Why is Aged Acetylcholinesterase So Difficult to Reactivate?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
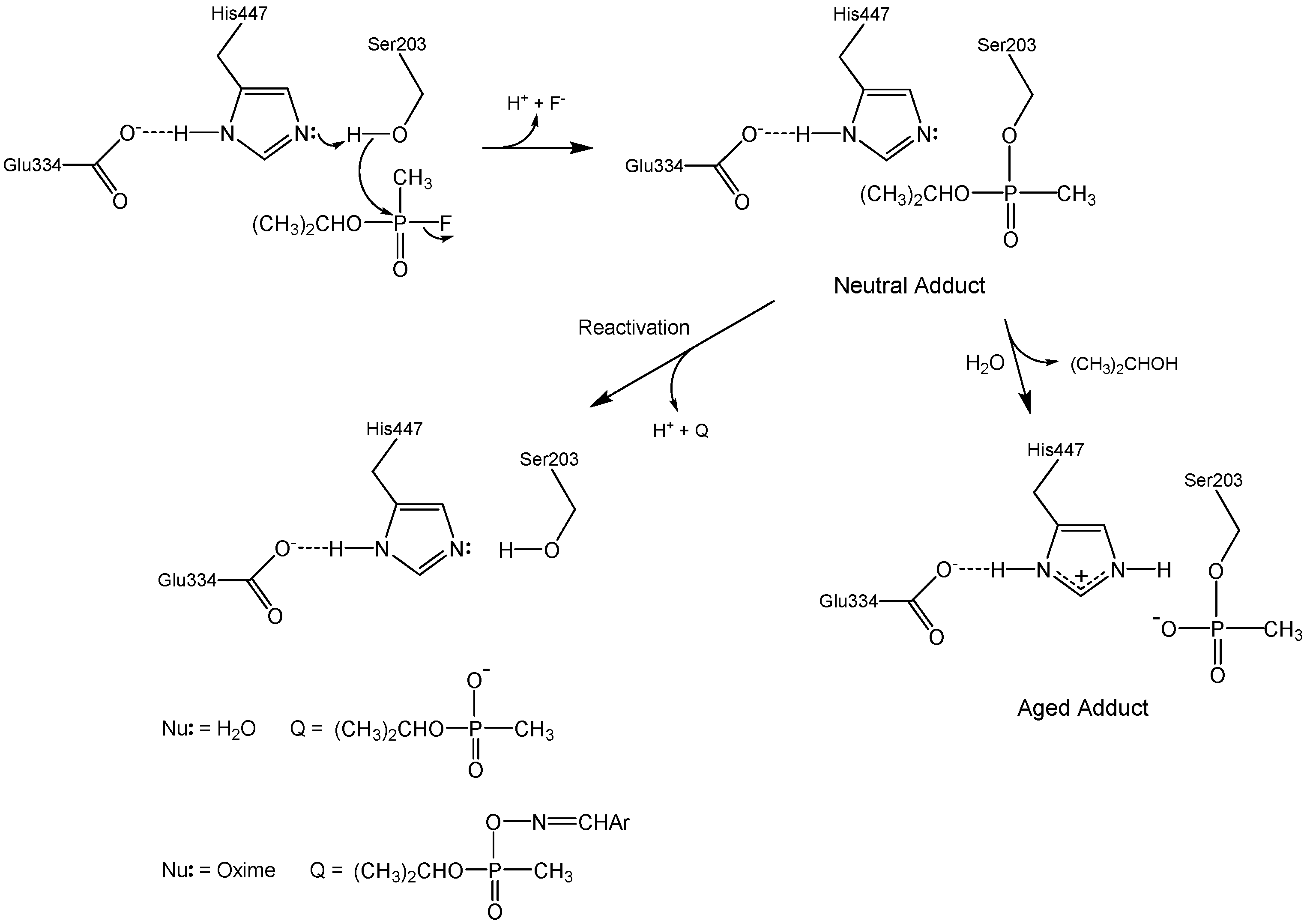
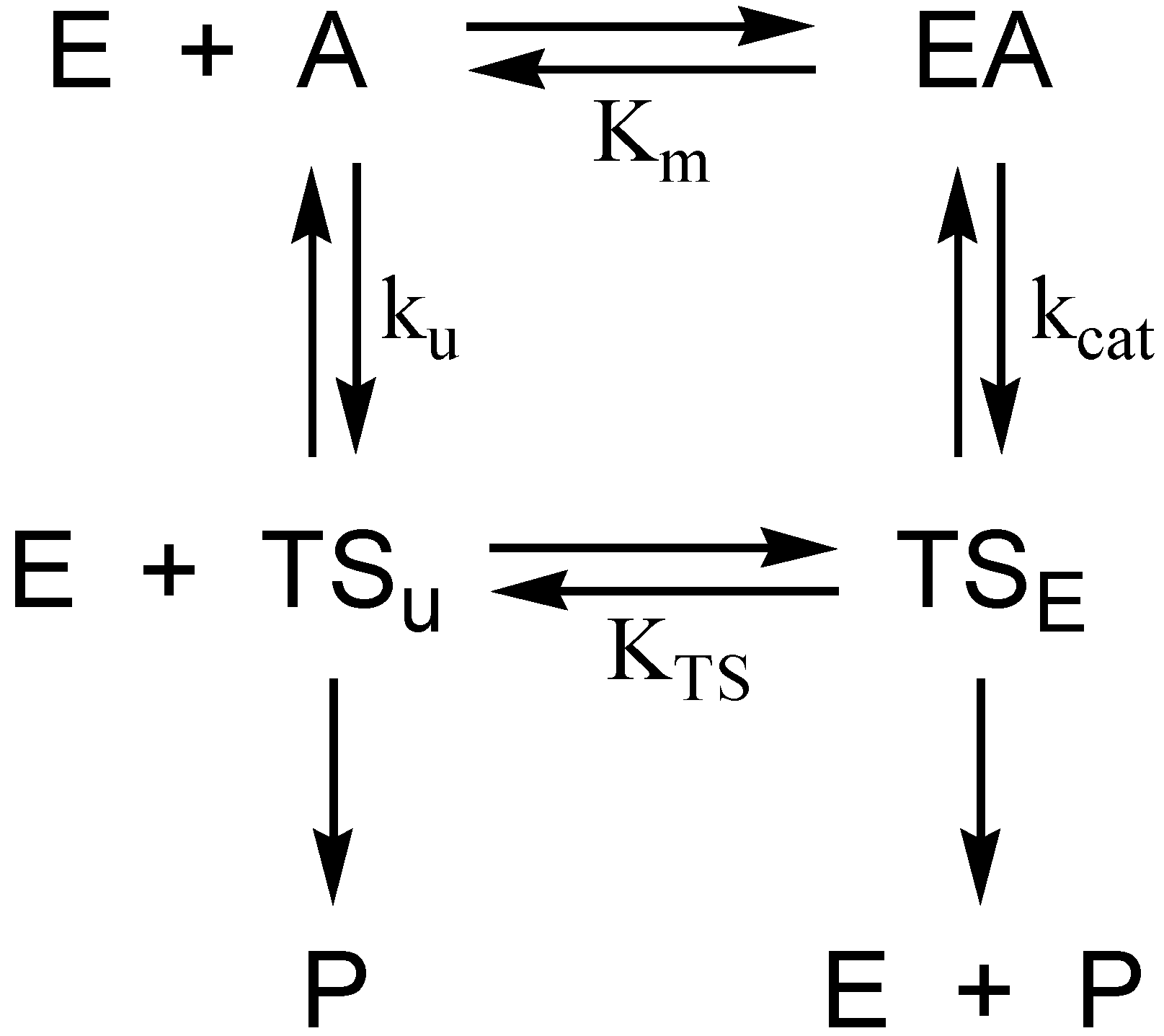
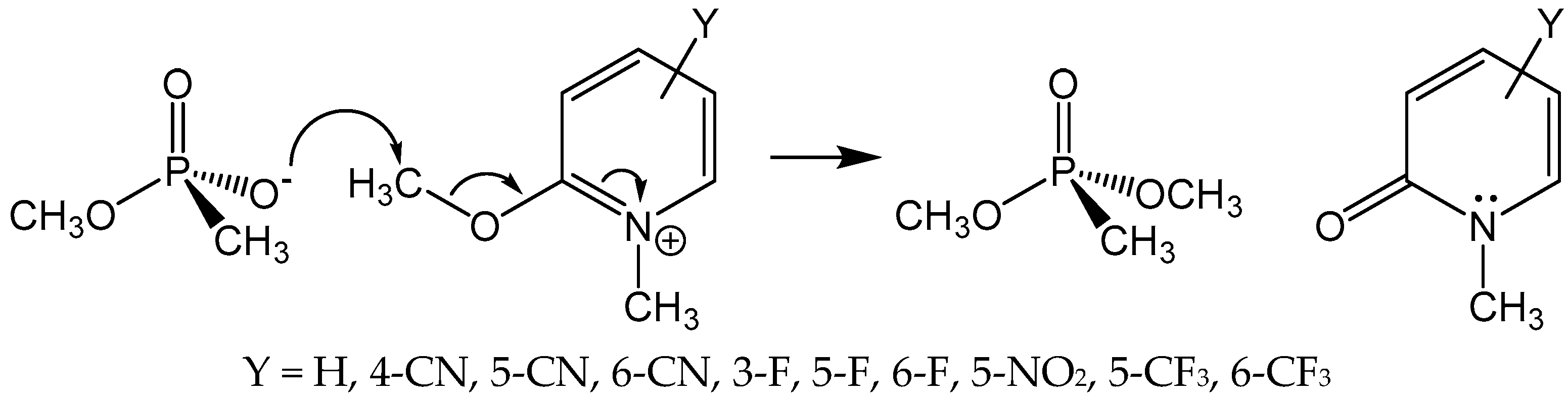
2. Discussion
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dingeman, J.; Jupa, R. Chemical warfare in the Iran-Iraq conflict. Strategy Tactics 1987, 113, 51–52. [Google Scholar]
- Tu, A.T. Overview of sarin terrorist attacks in Japan. ACS Symp. Ser. 2000, 745, 304–317. [Google Scholar]
- Dolgin, E. Syrian gas attack reinforces need for better anti-sarin drugs. Nat. Med. 2013, 19, 1194–1195. [Google Scholar] [CrossRef] [PubMed]
- Paddock, R.C.; Sang-Hun, C. Kim Jong-nam was killed by VX nerve agent, Malaysians say. New York Times, 23 February 2017. [Google Scholar]
- Malany, S.; Sawai, M.; Sikorski, R.S.; Seravalli, J.; Quinn, D.M.; Radić, Z.; Taylor, P.; Kronman, C.; Velan, B.; Shafferman, A. Transition state structure and rate determination for the acylation stage of acetylcholinesterase catalyzed hydrolysis of (acetylthio)choline. J. Am. Chem. Soc. 2000, 122, 2981–2987. [Google Scholar] [CrossRef]
- Harel, M.; Quinn, D.M.; Nair, H.K.; Silman, I.; Sussman, J.L. The X-ray structure of a transition state analog complex reveals the molecular origins of the catalytic power and substrate specificity of acetylcholinesterase. J. Am. Chem. Soc. 1996, 118, 2340–2346. [Google Scholar] [CrossRef]
- Kovach, I.M.; Huber, J.H.-A.; Schowen, R.L. Catalytic recruitment in the inactivation of acetylcholinesterase by soman: Temperature dependence of the solvent isotope effect. J. Am. Chem. Soc. 1988, 110, 590–593. [Google Scholar] [CrossRef]
