97 (m, 2H, CH2O), 4.11–4.12 (m, 2H, CH2O), 6.49–7.17 (m, 26 H, ArH). 13C-NMR (CDCl3): δ 13.48, 13.52, 28.76, 28.90, 37.34, 37.40, 48.21, 66.08, 66.42, 112.95, 113.72, 118.83, 119.40, 125.86, 127.61, 127.69, 127.73, 129.56, 130.39, 130.52, 130.60, 131.68, 131.87, 131.95, 136.03, 136.50, 136.57, 137.67, 137.74, 140.95, 142.38, 153.36, 156.98, 172.99, 177.08. IR: νmax (KBr) cm−1: 3435.7, 2930.6, 1713.4, 1604.6, 1505.6, 1408.2, 1254.2, 1171.6, 912.1, 838.3. HRMS (EI): Found 496.2115 [M + H]+, C29H31NO5Na requires 496.2100.
N-{2-[4-((Z)-1,2-Diphenylbut-1-enyl)phenoxy]ethyl}-N-methylsuccinamic acid 2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)vinyl]phenyl ester (16a). N-{2-[4-(1,2-Diphenylbut-1-enyl)phenoxy]ethyl}-N-methyl-succinamic acid 13a (0.11 g, 0.24 mmol), dicyclohexylcarbodiimide (0.05 g, 0.24 mmol) and 1-hydroxybenzotriazole hydrate (0.03 g, 0.24 mmol) were dissolved in dry DCM (5 mL) under nitrogen atmosphere. The mixture was stired for 20 min before adding a solution of combretastatin A-4 15 (0.15 g, 0.42 mmol) in dry DCM (3 mL). The reaction was stirred at room temperature for 24 h until no starting material was visible by TLC (DCM:MeOH = 4:1). The reaction mixture was filtered to remove the dicyclohexylurea byproduct. The mixture was evaporated to dryness in vacuo and the material was purified via flash chromatography on silica gel (DCM:MeOH = 20:1) to afford the product as an isomeric mixture (E:Z = 1:1) as a light yellow oil (150 mg, 83% yield). 1H-NMR (CDCl3): δ 0.92–0.98 (m, 6H, CH3), 2.45–2.55 (m, 4H, CH2), 2.67–3.24 (m, 14H, succinic CH2, NCH3), 3.69–4.26 (m, 32H, OCH3, OCH3, NCH3), 6.47–7.39 (m, 42H, ArH). 13C-NMR (CDCl3): δ 13.17, 24.53, 25.18, 25.68, 28.58, 33.51, 36.95, 47.67, 48.63, 55.47, 55.47, 55.64, 60.46, 60.53, 102.86, 105.39, 106.08, 111.50, 111.92, 112.76, 113.50, 122.89, 124.82, 125.21, 125.61, 125.99, 126.52, 126.85, 127.09, 127.33, 127.43, 127.67, 128.19, 129.00, 129.24, 129.65, 130.20, 130.35, 131.49, 132.01, 135.82, 139.55, 149.78, 152.48, 152.92, 170.91, 174.44. IR: νmax (KBr) cm−1: 3434.8, 3326.8, 2929.3, 2850.8, 1764.3, 1626.5, 1578.1, 1508.5, 1243.5, 1127.1, 701.3. HRMS (EI): Found 778.3353 [M + Na]+, C47H49NO8Na requires 778.3356.
N-(2-{4-[(Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl succinamic acid 2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)vinyl]phenyl ester 16b. N-[2-(4-{1-[4-(tert-Butyldimethylsilanyloxy)-phenyl]-2-phenylbut-1-enyl}phenoxy)ethyl]-N-methylsuccinamic acid (14b, 0.10 g, 0.17 mmol), dicyclohexylcarbodiimide (0.04 g, 0.17 mmol) and 1-hydroxybenzotriazole hydrate (0.02 g, 0.17 mmol) were dissolved in dry DCM (5 mL) under a nitrogen atmosphere. The mixture was allowed to stir for 20 minutes before adding a solution of combretastatin A-4 14 (0.05 g, 0.17 mmol) in dry DCM (3 mL). The reaction was allowed stir at room temperature for 24 h until no starting material was visible by TLC (DCM:MeOH = 4:1). The reaction mixture was filtered to remove the dicyclohexylurea byproduct. The mixture was evaporated to dryness in vacuo. The residue was dissolved in 3 mL anhydrous THF and stirred under a nitrogen atmosphere. A quantity of 0.1 M TBAF (0.20 mL, 0.02 mmol) was added to the mixture and allowed stir for 24 h. The mixture was evaporated to dryness under reduced pressure. The residue was dissolved in DCM and washed with 10% HCl solution. The resulting organic phase was dried over sodium sulfate and evaporated to dryness under vacuum. The residue was purified via flash chromatography on silica gel (DCM:MeOH, 20:1) to yield an isomeric mixture (E:Z = 1:1) of the product 16b (115 mg, 88%). 1H-NMR (CDCl3): δ 0.92 −0.96 (m, 6H, CH3), 2.46–2.54 (m, 4H, CH2), 2.61–3.20 (m, 14H, 4 × CH2, NCH3), 3.67–3.88 (m, 28H, OCH3, NCH3), 3.97 (t, 0.48 × 4H, J = 5.0 Hz, CH2O), 4.13 (t, 0.52 × 4H, J = 5.0 Hz, CH2O), 6.42–6.56 (m, 10H, ArH), 6.42–6.56 (m, 14H, ArH), 7.07–7.18 (m, 16H, ArH). 13C-NMR (CDCl3): δ 13.22, 24.46, 25.13, 27.95, 28.58, 28.82, 33.43, 36.91, 36.98, 47.73, 48.68, 51.40, 53.02, 55.47, 55.50, 60.49, 64.37, 65.91, 66.24, 105.41, 105.55, 109.91, 111.51, 112.65, 113.42, 113.91, 114.63, 120.65, 122.87, 125.41, 127.38, 128.53, 129.09, 129.28, 129.63, 130.11, 130.19, 130.28, 131.57, 132.31, 134.68, 135.79, 136.26, 136.57, 137.48, 142.21, 142.29, 144.81, 145.38, 149.74, 152.38, 152.45, 153.87, 154.84, 155.87, 156.72, 171.20, 173.27. IR: νmax (KBr) cm−1: 3429.3, 2930.5, 2347.8, 1735.7, 1627.6, 1580.6, 1508.3, 1264.8, 1240.1, 1171.1, 1127.2, 834.6, 732.1. HRMS (EI): Found 794.3306 [M + Na]+, C47H49NO9Na requires 794.3305.
N-{2-[4-((Z)-1,2-Diphenylbut-1-enyl)phenoxy]ethyl}-N-methylsuccinamic acid 2-methoxy-5-[(E)-2-methoxycarbonyl-2-(3,4,5-trimethoxyphenyl)vinyl]phenyl ester (16c). N-{2-[4-(1,2-Diphenylbut-1-enyl)-phenoxy]ethyl}-N-methylsuccinamic acid (14a, 0.06 g, 0.14 mmol), dicyclohexylcarbodiimide (0.03 g, 0.14 mmol) and 1-hydroxybenzo triazole hydrate (0.017 g, 0.136 mmol) were dissolved in dry DCM (5 mL) under a N2 environment. The mixture was allowed to stir for 20 min before adding a solution of the combretastatin analogue 1s (0.05 g, 0.14 mmol) in dry DCM (3 mL). The reaction was allowed stir at room temperature for 24 h until no starting material was visible by TLC (DCM:MeOH = 4:1). The reaction mixture was filtered to remove the dicyclohexylurea byproduct. The mixture was evaporated to dryness in vacuo and the material was purified via flash chromatography on silica gel (DCM:MeOH = 20:1) to afford an isomeric mixture (E:Z = 1:1) of the product as a light yellow oil (108 mg, 97% yield). 1H-NMR (CDCl3): δ 0.92 -0.98 (m, 6H, CH3), 2.44–2.55 (m, 4H, CH2), 2.65–3.22 (m, 14H, 4 × CH2, NCH3), 3.74–4.18 (m, 38H, OCH3, NCH3, CH2O), 6.45–7.38 (m, 40H, ArH). 13C-NMR (CDCl3): δ 13.16, 24.51, 25.17, 28.58, 33.50, 48.70, 51.95, 52.01, 55.45, 55.51, 55.65, 55.74, 60.48, 65.99, 66.31, 103.35, 106.00, 106.18, 109.71, 111.40, 111.71, 112.74, 113.48, 116.24, 122.66, 125.08, 125.21, 125.59, 126.30, 126.84, 127.14, 127.32, 128.99, 129.99, 130.12, 130.20, 130.26, 130.34, 130.73, 131.48, 133.08, 138.94, 141.87, 152.83, 153.19, 167.81, 170.63, 170.70. IR: νmax (KBr) cm−1: 3430.0, 3326.7, 2929.2, 2850.5, 1765.4, 1710.4, 1626.6, 1579.5, 1509.0, 1274.4, 1243.7, 1126.3, 1025.2, 771.1, 701.4. HRMS (EI): Found 836.3388 [M + Na]+, C49H51NO10Na requires 836.3411.
