Chlorophyll-Inspired Red-Region Fluorophores: Building Block Synthesis and Studies in Aqueous Media


Abstract
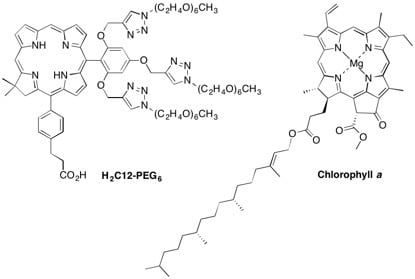
:
1. Introduction
2. Results and Discussion
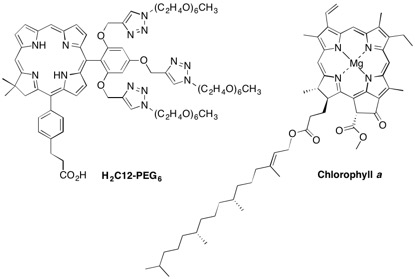
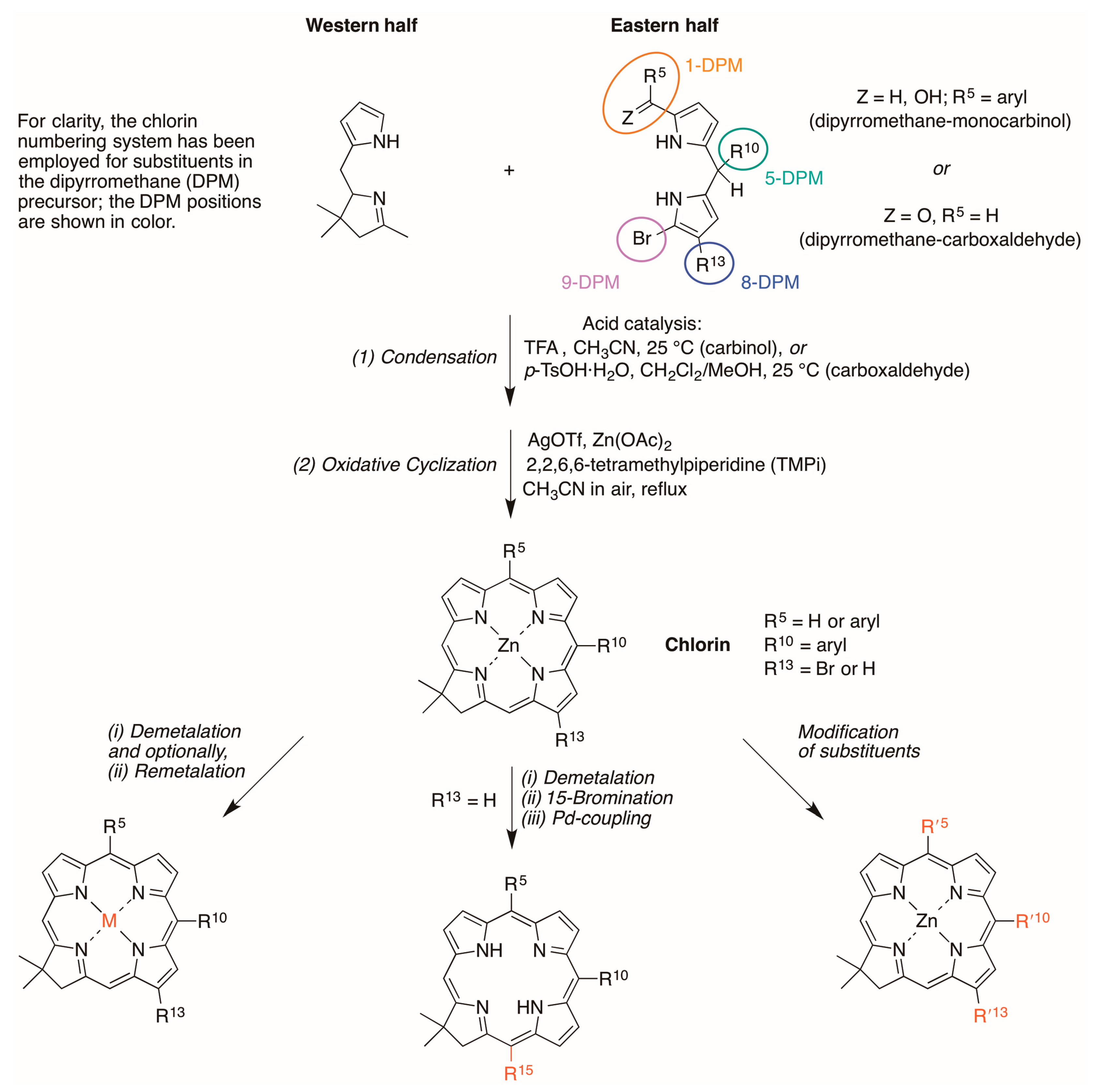
2.1. Reconnaissance
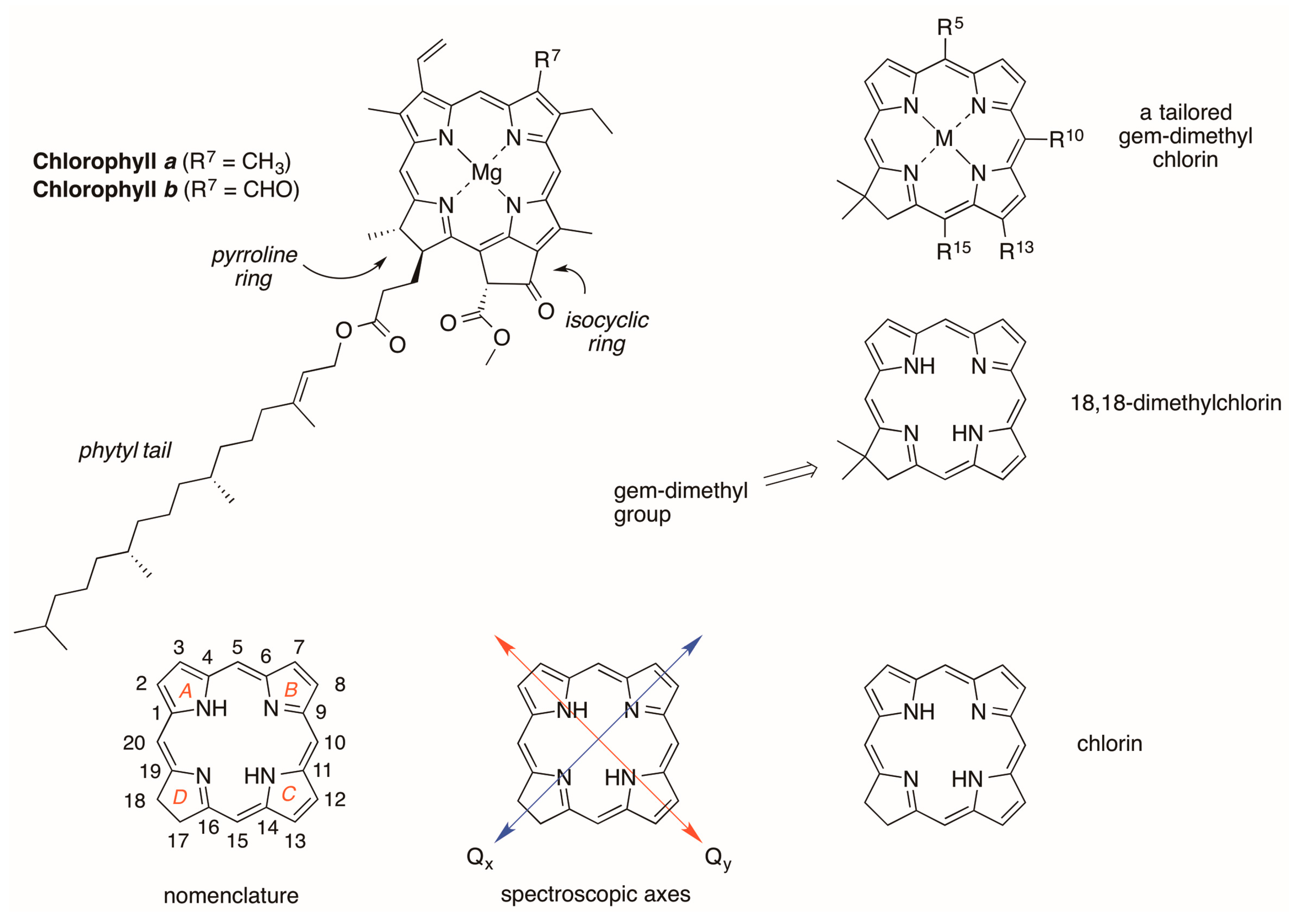
2.2. Synthesis of Chlorins
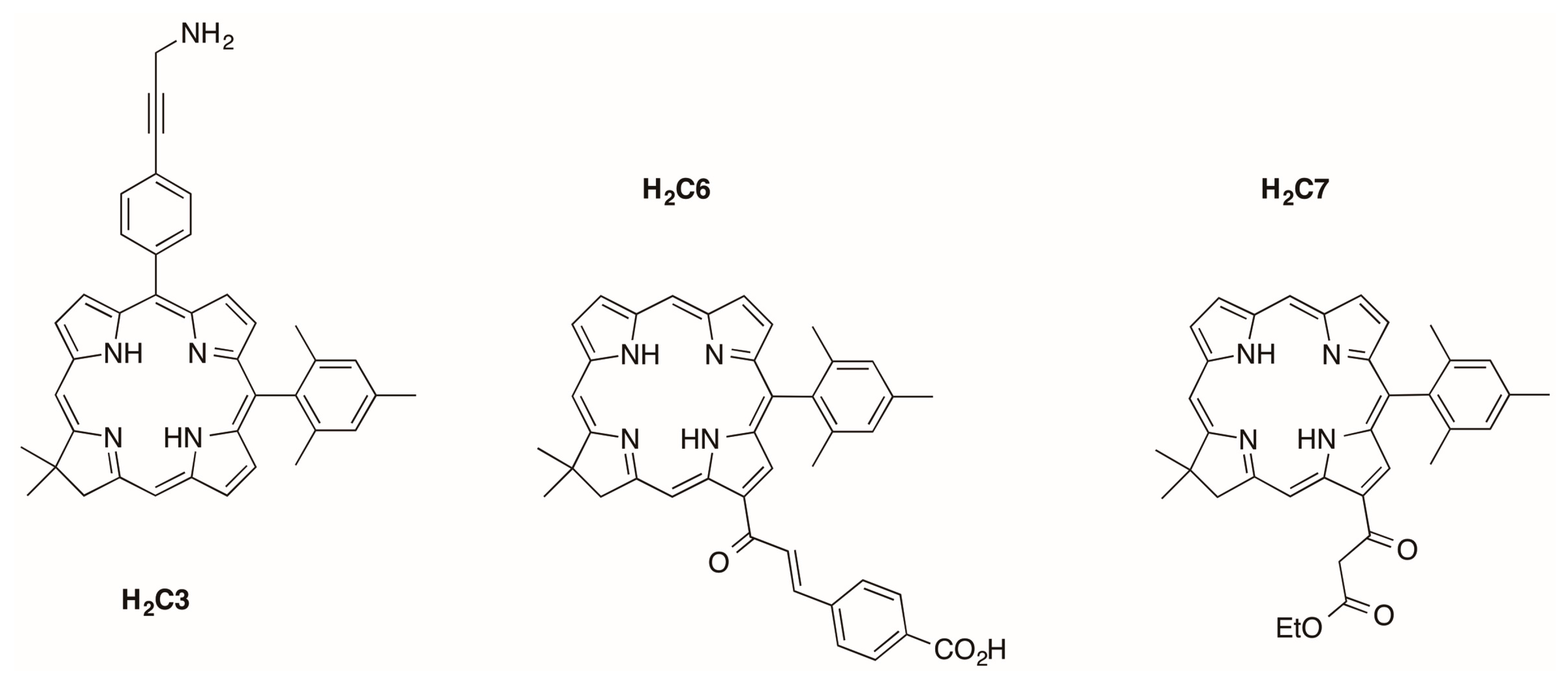
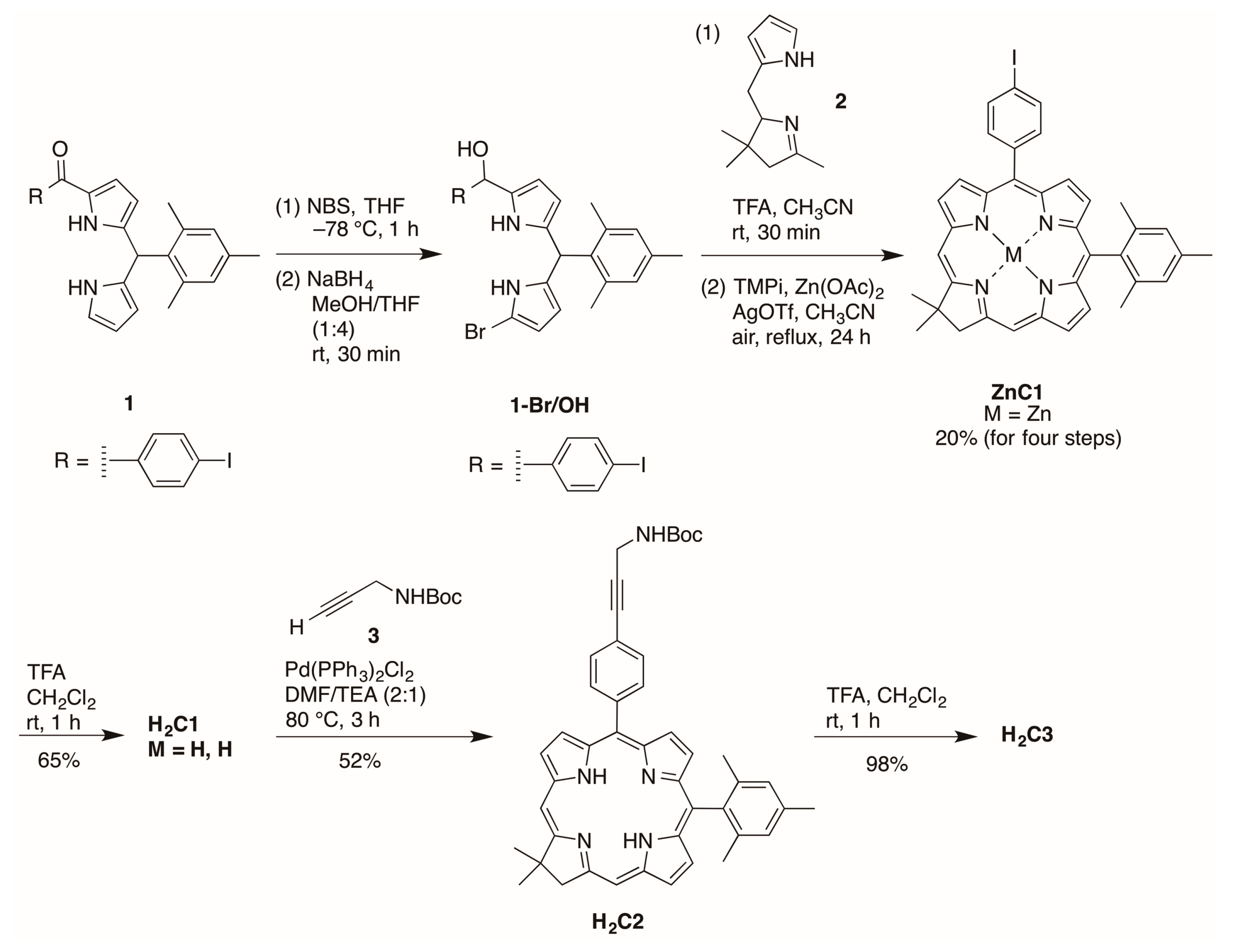
2.2.1. Amino Substitution at the 5-Position
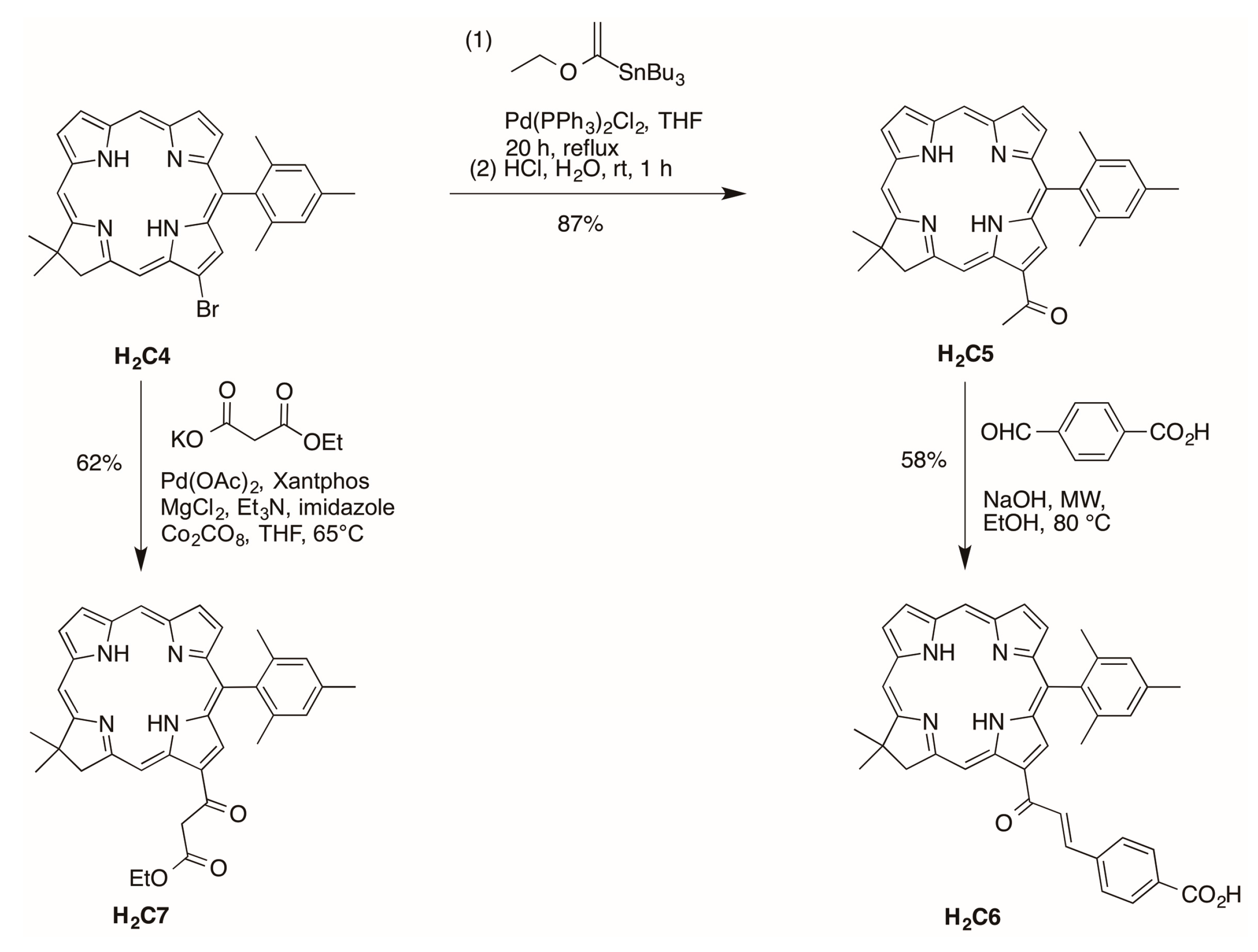
2.2.2. Chalcone or β-Ketoester Substitution at the 13-Position
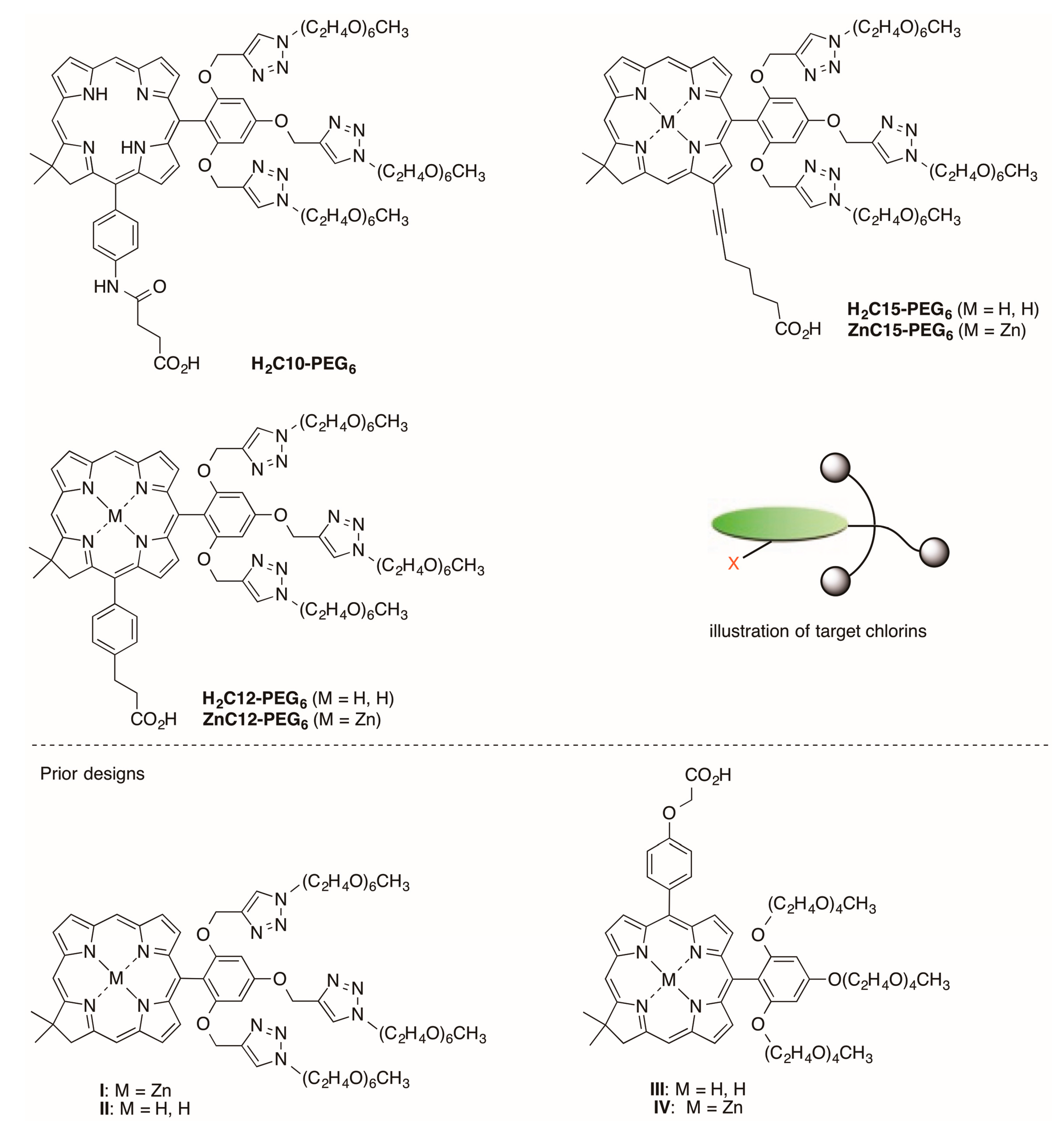
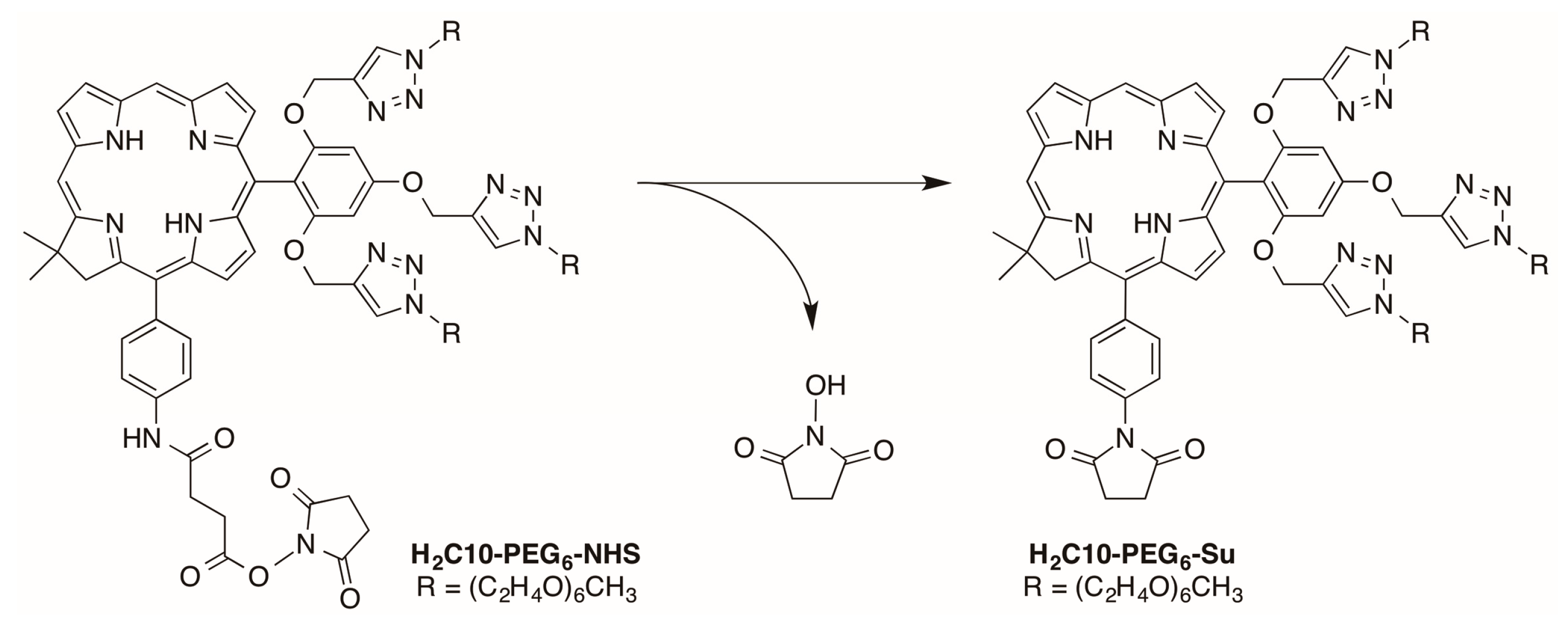
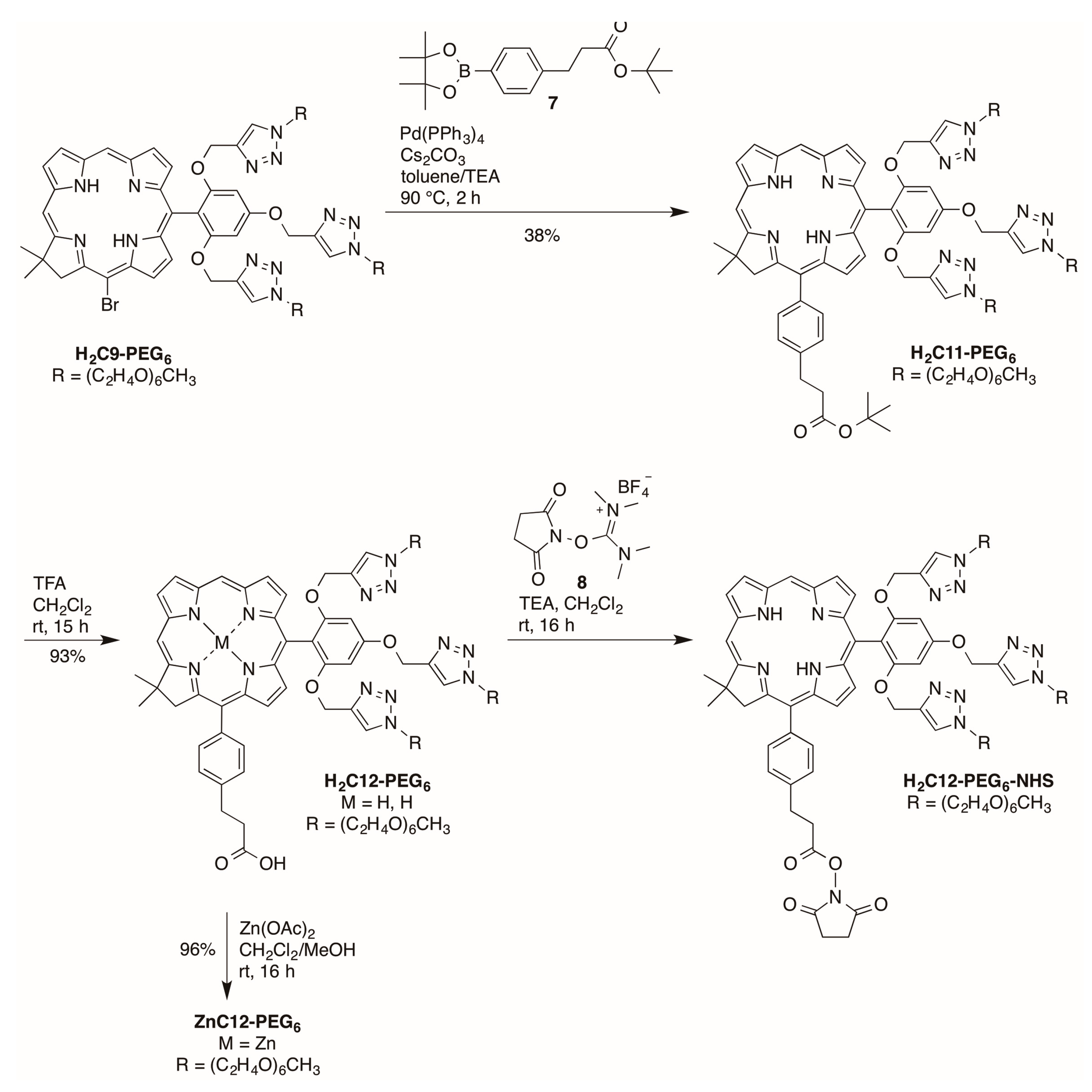
2.2.3. Carboxylic Acid (or NHS Ester) and Tris(PEGylation) at the Respective 15- and 10-Position
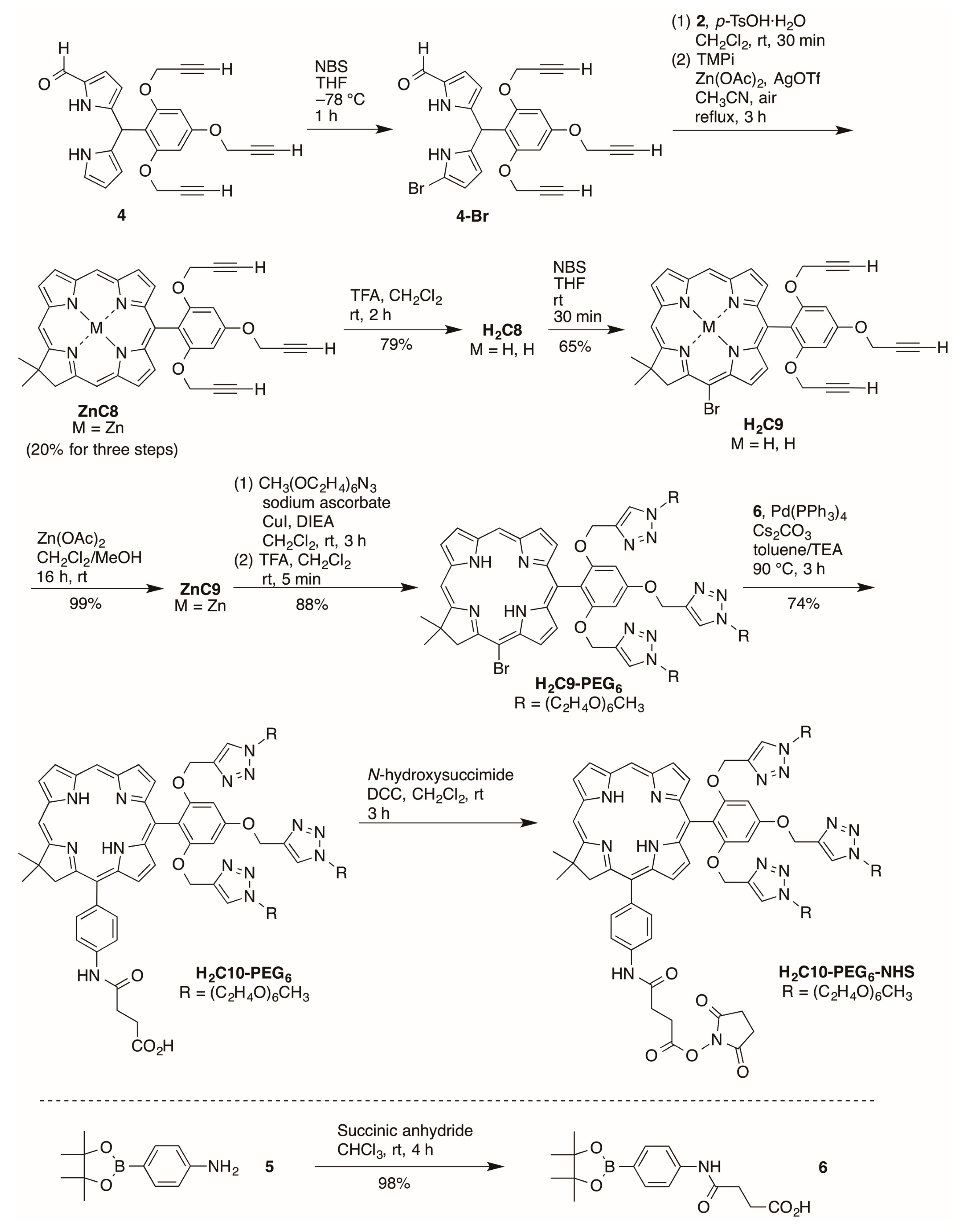
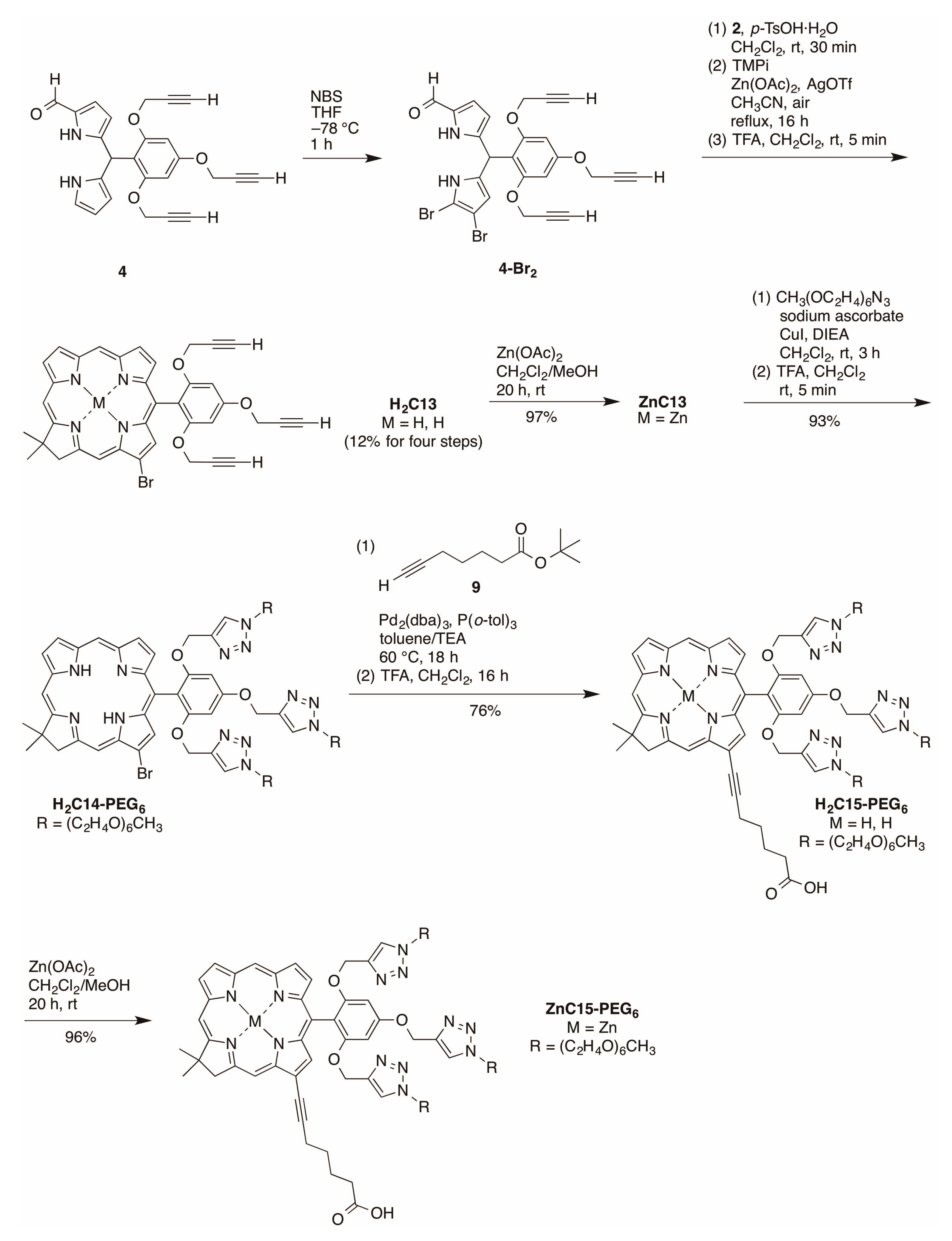