- Jodanović, M.; Prostran, M. Pyridinium oximes as cholinesterase reactivator. Structure-activity relationship and efficacy in the treatment of poisoning with organophosphorus compounds. Curr. Med. Chem. 2009, 16, 2177–2188. [Google Scholar] [CrossRef]
- Barak, D.; Ordentlich, A.; Segall, Y.; Velan, B.; Benschop, H.P.; De Jong, L.P.A.; Shafferman, A. Carbocation mediated processes in biocatalysts. Contribution of aromatic moieties. J. Am. Chem. Soc. 1997, 119, 3157–3158. [Google Scholar] [CrossRef]
- Nolte, H.-J.; Rosenberry, T.L.; Neumann, E. Effective charge on acetylcholinesterase active sites determined from the ionic strength dependence of association rate constants with cationic ligands. Biochemistry 1980, 19, 3705–3711. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L. Acetylcholinesterase. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 103–218. [Google Scholar] [PubMed]
- Quinn, D.M.; Pryor, A.N.; Selwood, T.; Lee, B.-H.; Acheson, S.A.; Barlow, P.N. The chemical mechanism of acetylcholinesterase reactions. Biological catalysis at the speed limit. In Cholinesterses: Structure, Function, Mechanism, Genetics, and Cell Biology; Massoulié, J., Bacou, F., Barnard, E., Chatonnet, A., Doctor, B.P., Quinn, D.M., Eds.; American Chemical Society: Washington, DC, USA, 1991; pp. 252–257. ISBN 0-8412-2008-5. [Google Scholar]
- Wolfenden, R.; Yuan, Y. The “neutral” hydrolysis of simple carboxylic esters in water and the rate enhancements produced by acetylcholinesterase and other carboxylic acid esterases. J. Am. Chem. Soc. 2011, 133, 13821–13823. [Google Scholar] [CrossRef] [PubMed]
- Radzicka, A.; Wolfenden, R. A proficient enzyme. Science 1995, 267, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Froede, H.C.; Wilson, I.B. Direct determination of acetyl-enzyme intermediate in the acetylcholinesterase-catalyzed hydrolysis of acetylcholine and acetylthiocholine. J. Biol. Chem. 1984, 259, 11010–11013. [Google Scholar] [PubMed]