3.2.10. Stability Study for Compounds 16a, 16b and 16c
Analytical high-performance liquid chromatography (HPLC) stability studies were performed using a Symmetry® column (C18, 5 µm, 4.6 × 150 mm), a Waters 2487 Dual Wavelength Absorbance detector, a Waters 1525 binary HPLC pump and a Waters 717 plus Autosampler. Samples were detected at wavelength of 254 nm. All samples were analysed using acetonitrile (80%):water (20%) as the mobile phase over 10 min and a flow rate of 1 mL/min. Stock solutions are prepared by dissolving 5mg of each of the compounds 16a, 16b and 16c in 10 mL of mobile phase. Phosphate buffers at the desired pH values (4, 7.4, and 9) were prepared in accordance with the British Pharmacopoeia monograph 2017. Thirty µL of stock solution was diluted with 1 mL of appropriate buffer, shaken and injected immediately. Samples were withdrawn and analysed at time intervals of t = 0 min, 5 min, 30 min, 60 min, 90 min, 120 min, 24 h and 48 h.

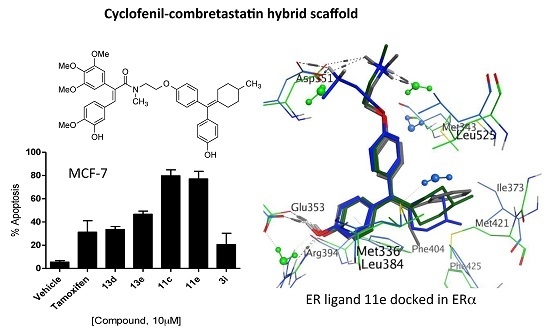
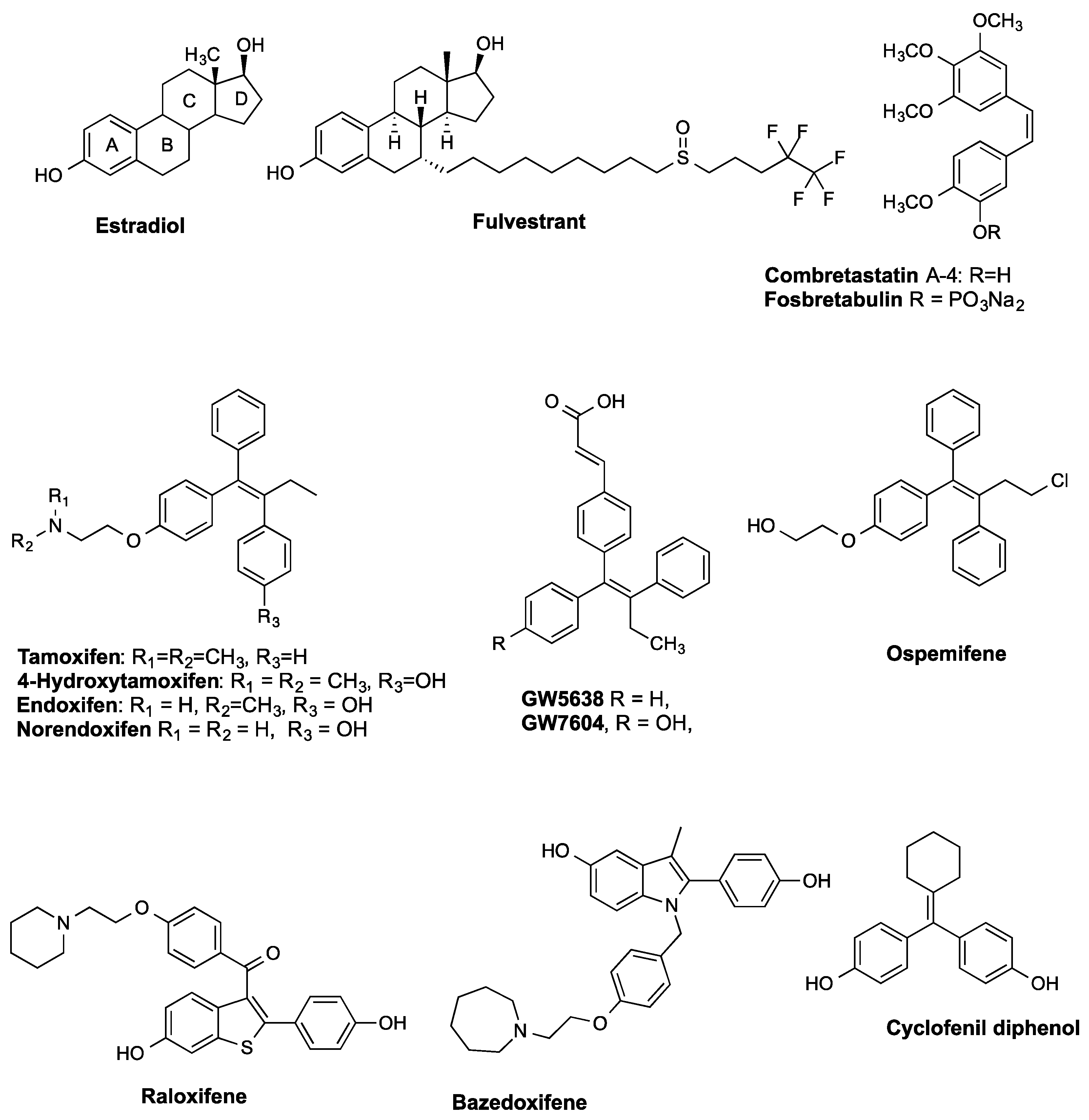