2.2.4. Heptynoic Acid and Tris(PEGylation) at the Respective 13- and 10-Position
2.3. Photophysical Characterization
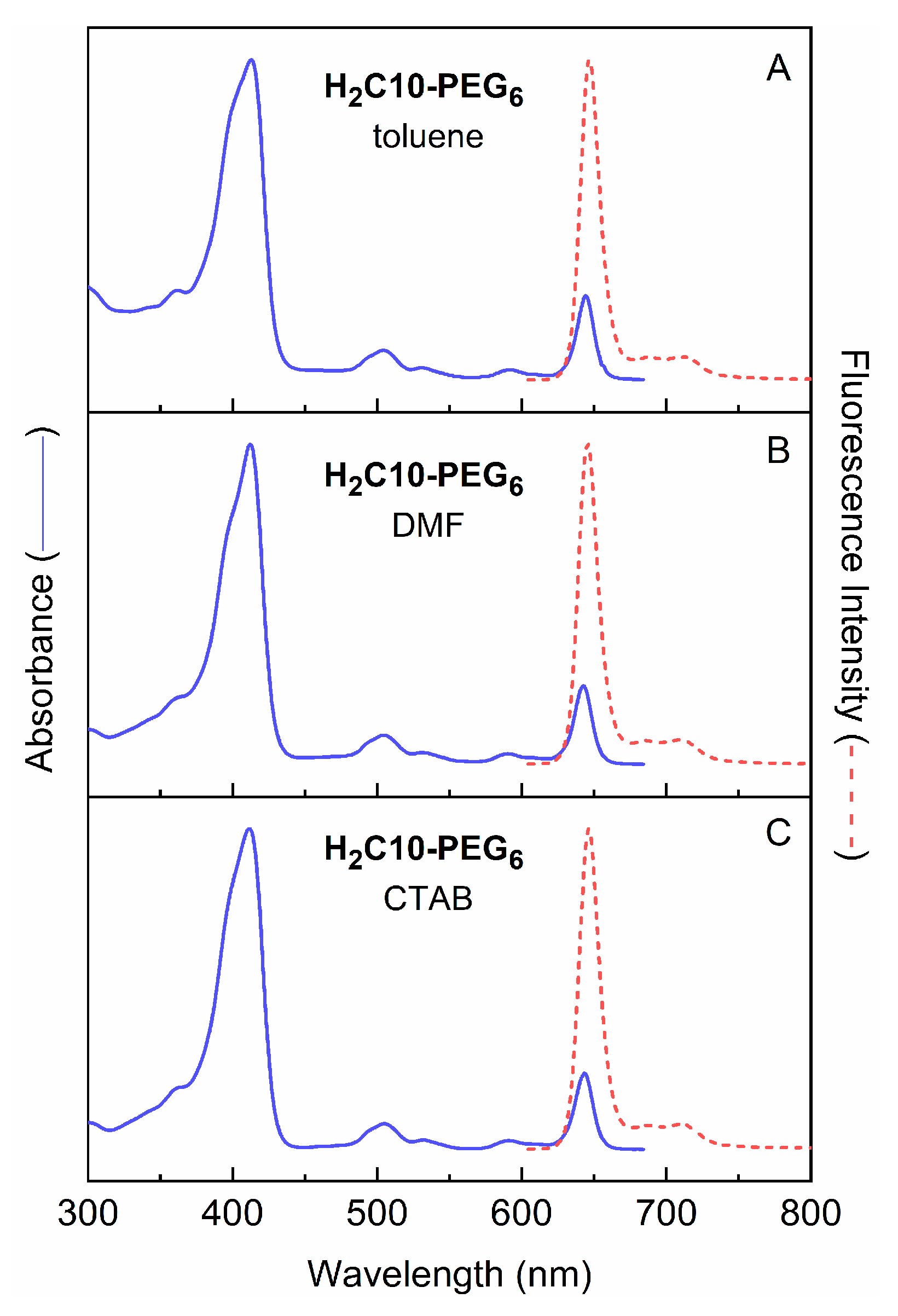
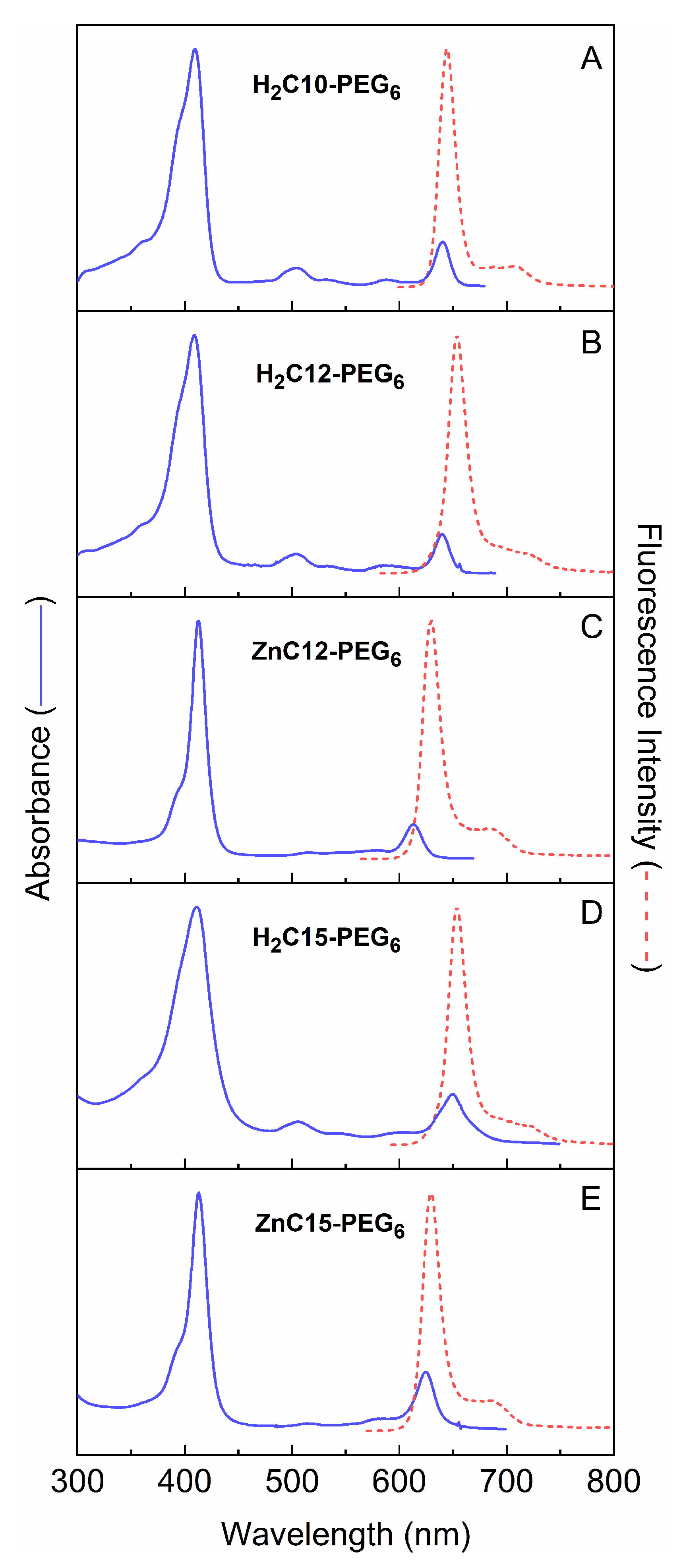
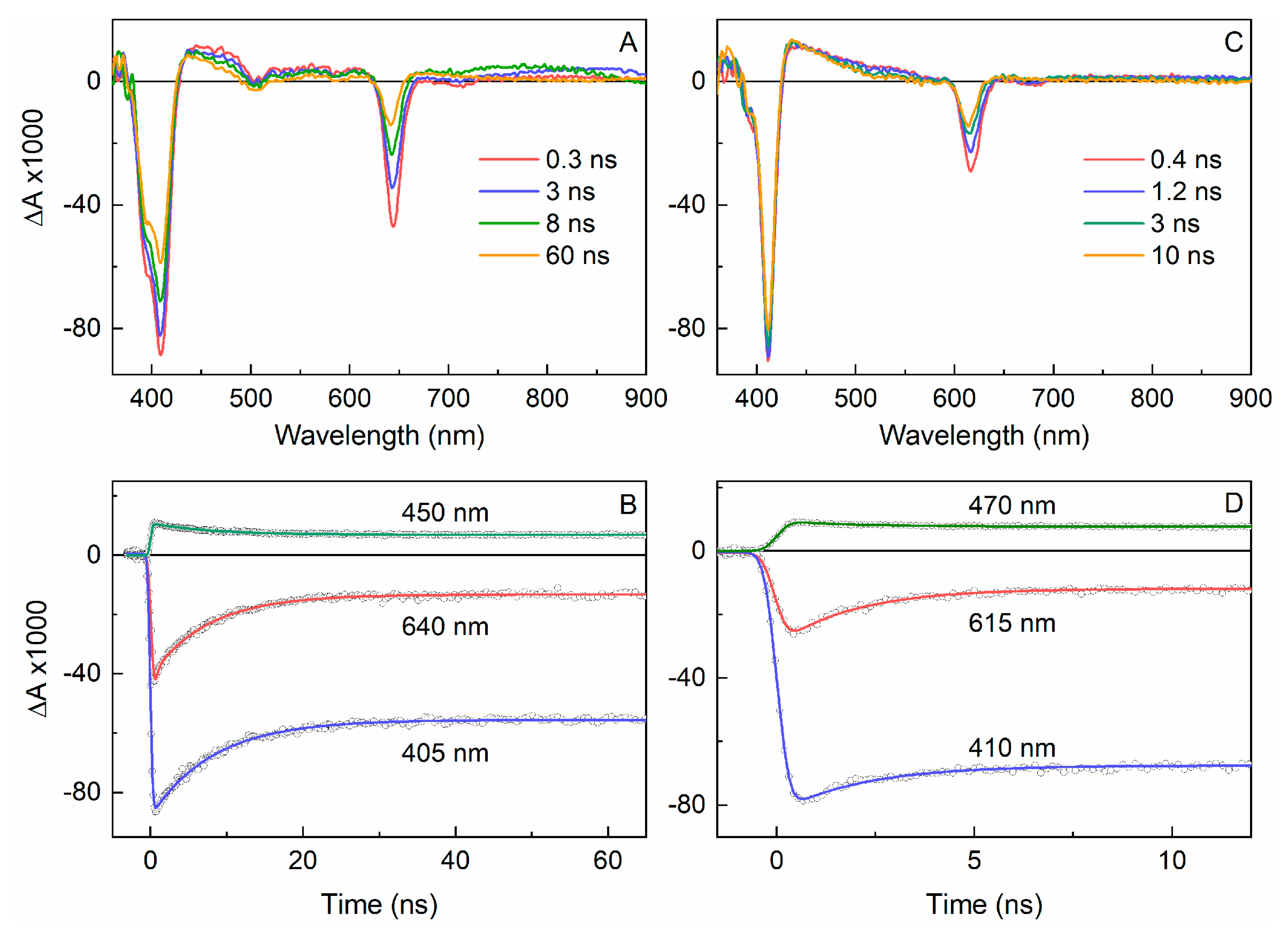
2.3.1. Spectral and Photophysical Properties of PEGylated Chlorins
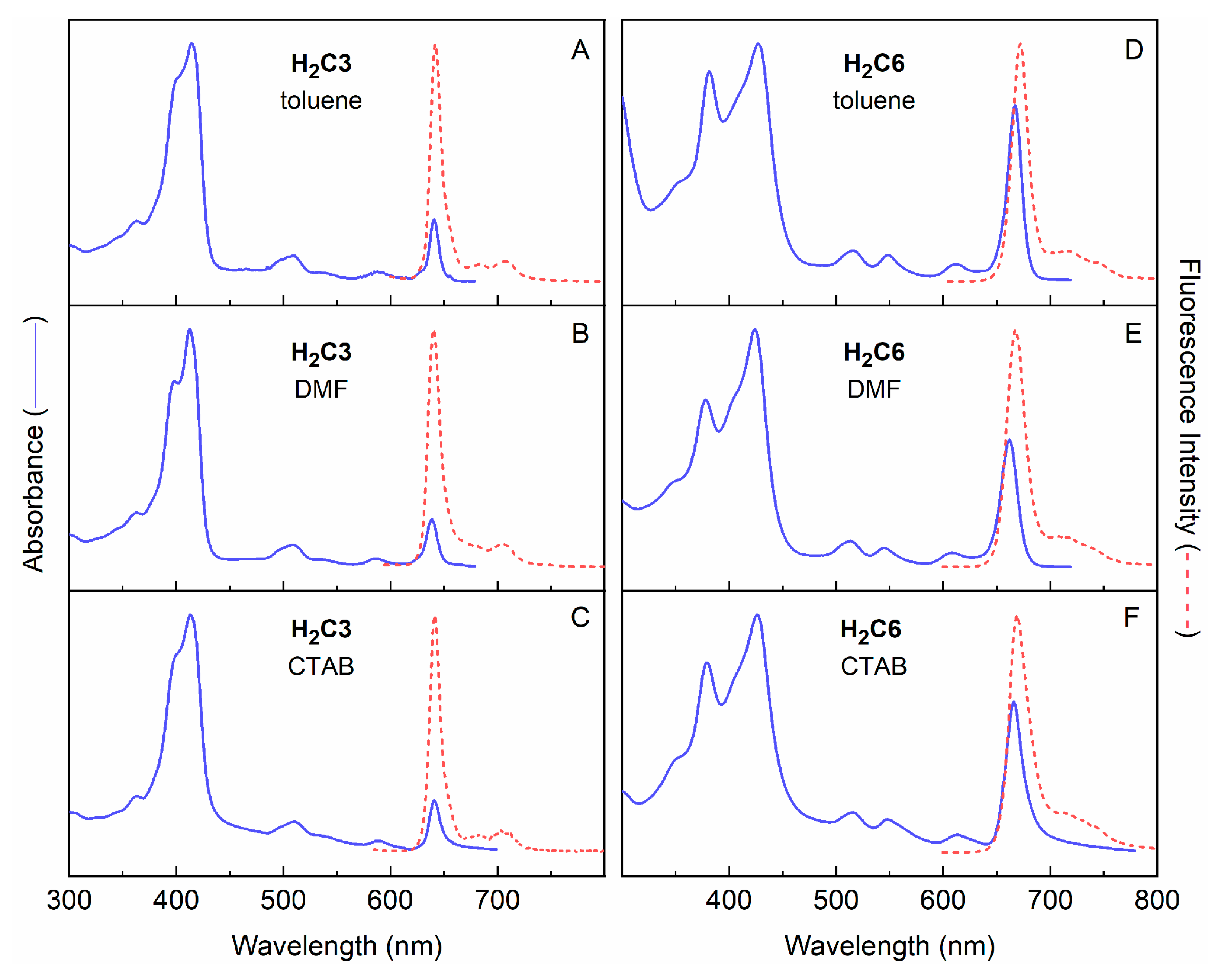
2.3.2. Amphiphilic Chlorins in Various Solvents Including Micellar Solution
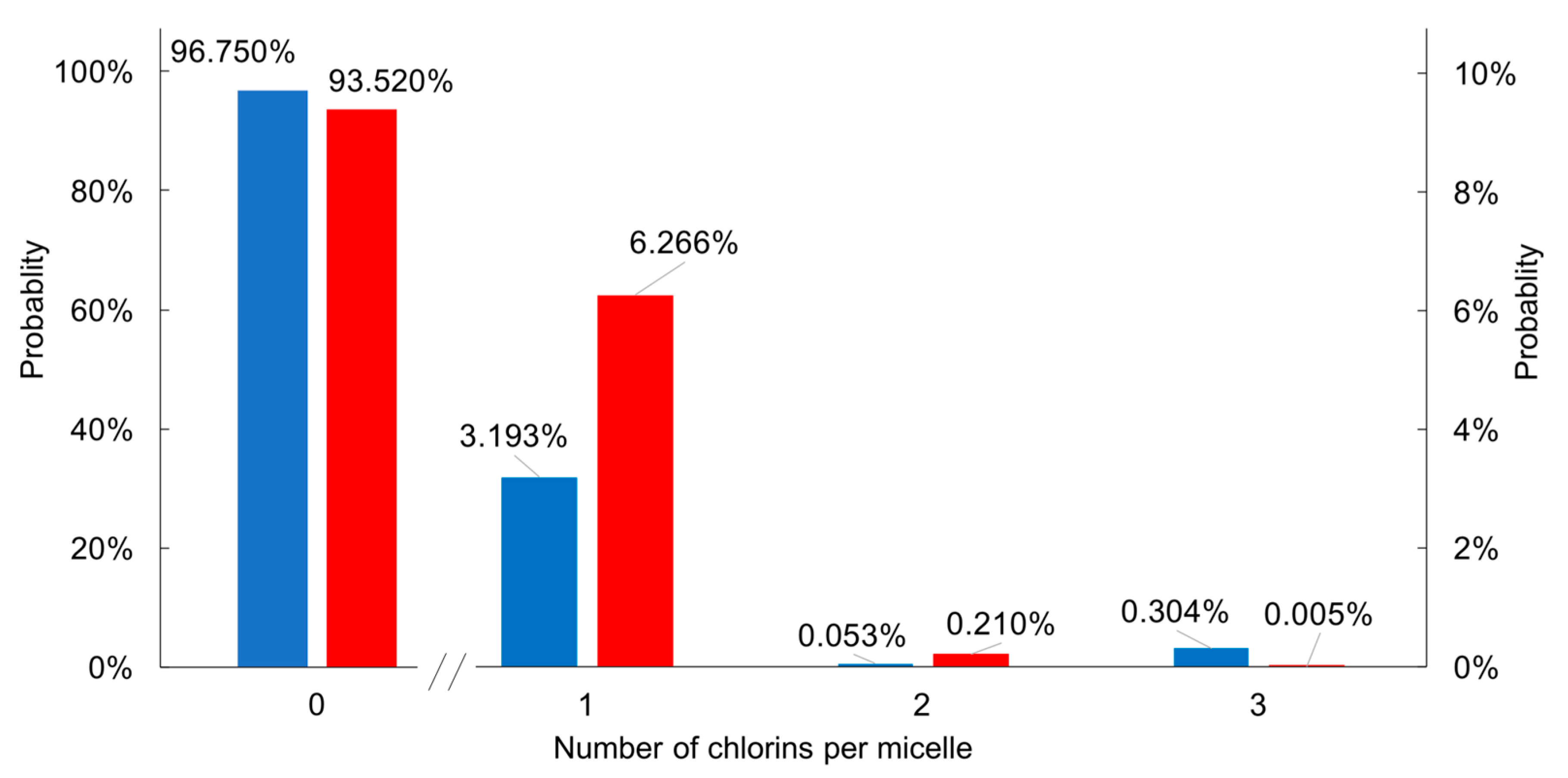
2.3.3. Poisson Distribution of H2C3 and H2C6 in CTAB Micelle Solution
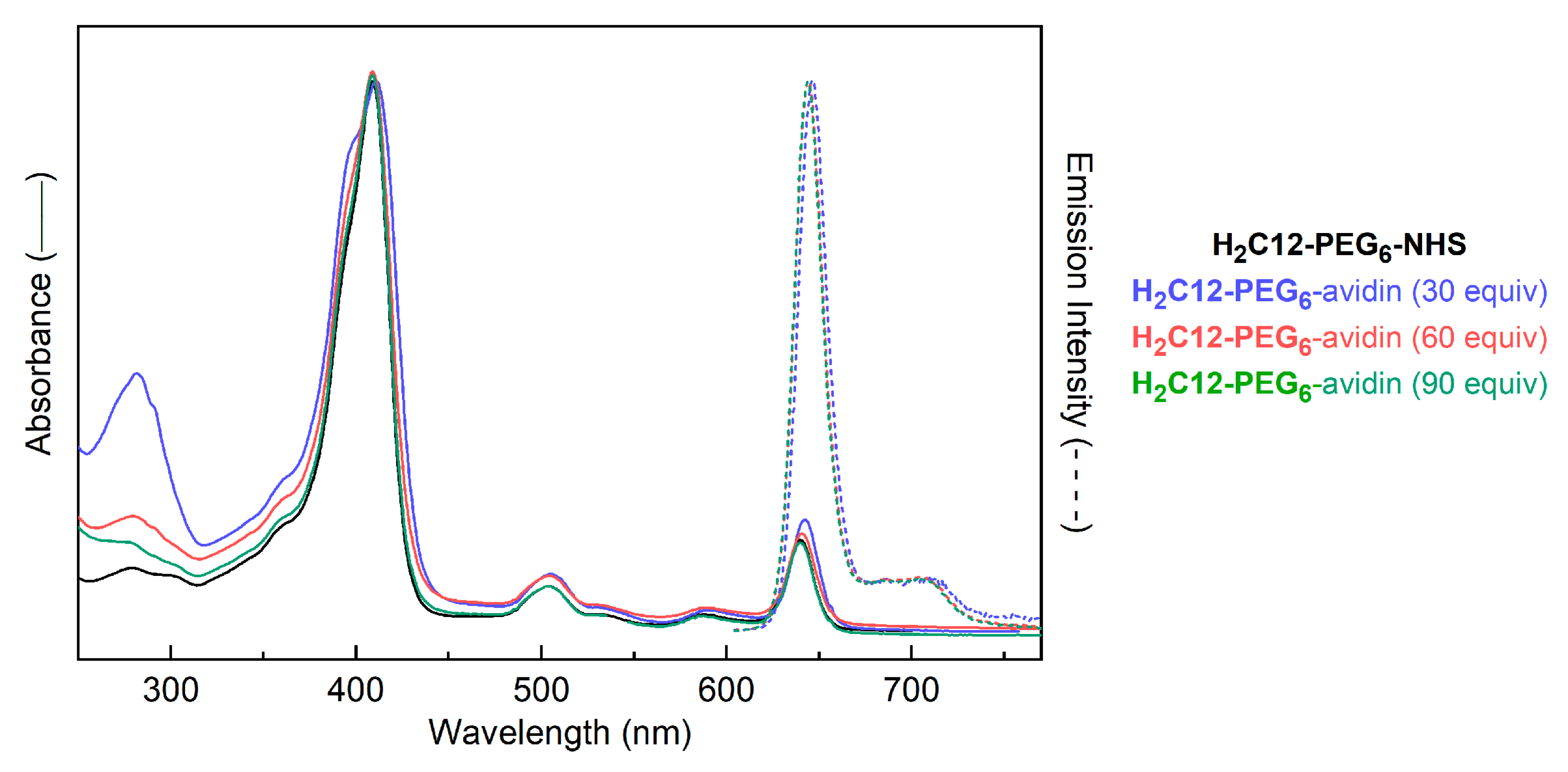
2.3.4. H2C12-PEG6-NHS Avidin Loading Experiment
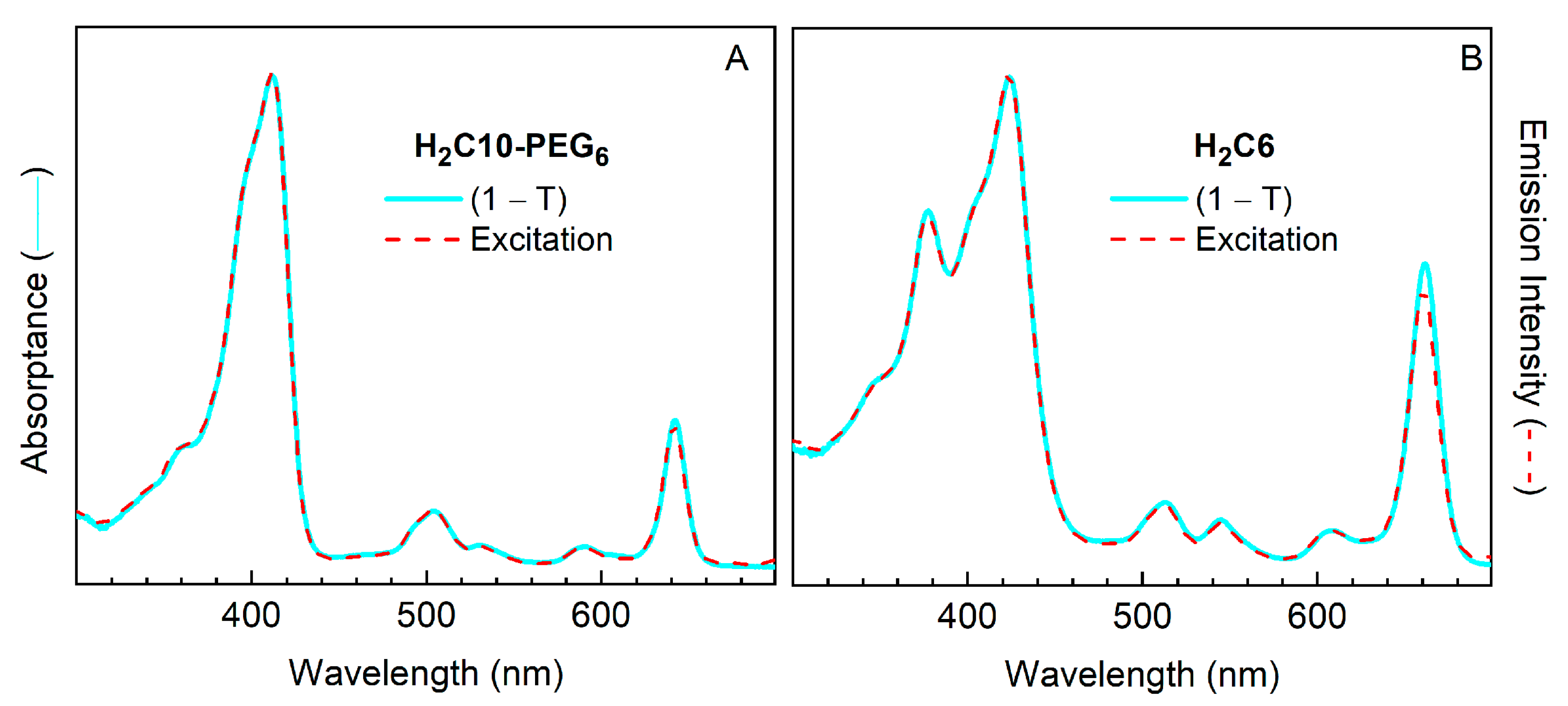
2.3.5. Comparison of Absorptance Spectra and Fluorescence Excitation Spectra
3. Materials and Methods
3.1. General Methods
3.2 Synthesis
3.3. Measuring Absorption and Emission of Chlorins in Micellar Solution
3.4. Experimental Method for Chlorin–Avidin Conjugation