- Ashani, Y.; Green, B.S. Are the organophosphorus inhibitors of acetylcholinesterase transition-state analogs? In Chemical Approaches to Understanding Enzyme Catalysis: Biomimetic Chemistry and Transition State Analogs; Green, B.S., Ashani, Y., Chipman, D., Eds.; Elsevier Scientific Publishing Co.: Amsterdam, The Netherlands; Oxford, UK; New York, NY, USA, 1982; pp. 169–188. [Google Scholar]
- Millard, C.B.; Kryger, G.; Ordentlich, A.; Greenblatt, H.M.; Harel, M.; Ravel, M.L.; Segall, Y.; Barak, D.; Shafferman, A.; Silman, I.; et al. Crystal structures of aged phosphonylated acetylcholinesterase: Nerve agent reaction products at the atomic level. Biochemistry 1999, 38, 7032–7039. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.B.; Koellner, G.; Ordentlich, A.; Shafferman, A.; Silman, I.; Sussman, J.L. Reaction products of acetylcholinesterase and VX indicated a mobile histidine mechanism. J. Am. Chem. Soc. 1999, 121, 9883–9884. [Google Scholar] [CrossRef]
- Carletti, E.; Colletier, J.P.; Dupeux, F.; Trovaslet, M.; Masson, P.; Nachon, F. Structural evidence that human acetylcholinesterase inhibited by tabun ages through O-dealkylation. J. Med. Chem. 2010, 53, 4002–4008. [Google Scholar] [CrossRef] [PubMed]
- Sanson, B.; Nachon, F.; Colletier, J.P.; Froment, M.T.; Toker, L.; Greenblatt, H.M.; Sussman, J.L.; Ashani, Y.; Masson, P.; Silman, I.; et al. Crystallographic snapshots of nonaged and aged conjugates of soman with acetylcholinesterase, and of a ternary complex of the aged conjugate with pralidoxime. J. Med. Chem. 2009, 52, 7593–7603. [Google Scholar] [CrossRef] [PubMed]