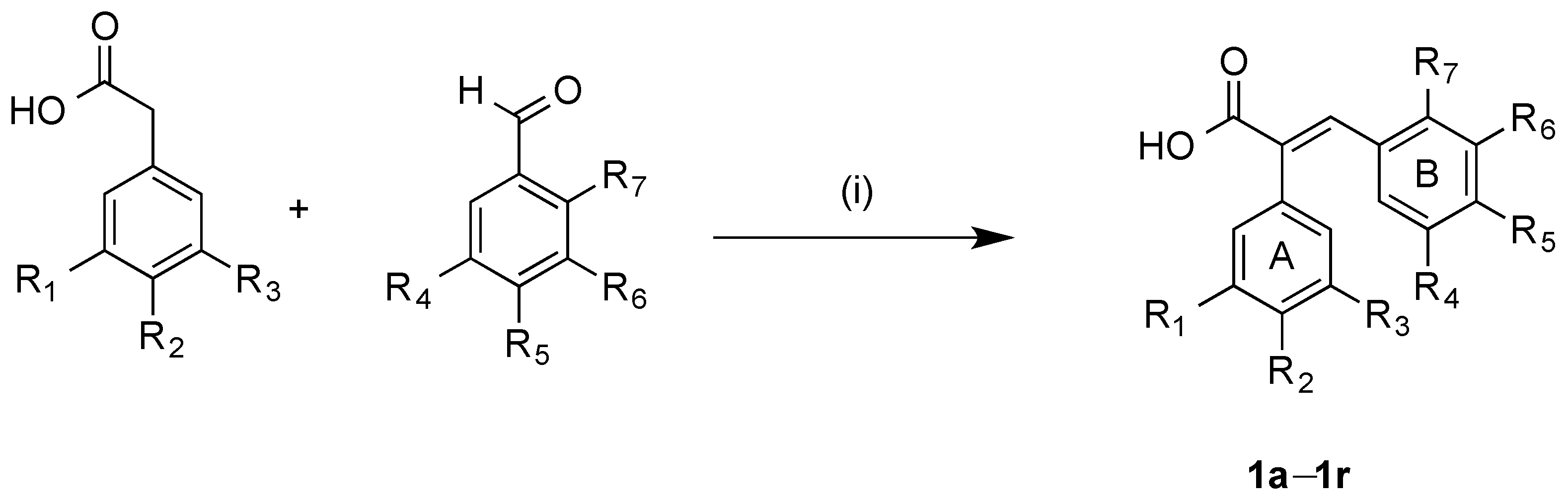
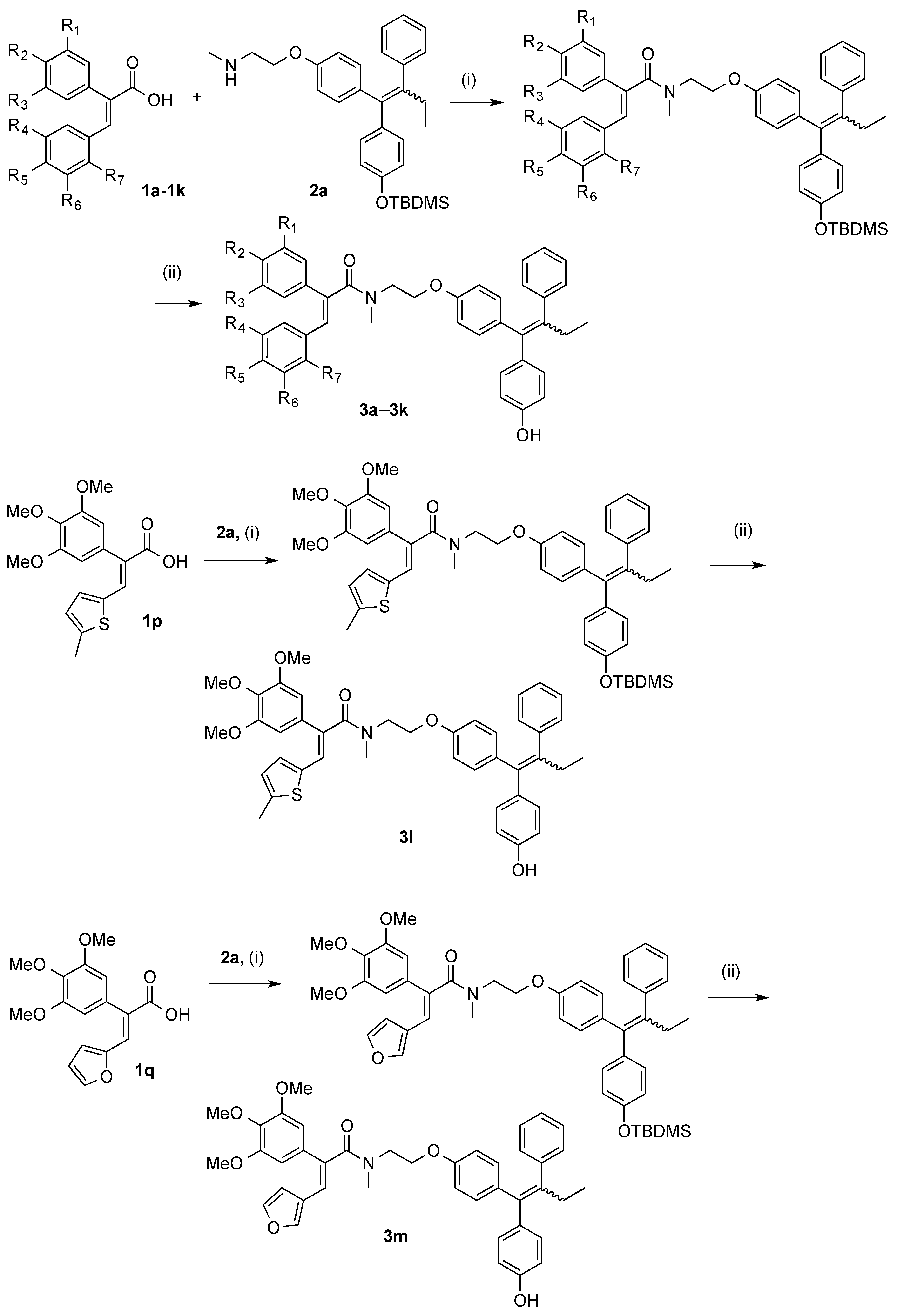

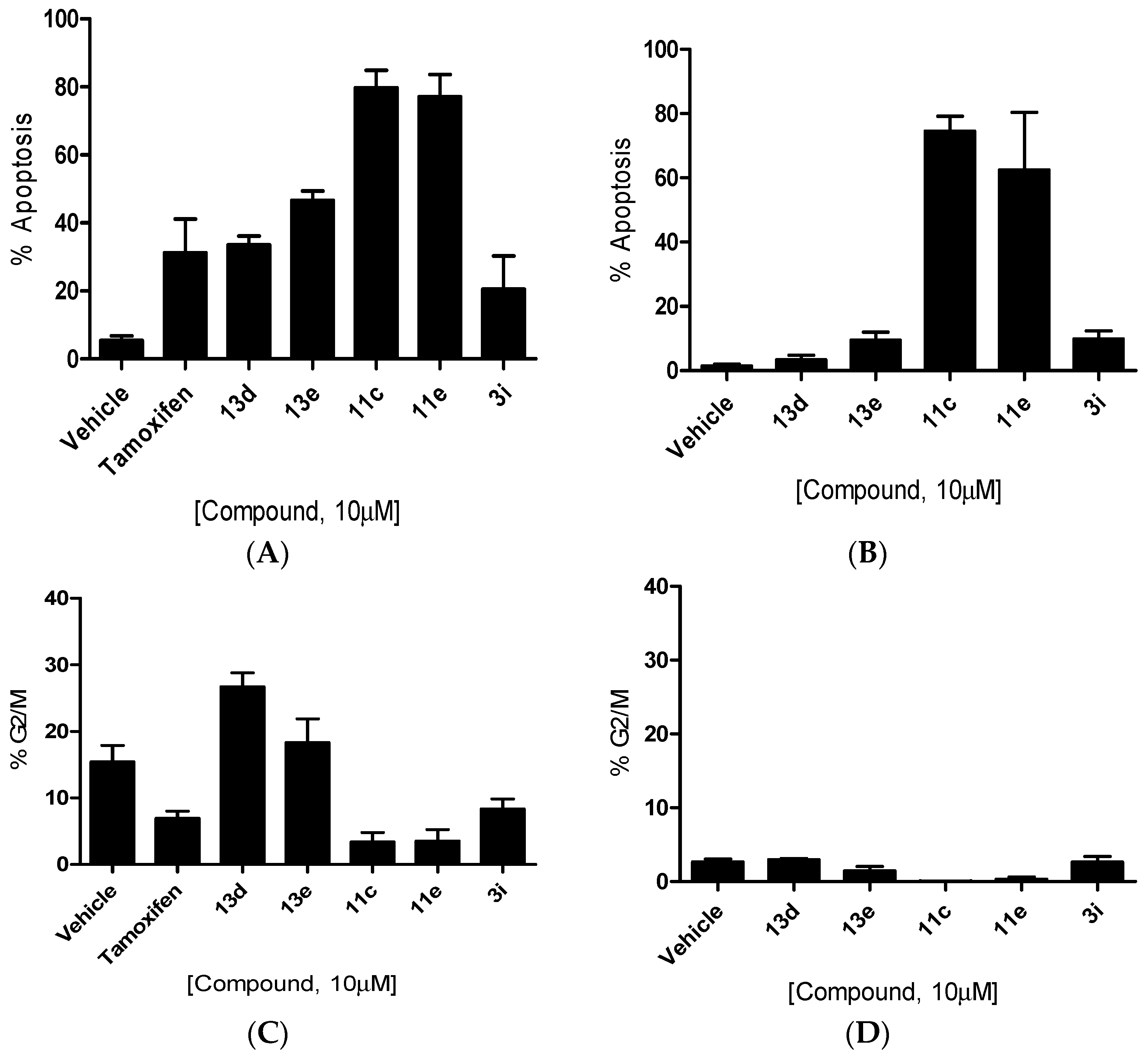
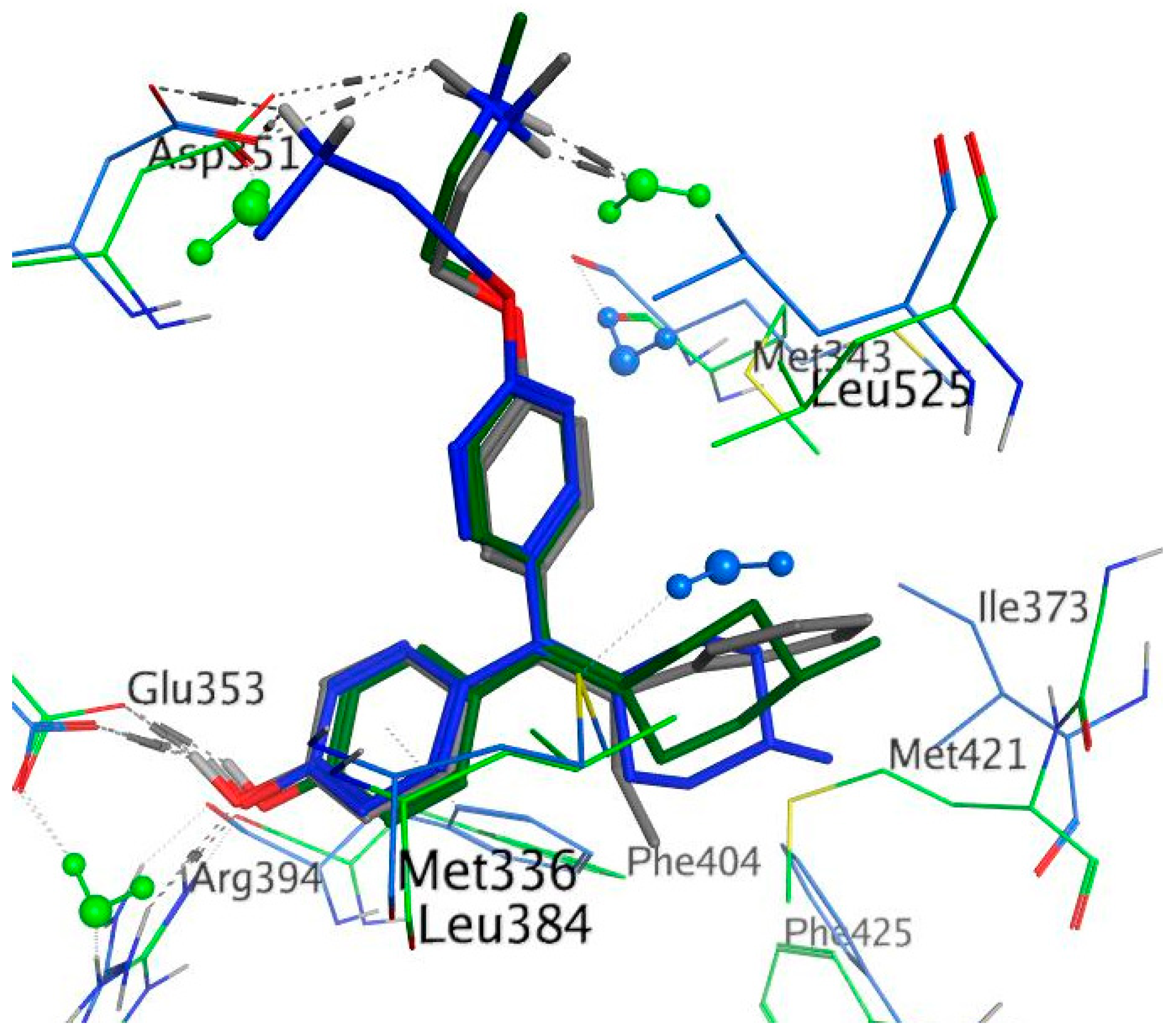
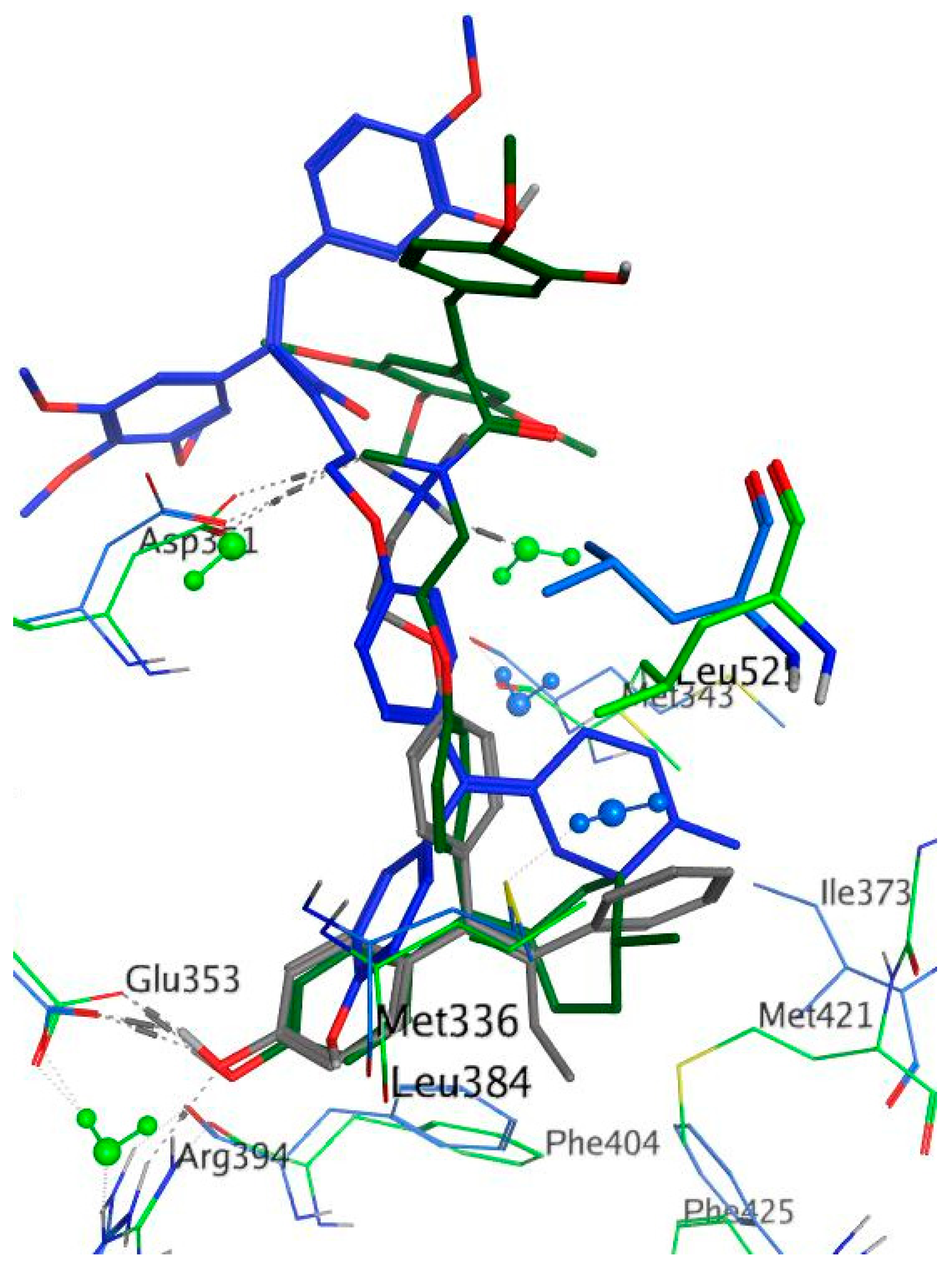
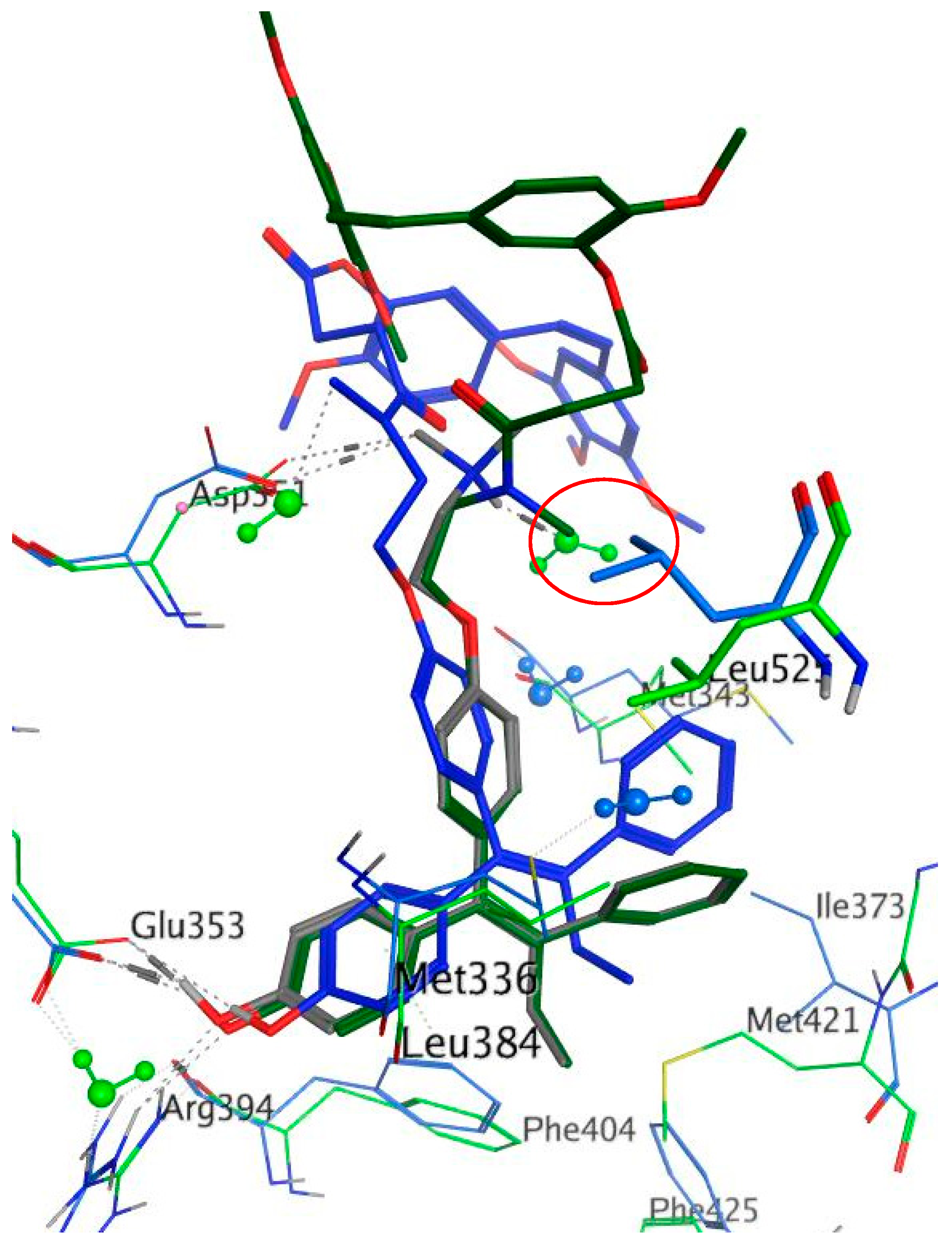