3.5. Photophysical Properties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mauzerall, D. Why Chlorophyll? Ann. N. Y. Acad. Sci. 1973, 206, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Mauzerall, D. Porphyrins, chlorophyll, and photosynthesis. In Photosynthesis I. Photosynthetic Electron Transport and Photophosphorylation; Trebst, A., Avron, M., Eds.; Springer: Berlin, Germany, 1977; pp. 117–124. [Google Scholar]
- Mauzerall, D. The Photosynthetic Bacteria; Clayton, R.K., Sistrom, W.R., Eds.; Plenum Press: New York, NY, USA, 1978; pp. 223–231. [Google Scholar]
- Mauzerall, D.; Ballard, S.G. Ionization in Solution by Photoactivated Electron Transfer. Ann. Rev. Phys. Chem. 1982, 33, 377–407. [Google Scholar] [CrossRef]
- Mauzerall, D. Light, Iron, Sam Granick and the Origin of Life. Photosynth. Res. 1992, 33, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Enumeration of isomers of substituted tetrapyrrole macrocycles: From classical problems in biology to modern combinatorial libraries. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2012; Volume 23, pp. 1–80. [Google Scholar]
- Scheer, H. An overview of chlorophylls and bacteriochlorophylls: Biochemistry, biophysics, functions and applications. In Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 25, pp. 1–26. [Google Scholar]
- Lindsey, J.S. De Novo Synthesis of Gem-Dialkyl Chlorophyll Analogues for Probing and Emulating our Green World. Chem. Rev. 2015, 115, 6534–6620. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Dudkin, S.V.; Makarova, E.A.; Lukyanets, E.A. Synthesis of Chlorins, Bacteriochlorins and Their Tetraaza Analogues. Russ. Chem. Rev. 2016, 85, 700–730. [Google Scholar] [CrossRef]
- Pavlov, V.Y.; Ponomarev, G.V. Modification of the Peripheral Substituents in Chlorophylls a and b and Their Derivatives. Chem. Heterocycl. Compd. 2004, 40, 393–425. [Google Scholar] [CrossRef]