- Tormos, J.R.; Wiley, K.L.; Seravalli, J.; Nachon, F.; Masson, P.; Nicolet, T.; Quinn, D.M. The reactant state for substrate-activated turnover of acetylthiocholine by butyrylcholinesterase is a tetrahedral intermediate. J. Am. Chem. Soc. 2005, 127, 14538–14539. [Google Scholar] [CrossRef] [PubMed]
- Wiley, K.L.; Tormos, J.R.; Quinn, D.M. A secondary isotope effect study of equine serum butyrylcholinesterase-catalyzed hydrolysis of acetylthiocholine. Chem. Biol. Interact. 2010, 187, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Tormos, J.R.; Wiley, K.L.; Wang, Y.; Fournier, D.; Masson, P.; Nachon, F.; Quinn, D.M. Accumulation of tetrahedral intermediates in cholinesterase catalysis: A secondary isotope effect study. J. Am. Chem. Soc. 2010, 132, 17751–17759. [Google Scholar] [CrossRef] [PubMed]
- Topczewski, J.J.; Quinn, D.M. Kinetic assessment of N-methyl-2-methoxypyridinium species as phosphonate anion methylating agents. Org. Lett. 2013, 15, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- March, J. Advanced Organic Chemistry, 4th ed.; John Wiley & Sons: New York, NY, USA, 1992; pp. 258–259. ISBN 0-471-60180-2. [Google Scholar]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, D.M.; Topczewski, J.; Yasapala, N.; Lodge, A. Why is Aged Acetylcholinesterase So Difficult to Reactivate? Molecules 2017, 22, 1464. https://doi.org/10.3390/molecules22091464
Quinn DM, Topczewski J, Yasapala N, Lodge A. Why is Aged Acetylcholinesterase So Difficult to Reactivate? Molecules. 2017; 22(9):1464. https://doi.org/10.3390/molecules22091464
Chicago/Turabian StyleQuinn, Daniel M., Joseph Topczewski, Nilanthi Yasapala, and Alexander Lodge. 2017. "Why is Aged Acetylcholinesterase So Difficult to Reactivate?" Molecules 22, no. 9: 1464. https://doi.org/10.3390/molecules22091464