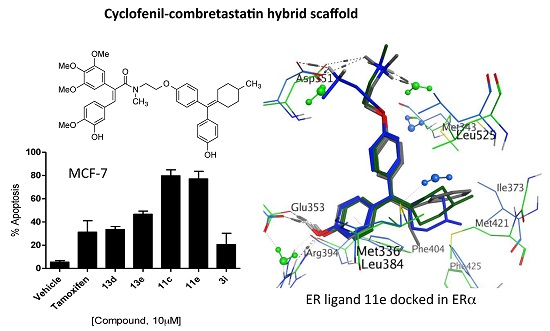{kind=link}
{kind=link}
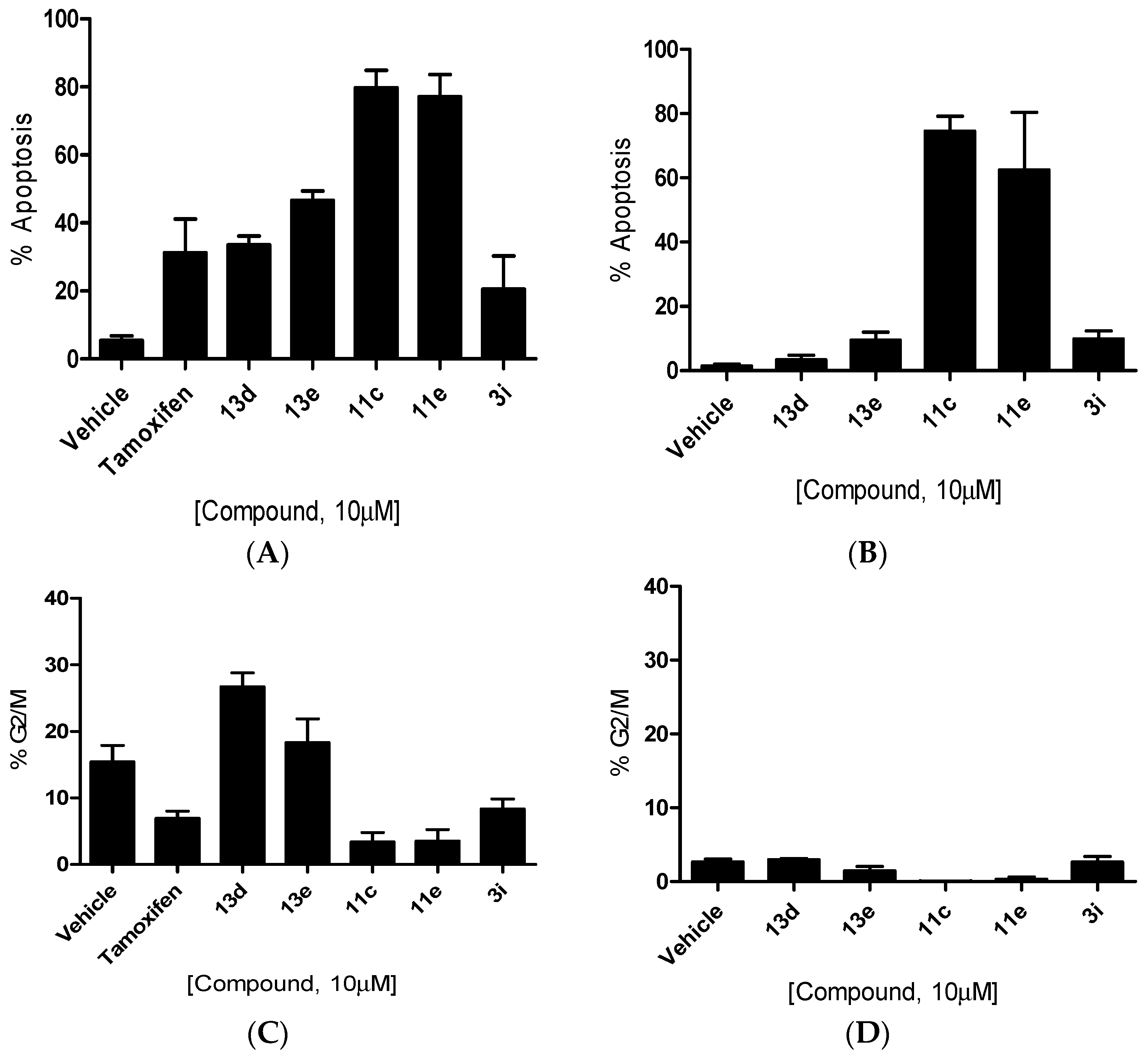{kind=link}
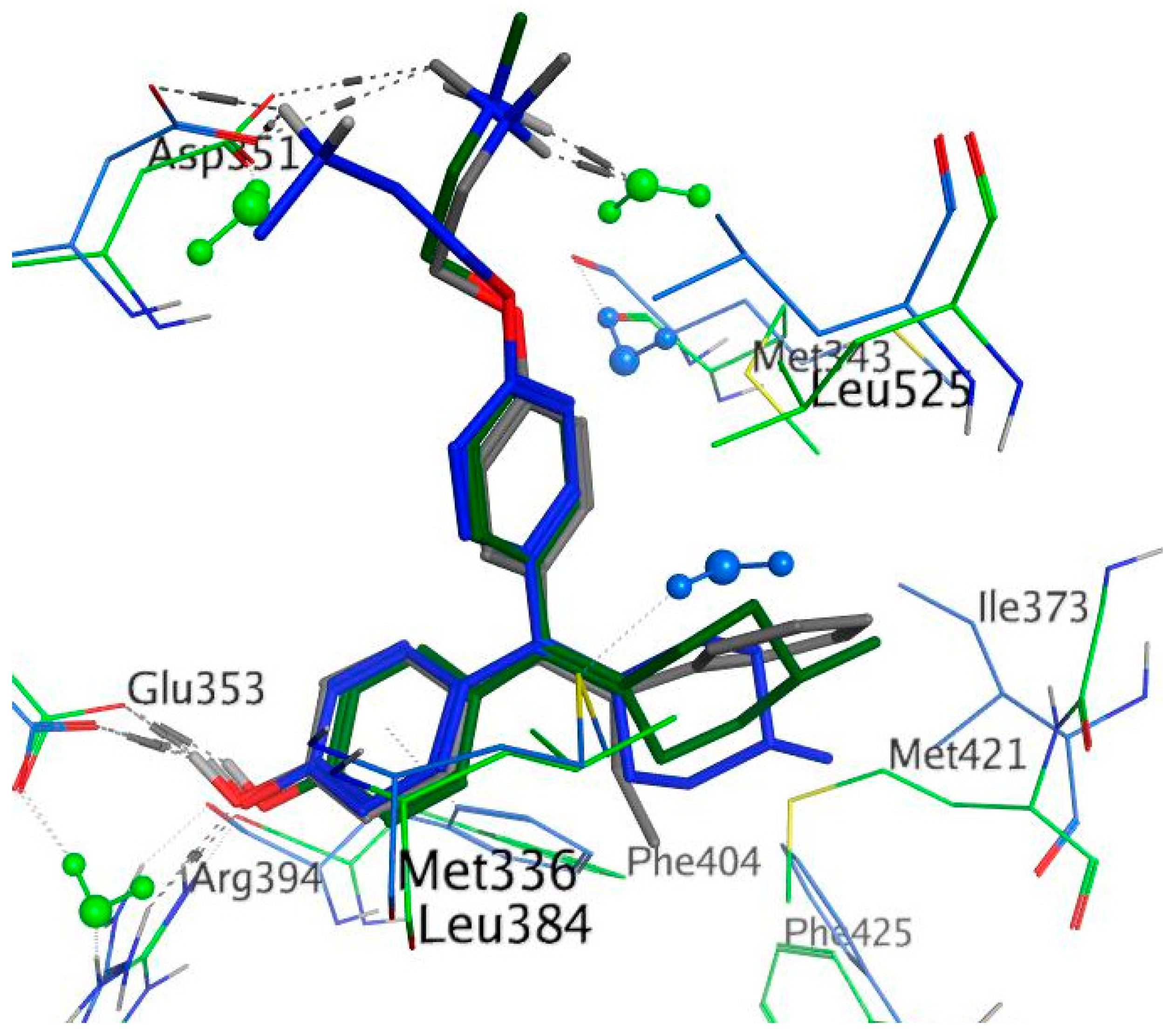{kind=link}
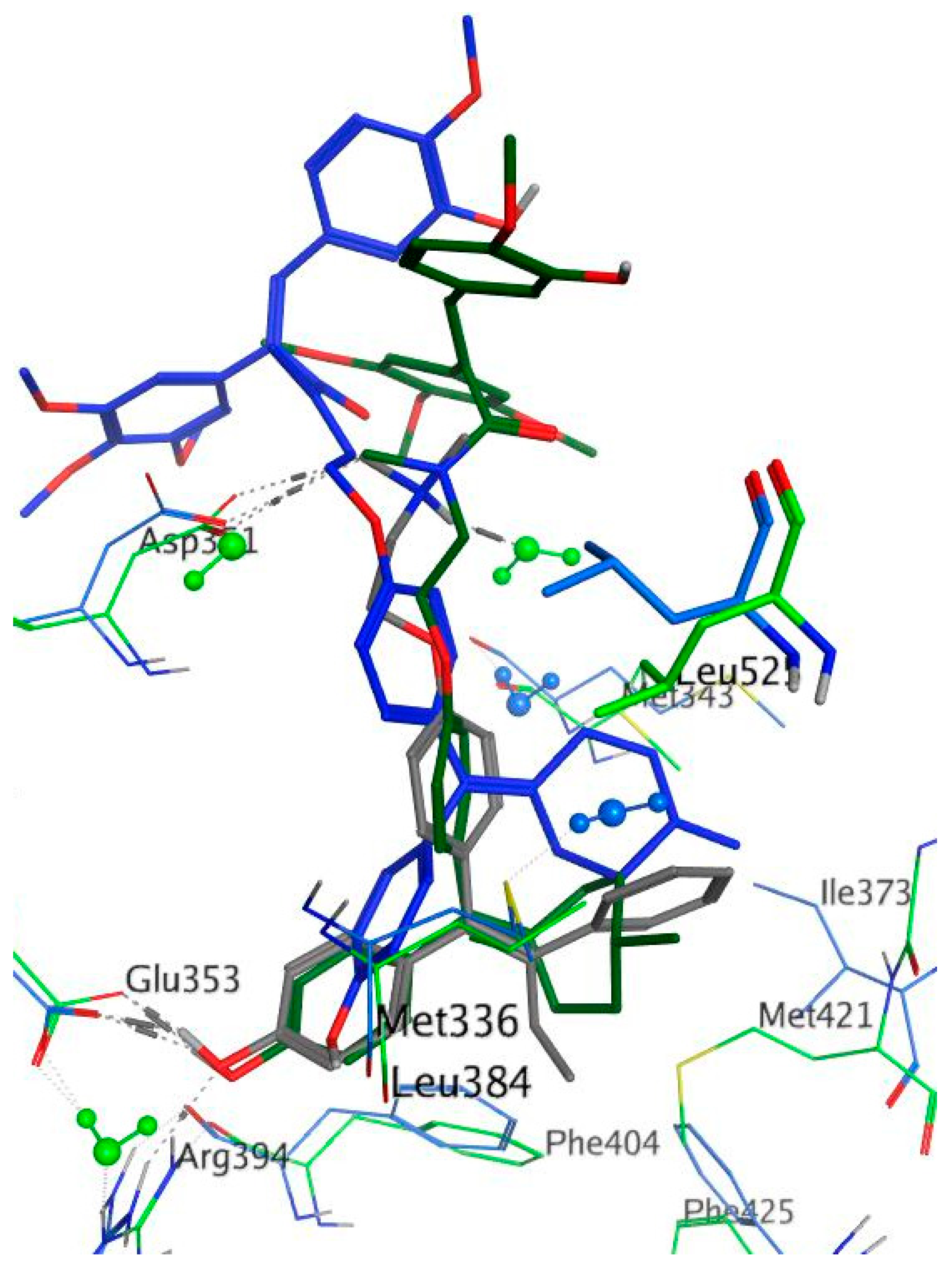{kind=link}
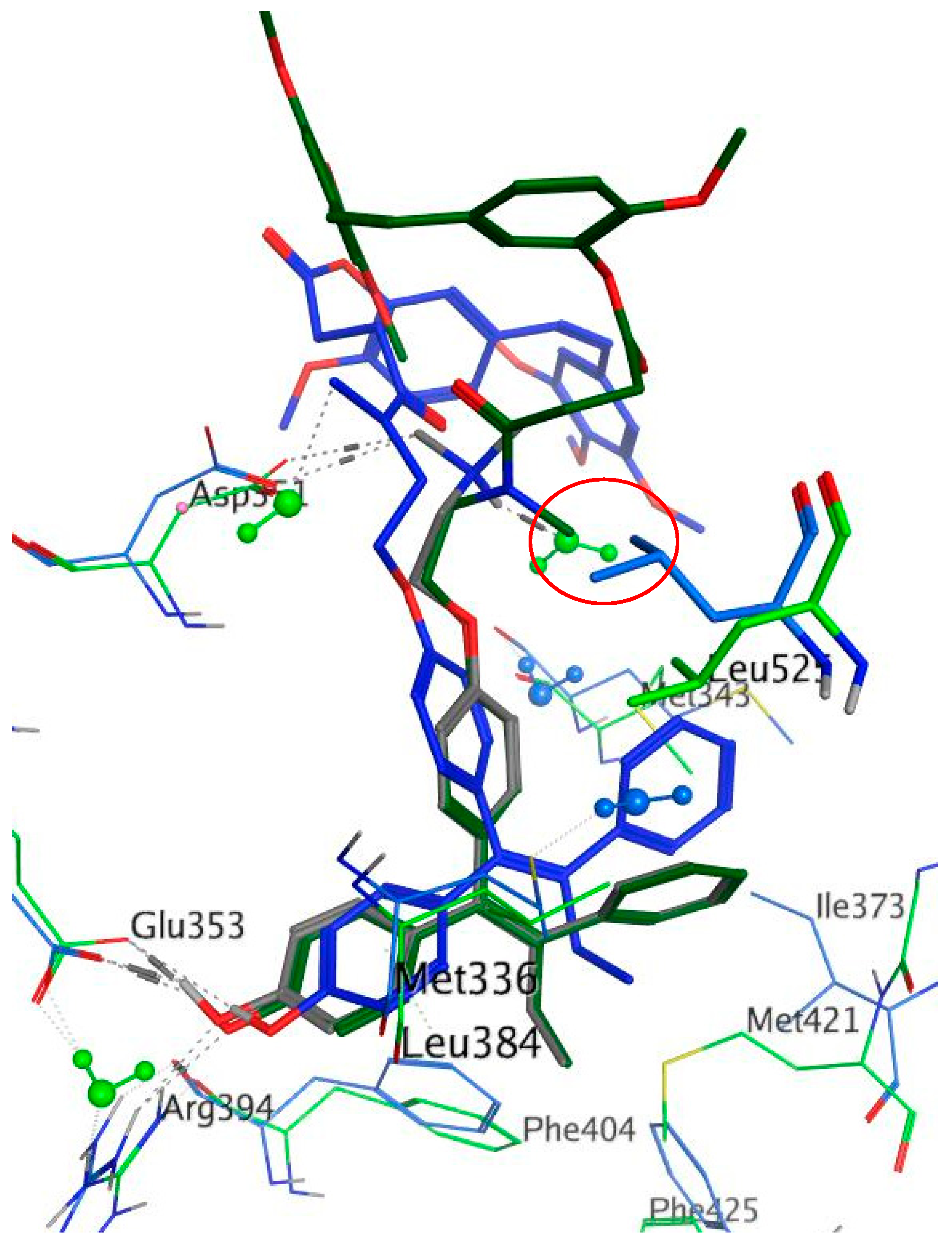{kind=link}