- Borbas, K.E. Chlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2016; Volume 36, pp. 1–149. [Google Scholar]
- Aravindu, K.; Mass, O.; Vairaprakash, P.; Springer, J.W.; Yang, E.; Niedzwiedzki, D.M.; Kirmaier, C.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Amphiphilic Chlorins and Bacteriochlorins in Micellar Environments. Molecular Design, de Novo Synthesis, and Photophysical Properties. Chem. Sci. 2013, 4, 3459–3477. [Google Scholar] [CrossRef]
- Zhang, S.; Lindsey, J.S. Construction of the Bacteriochlorin Macrocycle with Concomitant Nazarov Cyclization to Form the Annulated Isocyclic Ring: Analogues of Bacteriochlorophyll a. J. Org. Chem. 2017, 82, 2489–2504. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, C.-Y.; Mandal, A.K.; Chandrashaker, V.; Evans-Storms, R.B.; Pitner, J.B.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Bioconjugatable, PEGylated Hydroporphyrins for Photochemistry and Photomedicine. Narrow-Band, Red-Emitting Chlorins. New J. Chem. 2016, 40, 7721–7740. [Google Scholar] [CrossRef] [PubMed]
- Meares, A.; Satraitis, A.; Akhigbe, J.; Santhanam, N.; Swaminathan, S.; Ehudin, M.; Ptaszek, M. Amphiphilic BODIPY-Hydroporphyrin Energy Transfer Arrays with Broadly Tunable Absorption and Deep Red/Near-infrared Emission in Aqueous Micelles. J. Org. Chem. 2017, 82, 6054–6070. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Loewe, R.S.; Tomizaki, K.-Y.; Youngblood, W.J.; Bo, Z.; Lindsey, J.S. Synthesis of Perylene–Porphyrin Building Blocks and Rod-Like Oligomers for Light-Harvesting Applications. J. Mater. Chem. 2002, 12, 3438–3451. [Google Scholar] [CrossRef]
- Laysan Bio Inc. Hydrolysis Half-Lives at pH 8, 25 °C. Available online: http://laysanbio.com/index.php?submenu=Links&src=gendocs&link=Links_Downloads&category=Main (accessed on 6 December 2017).