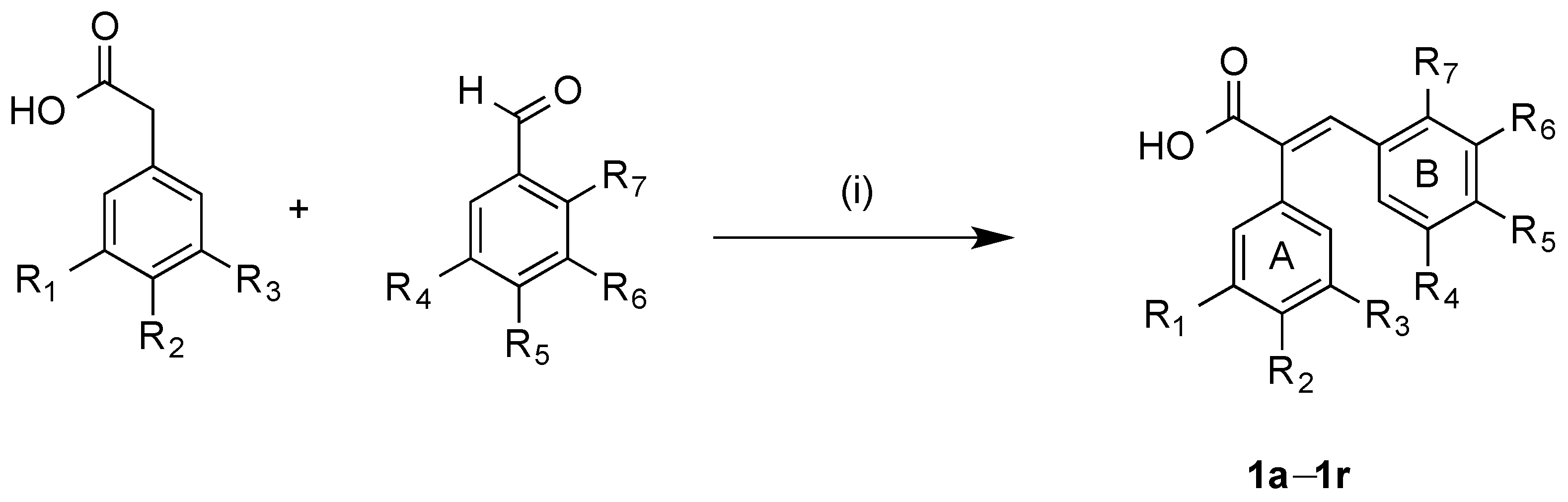{kind=link}
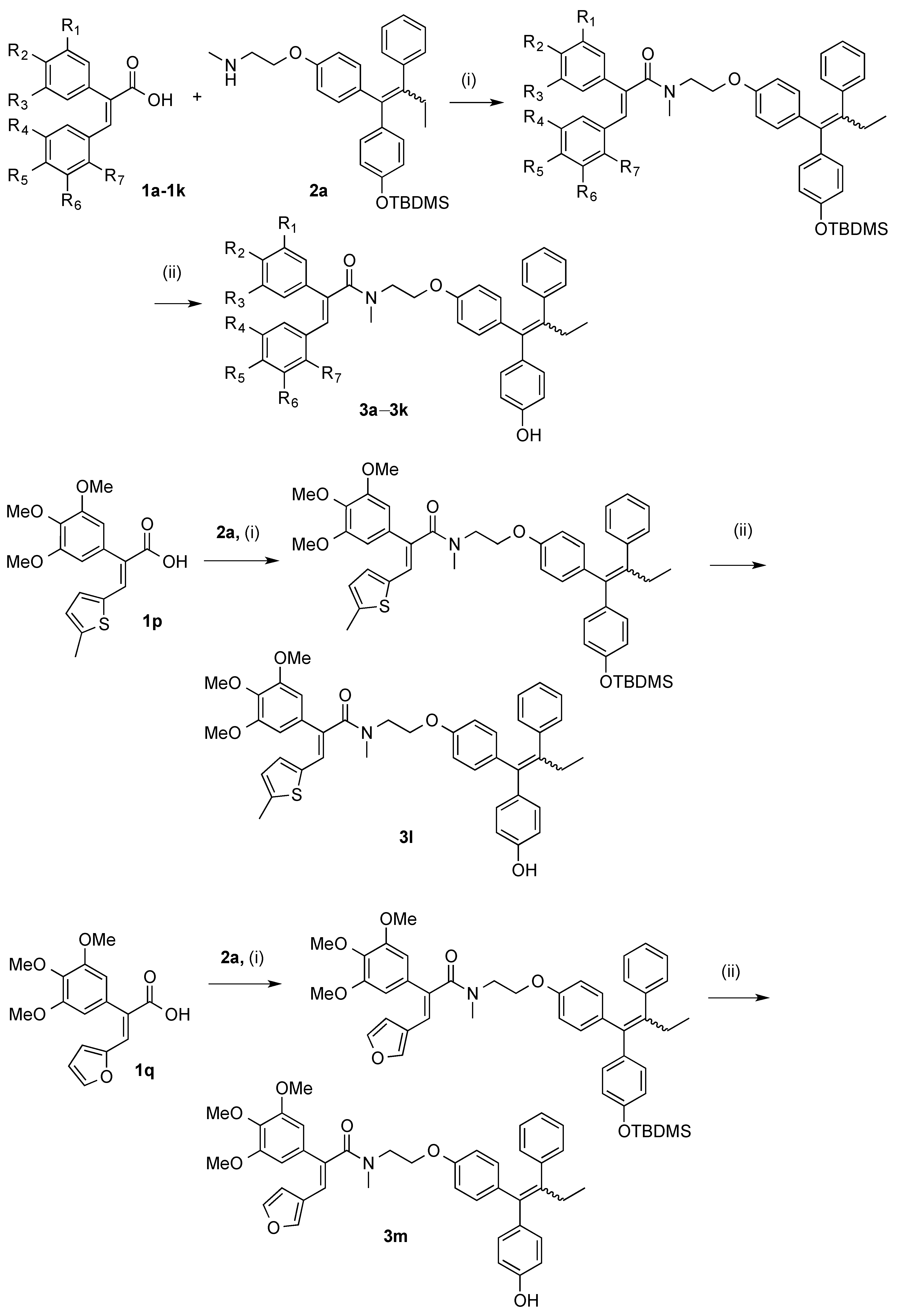{kind=link}
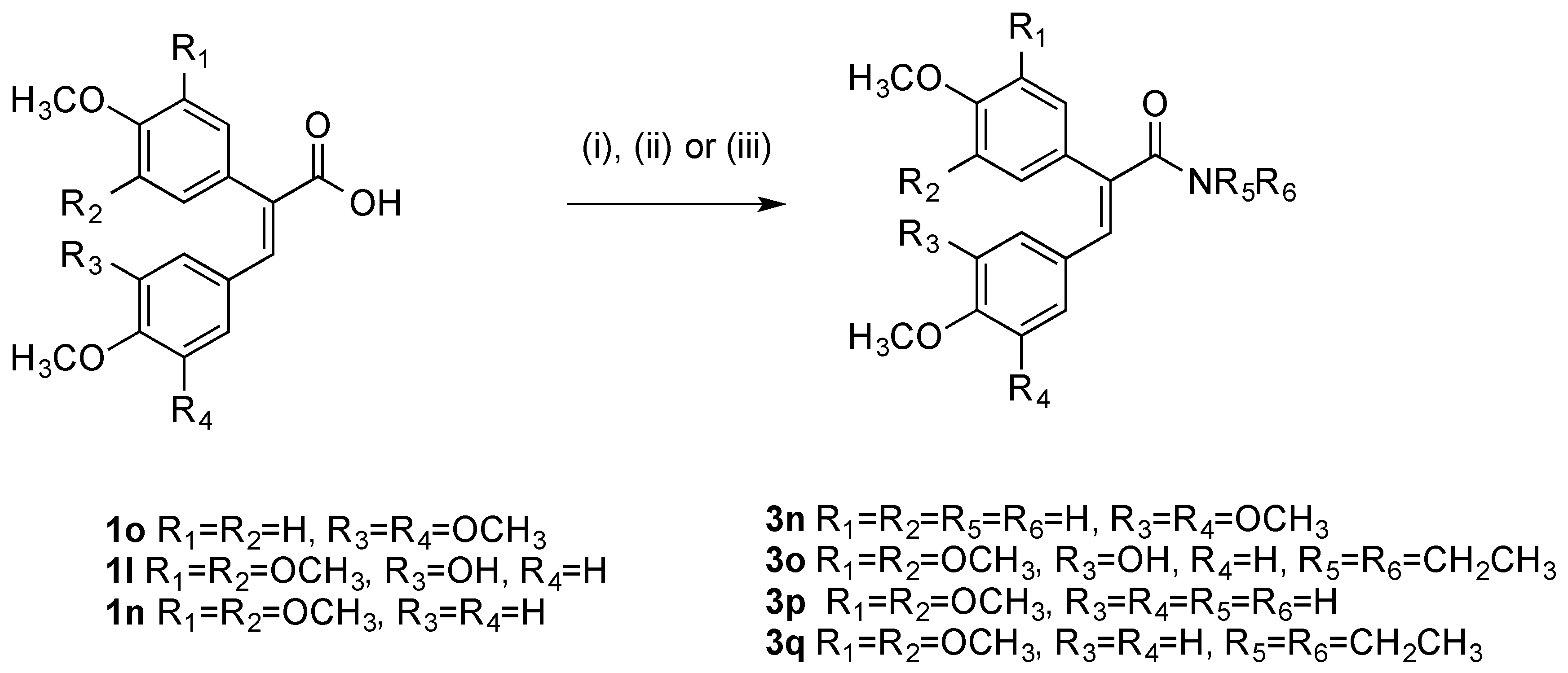{kind=link}
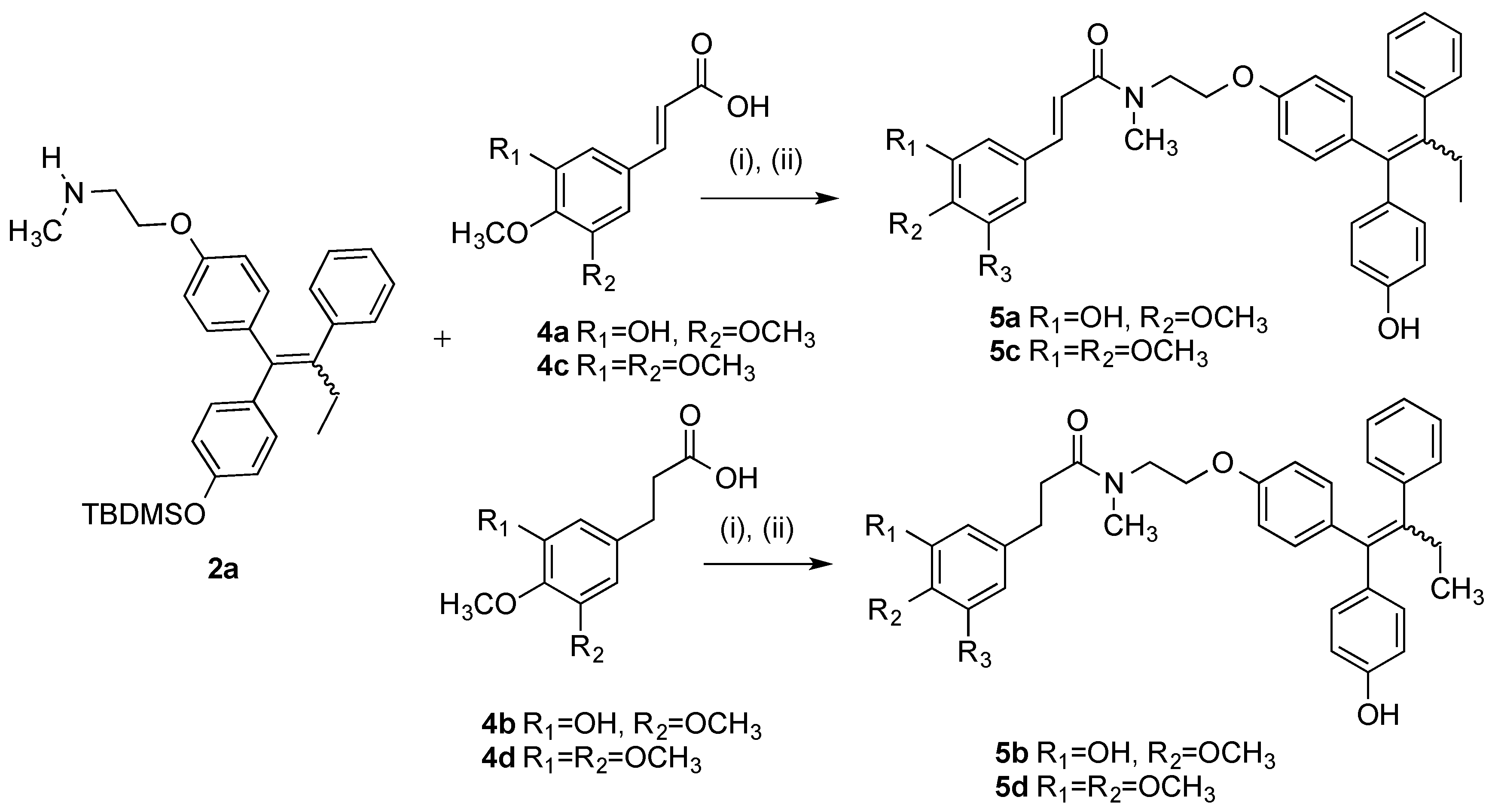{kind=link}
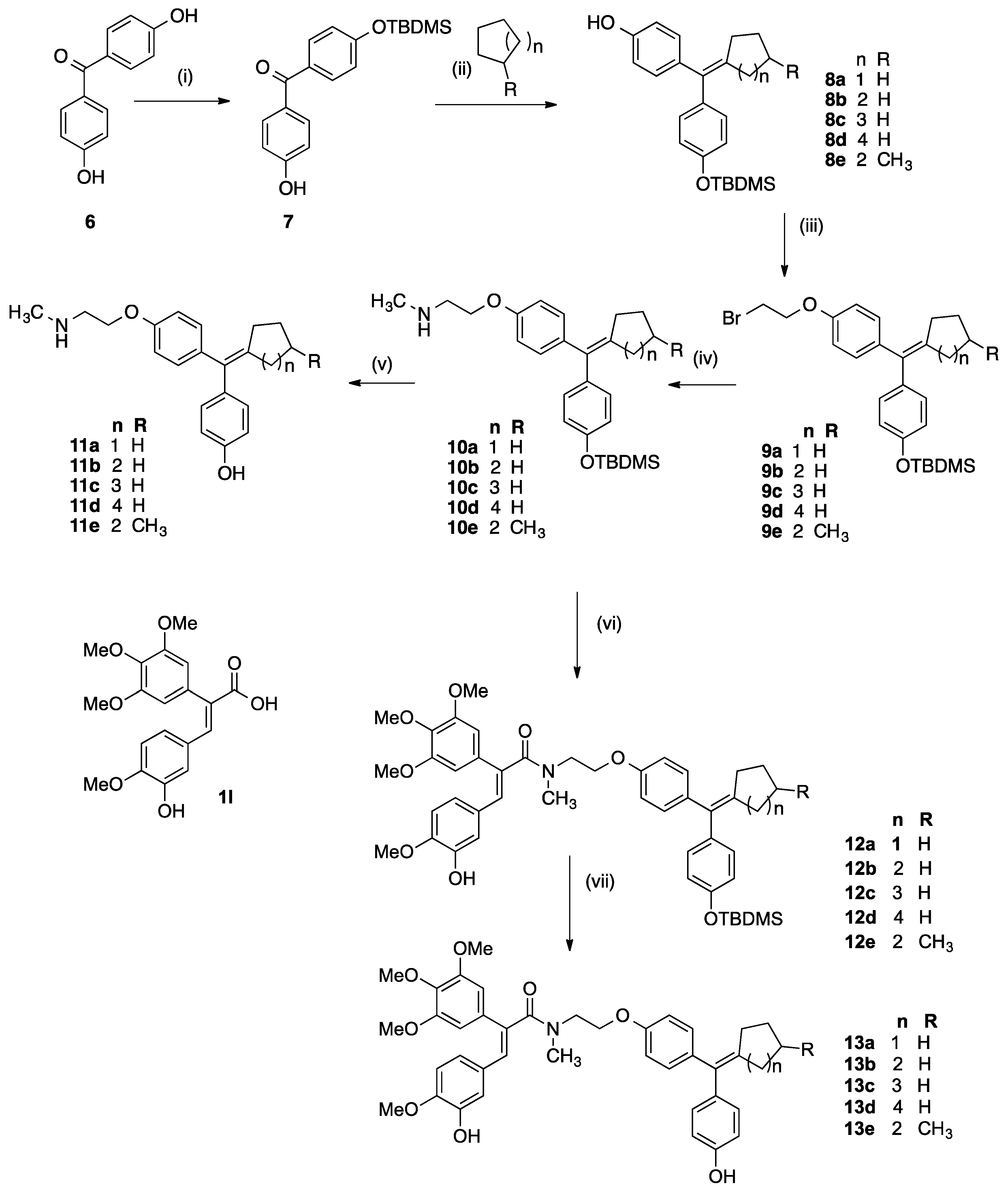{kind=link}
{kind=link}