- Taniguchi, M.; Kim, H.-J.; Ra, D.; Schwartz, J.K.; Kirmaier, C.; Hindin, E.; Diers, J.R.; Prathapan, S.; Bocian, D.F.; Holten, D.; et al. Synthesis and Electronic Properties of Regioisomerically Pure Oxochlorins. J. Org. Chem. 2002, 67, 7329–7342. [Google Scholar] [CrossRef] [PubMed]
- Muthukumaran, K.; Ptaszek, M.; Noll, B.; Scheidt, W.R.; Lindsey, J.S. Boron-Complexation Strategy for Use with 1-Acyldipyrromethanes. J. Org. Chem. 2004, 69, 5354–5364. [Google Scholar] [CrossRef] [PubMed]
- Ptaszek, M.; Bhaumik, J.; Kim, H.-J.; Taniguchi, M.; Lindsey, J.S. Refined Synthesis of 2,3,4,5-Tetrahydro-1,3,3-trimethyldipyrrin, a Deceptively Simple Precursor to Hydroporphyrins. Org. Process Res. Dev. 2005, 9, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.W.; Ciringh, Y.; Clausen, C.; Lindsey, J.S. Investigation and Refinement of Palladium-Coupling Conditions for the Synthesis of Diarylethyne-Linked Multiporphyrin Arrays. Chem. Mater. 1999, 11, 2974–2983. [Google Scholar] [CrossRef]
- Hu, G.; Liu, R.; Alexy, E.J.; Mandal, A.K.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Panchromatic Chromophore–Tetrapyrrole Light-Harvesting Arrays Constructed from Bodipy, Perylene, Terrylene, Porphyrin, Chlorin, and Bacteriochlorin Building Blocks. New J. Chem. 2016, 40, 8032–8052. [Google Scholar] [CrossRef]
- Muthiah, C.; Lahaye, D.; Taniguchi, M.; Ptaszek, M.; Lindsey, J.S. Regioselective Bromination Tactics in the de Novo Synthesis of Chlorophyll b Analogues. J. Org. Chem. 2009, 74, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Ruzié, C.; Krayer, M.; Lindsey, J.S. Fast and Robust Route to Hydroporphyrin–Chalcones with Extended Red or Near-Infrared Absorption. Org. Lett. 2009, 11, 1761–1764. [Google Scholar] [CrossRef] [PubMed]
- Baburajan, P.; Elango, K.P. One Pot Direct Synthesis of β-Ketoesters via Carbonylation of Aryl Halides using Cobalt Carbonyl. Tetrahedron Lett. 2014, 55, 3525–3528. [Google Scholar] [CrossRef]
- Laha, J.K.; Muthiah, C.; Taniguchi, M.; McDowell, B.E.; Ptaszek, M.; Lindsey, J.S. Synthetic Chlorins Bearing Auxochromes at the 3- and 13-Positions. J. Org. Chem. 2006, 71, 4092–4102. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, C.; Ptaszek, M.; Nguyen, T.M.; Flack, K.M.; Lindsey, J.S. Two Complementary Routes to 7-Substituted Chlorins. Partial Mimics of Chlorophyll b. J. Org. Chem. 2007, 72, 7736–7749. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Ptaszek, M.; McDowell, B.E.; Lindsey, J.S. Sparsely Substituted Chlorins as Core Constructs in Chlorophyll Analogue Chemistry. Part 2: Derivatization. Tetrahedron 2007, 63, 3840–3849. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Synthesis of Oligo(p-Phenylene)-linked Dyads Containing Free Base, Zinc(II) or Thallium(III) Porphyrins for Studies in Artificial Photosynthesis. Tetrahedron 2010, 66, 5549–5565. [Google Scholar] [CrossRef]
- Ikawa, Y.; Harada, H.; Toganoh, M.; Furuta, H. Synthesis and Protonation Behavior of a Water-soluble N-fused Porphyrin: Conjugation with an Oligoarginine by Click Chemistry. Bioorg. Med. Chem. Lett. 2009, 19, 2448–2452. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.W.; Johnson, T.E.; Lindsey, J.S. Soluble Synthetic Multiporphyrin Arrays. 1. Modular Design and Synthesis. J. Am. Chem. Soc. 1996, 118, 11166–11180. [Google Scholar] [CrossRef]
- Zhang, N.; Reddy, K.R.; Jiang, J.; Taniguchi, M.; Sommer, R.D.; Lindsey, J.S. Elaboration of an Unexplored Substitution Site in Synthetic Bacteriochlorins. J. Porphyrins Phthalocyanines 2015, 19, 887–902. [Google Scholar] [CrossRef]
- Aravindu, K.; Kim, H.-J.; Taniguchi, M.; Dilbeck, P.L.; Diers, J.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthesis and Photophysical Properties of Chlorins Bearing 0–4 Distinct meso-Substituents. Photochem. Photobiol. Sci. 2013, 12, 2089–2109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chen, C.-Y.; Zhang, N.; Vairaprakash, P.; Lindsey, J.S. Polarity-Tunable and Wavelength-Tunable Bacteriochlorins Bearing a Single Carboxylic Acid or NHS Ester. Use in a Protein Bioconjugation Model System. New J. Chem. 2015, 39, 403–419. [Google Scholar] [CrossRef]
- Kaválek, J.; Machácek, V.; Svobodová, G. Kinetics and Mechanism of Reversible, Base-Catalyzed Ring Closure of 3-(Methoxycarbonyl)propionanilide and O-(methoxycarbonylmethyl)-N-phenylcarbamate. Collect. Czechoslov. Chem. Commun. 1989, 54, 1005–1011. [Google Scholar] [CrossRef]
- Kumar, P.P.; Devi, B.R.; Dubey, P.K. A Facile and Green Synthesis of N-substituted Imides. Ind. J. Chem. 2013, 52B, 1166–1171. [Google Scholar]
- Sutton, J.M.; Clarke, O.J.; Fernandez, N.; Boyle, R.W. Porphyrin, Chlorin and Bacteriochlorin Isothiocyanates: Useful Reagents for the Synthesis of Photoactive Bioconjugates. Bioconj. Chem. 2002, 13, 249–263. [Google Scholar] [CrossRef]
- Knorr, R.; Trzeciak, A.; Bannwarth, W.; Gilleseen, D. New Coupling Reagents in Peptide Chemistry. Tetrahedron Lett. 1989, 30, 1927–1930. [Google Scholar] [CrossRef]
- Bannwarth, W.; Knorr, R. Formation of Carboxamides with N,N,N′,N′-Tetramethyl (Succinimido) Uronium Tetrafluoroborate in Aqueous/Organic Solvent Systems. Tetrahedron Lett. 1991, 32, 1157–1160. [Google Scholar] [CrossRef]
- Mass, O.; Ptaszek, M.; Taniguchi, M.; Diers, J.R.; Kee, H.L.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthesis and Photochemical Properties of 12-Substituted versus 13-Substituted Chlorins. J. Org. Chem. 2009, 74, 5276–5289. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.W.; Johnson, T.E.; Li, F.; Lindsey, J.S. Synthesis of Ethyne-Linked or Butadiyne-Linked Porphyrin Arrays Using Mild, Copper-Free, Pd-Mediated Coupling Reactions. J. Org. Chem. 1995, 60, 5266–5273. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent Advances in the Transition Metal-catalyzed Twofold Oxidative C–H Bond Activation Strategy for C–C and C–N Bond Formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Jiang, J.; Liu, M.; Taniguchi, M.; Mandal, A.K.; Evans-Storms, R.B.; Pitner, J.B.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Bioconjugatable, PEGylated Hydroporphyrins for Photochemistry and Photomedicine. Narrow-Band, Near-Infrared-Emitting Bacteriochlorins. New J. Chem. 2016, 40, 7750–7767. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.L.; Kirmaier, C.; Tang, Q.; Diers, J.R.; Muthiah, C.; Taniguchi, M.; Laha, J.K.; Ptaszek, M.; Lindsey, J.S.; Bocian, D.F.; et al. Effects of Substituents on Synthetic Analogs of Chlorophylls. Part 2: Redox Properties, Optical Spectra and Electronic Structure. Photochem. Photobiol. 2007, 83, 1125–1143. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.B. Photophysics of Aromatic Molecules; Wiley-Interscience: London, UK, 1970; pp. 142–192. [Google Scholar]
- Batschelet, E. Introduction to Mathematics for Life Scientists, 3rd ed.; Springer: New York, NY, USA, 1979; pp. 446–452. [Google Scholar]
- Mauzerall, D. Statistical theory of the effect of mutiple excitation in photosynthetic systems. In Biological Events Probed by Ultrafast Laser Spectroscopy; Alfano, R.R., Ed.; Academic Press: New York, NY, USA, 1982; pp. 215–235. [Google Scholar]
- Anachkov, S.E.; Danov, K.D.; Basheva, E.S.; Kralchevsky, P.A.; Ananthapadmanabhan, K.P. Determination of the Aggregation Number and Charge of Ionic Surfactant Micelles from the Stepwise Thinning of Foam Films. Adv. Colloid Interface Sci. 2012, 183–184, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, P.; Mysels, K.J. Critical Micelle Concentrations in Aqueous Surfactant Systems. Natl. Stand. Ref. Data Ser. 1971, 36, 1–222. [Google Scholar]
- Scherz, A.; Rosenbach-Belkin, V.; Fisher, J.R.E. Distribution and Self-organization of Photosynthetic Pigments in Micelles: Implication for the Assembly of Light-Harvesting Complexes and Reaction Centers in the Photosynthetic Membrane. Proc. Natl. Acad. Sci. USA 1990, 87, 5430–5434. [Google Scholar] [CrossRef] [PubMed]
- Avital, S.; Malkin, S. Quenching of Chlorophyl Fluorescence by Carotenoids in a Micellar Model System. In Photosynthesis: Mechanisms and Effects; Garab, G., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1998; Volume 1, pp. 477–482. [Google Scholar]
- Alexy, E.J.; Hintz, C.W.; Hughes, H.M.; Taniguchi, M.; Lindsey, J.S. Paley’s Watchmaker Analogy and Prebiotic Synthetic Chemistry in Surfactant Assemblies. Formaldehyde Scavenging by Pyrroles Leading to Porphyrins as a Case Study. Org. Biomol. Chem. 2015, 13, 10025–10031. [Google Scholar] [CrossRef] [PubMed]
- Agostiano, A.; Catucci, L.; Colafemmina, G.; Scheer, H. Role of Functional Groups and Surfactant Charge in Regulating Chlorophyll Aggregation in Micellar Solutions. J. Phys. Chem. B 2002, 106, 1446–1454. [Google Scholar] [CrossRef]
- Agostiano, A.; Catucci, L.; Colafemmina, G.; Della Monica, M. Chlorophyll a Self-Organization in Microheterogeneous Surfactant Systems. Biophys. Chem. 1996, 60, 17–27. [Google Scholar] [CrossRef]
- Ricchelli, F. Photophysical Properties of Porphyrins in Biological Membranes. J. Photochem. Photobiol. B Biol. 1995, 29, 109–118. [Google Scholar] [CrossRef]
- Nantes, I.L.; Durán, N.; Pinto, S.M.S.; da Silva, F.B.; de Souza, J.S.; Isoda, N.; Luz, R.A.S.; de Oliveira, T.G.; Fernandes, V.G. Modulation of the Catalytic Activity of Porphyrins by Lipid- and Surfactant-Containing Nanostructures. J. Braz. Chem. Soc. 2011, 22, 1621–1633. [Google Scholar] [CrossRef]
- Bohne, C.; Konuk, R.; Scaiano, J.C. Dynamics of the Redistribution of 1-Dodecylpyrene Aggregates in Micellar Solution. Chem. Phys. Lett. 1988, 152, 156–159. [Google Scholar] [CrossRef]
- Rharbi, Y.; Winnik, M.A. Salt Effects on Solute Exchange in Sodium Dodecyl Sulfate Micelles. J. Am. Chem. Soc. 2002, 124, 2082–2083. [Google Scholar] [CrossRef] [PubMed]
- Rharbi, Y.; Winnik, M.A. Salt Effects on Solute Exchange and Micelle Fission in Sodium Dodecyl Sulfate Micelles below the Micelle-to-Rod Transition. J. Phys. Chem. B 2003, 107, 1491–1501. [Google Scholar] [CrossRef]
- Rharbi, Y.; Chen, L.; Winnik, M.A. Exchange Mechanisms for Sodium Dodecyl Sulfate Micelles: High Salt Concentration. J. Am. Chem. Soc. 2004, 126, 6025–6034. [Google Scholar] [CrossRef] [PubMed]
- Rharbi, Y.; Karrouch, M.; Richardson, P. Fusion and Fission Inhibited by the Same Mechanism in Electrostatically Charged Surfactant Micelles. Langmuir 2014, 30, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Thayumanavan, S. Environment-Dependent Guest Exchange in Supramolecular Hosts. Langmuir 2014, 30, 12384–12390. [Google Scholar] [CrossRef] [PubMed]
- Green, N.M. Avidin. 1. The Use of [14C]Biotin for Kinetic Studies and for Assay. Biochem. J. 1963, 89, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Green, N.M. The Molecular Weight of Avidin. Biochem. J. 1964, 92, 16C–17C. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-M.; Raine, L.; Fanger, H. Use of Avidin-Biotin-Peroxidase Complex (ABC) in Immunoperoxidase Techniques: A Comparison between ABC and Unlabeled Antibody (PAP) Procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Wilchek, M. Biotin-binding proteins: Overview and prospects. In Methods of Enzymology; Wilchek, M., Bayer, E.A., Eds.; Academic Press: San Diego, CA, USA, 1990; Volume 184, pp. 49–51. [Google Scholar]
- Green, N.M. Avidin and streptavidin. In Methods in Enzymology; Wilchek, M., Bayer, E.A., Eds.; Academic Press: San Diego, CA, USA, 1990; Volume 184, pp. 51–67. [Google Scholar]
- Delange, R.J.; Huang, T.-S. Egg White Avidin. III. Sequence of the 78-Residue Middle Cyanogen Bromide Peptide. Complete Amino Acid Sequence of the Protein Subunit. J. Biol. Chem. 1971, 246, 698–709. [Google Scholar] [PubMed]
- Song, F. A Study of Noncovalent Protein Complexes by Matrix-Assisted Laser Desorption/Ionization. J. Am. Soc. Mass Spectrom. 2007, 18, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, N.; Haney, C.A.; Lindsey, J.S.; Zhang, W.; Chait, B.T. Investigation of MALDI-TOF Mass Spectrometry of Diverse Synthetic Metalloporphyrins, Phthalocyanines, and Multiporphyrin Arrays. J. Porphyrins Phthalocyanines 1999, 3, 283–291. [Google Scholar] [CrossRef]
- Mandal, A.K.; Taniguchi, M.; Diers, J.R.; Niedzwiedzki, D.M.; Kirmaier, C.; Lindsey, J.S.; Bocian, D.F.; Holten, D. Photophysical Properties and Electronic Structure of Porphyrins Bearing Zero to Four meso-Phenyl Substituents: New Insights into Seemingly Well Understood Tetrapyrroles. J. Phys. Chem. A 2016, 120, 9719–9731. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, J.; Bhupathiraju, N.V.S.D.K.; Perea, W.; Chu, H.; Berisha, N.; Bueno, V.; Dodic, N.; Rozenberg, J.; Greenbaum, N.L.; Drain, C.M. Facile Synthesis of Chlorin Bioconjugates by a Series of Click Reactions. Chem. Commun. 2017, 53, 3773–3776. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chlorin | Solvent | Bmax Abs (nm) | Qy Abs (fwhm) in nm | Qy Em (fwhm) in nm | τS (ns) | Φf | Φisc | Φic | kf−1 (ns) | kisc−1 (ns) | kic−1 (ns) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2C10-PEG6 | toluene b | 413 | 644 (14) | 647 (15) | 9.3 | 0.25 | 0.61 | 0.14 | 37 | 15 | 66 |
| H2C10-PEG6 | DMF | 412 | 642 (14) | 645 (16) | 9.3 | 0.26 | 0.62 | 0.12 | 36 | 15 | 78 |
| H2C10-PEG6 | water | 409 | 640 (16) | 644 (18) | 7.4 c | 0.20 d | 0.52 | 0.28 | 37 | 14 | 26 |
| H2C12-PEG6 | water | 409 | 640 (17) | 644 (18) | 7.6 b | 0.23 | 0.70 | 0.07 | 33 | 11 | 110 |
| ZnC12-PEG6 | toluene b | 415 | 612 (18) | 616 (20) | 2.4 | 0.092 | 0.87 | 0.04 | 22 | 2.8 | 63 |
| ZnC12-PEG6 | DMF | 415 | 613 (20) | 617 (21) | 1.9 | 0.065 | 0.84 | 0.10 | 29 | 2.3 | 20 |
| ZnC12-PEG6 | water | 413 | 613 (20) | 619 (20) | 2.0 c | 0.060 | 0.80 | 0.14 | 33 | 2.5 | 14 |
| H2C15-PEG6 | water | 411 | 650 (31) | 654 (20) | 6.2 c | 0.20 | 0.67 | 0.13 | 31 | 9.3 | 48 |
| ZnC15-PEG6 | toluene b | 416 | 625 (26) | 627 (22) | 2.8 | 0.11 | 0.74 | 0.15 | 25 | 3.8 | 19 |
| ZnC15-PEG6 | DMF | 415 | 625 (18) | 628 (22) | 2.4 | 0.090 | 0.72 | 0.18 | 27 | 3.3 | 13 |
| ZnC15-PEG6 | water | 413 | 625 (22) | 630 (19) | 2.8 c | 0.11 | 0.67 | 0.22 | 25 | 4.2 | 13 |
| Chlorin | Solvent | Bmax Abs (nm) | Qy Abs (fwhm) in nm | Qy Em (fwhm) in nm | τS (ns) | Φf | Φisc | Φic | kf−1 (ns) | kisc−1 (ns) | kic−1 (ns) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2C2 | toluene | 415 | 641 (10) | 643 (13) | 9.3 | 0.23 | 0.70 | 0.07 | 40 | 13 | 130 |
| H2C2 | DMF | 413 | 638 (11) | 641 (16) | 9.8 | 0.18 | 0.74 | 0.08 | 54 | 13 | 120 |
| H2C3 | toluene | 414 | 641 (10) | 642 (12) | 7.1 | 0.18 | 0.72 | 0.10 | 39 | 10 | 71 |
| H2C3 | DMF | 412 | 639 (12) | 640 (13) | 8.4 | 0.18 | 0.71 | 0.11 | 47 | 12 | 76 |
| H2C3 | CTAB b | 413 | 641 (12) | 641 (20) | 7.9 | 0.16 | 0.73 | 0.11 | 49 | 11 | 72 |
| H2C6 | toluene | 427 | 667 (15) | 672 (18) | 6.4 | 0.29 | 0.62 | 0.090 | 22 | 10 | 71 |
| H2C6 | DMF | 424 | 662 (18) | 667 (20) | 6.5 | 0.26 | 0.63 | 0.11 | 25 | 10 | 59 |
| H2C6 | CTAB | 426 | 666 (19) | 669 (22) | 6.4 | 0.24 | 0.44 | 0.32 | 27 | 15 | 20 |
| Entry | [Chlorin] | [Avidin] | Ratio a | Loading b | Φf of Conjugate c |
|---|---|---|---|---|---|
| 1 | 2.4 mM | 79 µM | 30 | 2.3 | 0.075 |
| 2 | 4.8 mM | 79 µM | 60 | 6.2 | 0.13 |
| 3 | 7.2 mM | 79 µM | 90 | 12 | 0.17 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Liu, M.; Hood, D.; Chen, C.-Y.; MacNevin, C.J.; Holten, D.; Lindsey, J.S. Chlorophyll-Inspired Red-Region Fluorophores: Building Block Synthesis and Studies in Aqueous Media. Molecules 2018, 23, 130. https://doi.org/10.3390/molecules23010130
Liu R, Liu M, Hood D, Chen C-Y, MacNevin CJ, Holten D, Lindsey JS. Chlorophyll-Inspired Red-Region Fluorophores: Building Block Synthesis and Studies in Aqueous Media. Molecules. 2018; 23(1):130. https://doi.org/10.3390/molecules23010130
Chicago/Turabian StyleLiu, Rui, Mengran Liu, Don Hood, Chih-Yuan Chen, Christopher J. MacNevin, Dewey Holten, and Jonathan S. Lindsey. 2018. "Chlorophyll-Inspired Red-Region Fluorophores: Building Block Synthesis and Studies in Aqueous Media" Molecules 23, no. 1: 130. https://doi.org/10.3390/molecules23010130