CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives

 , ,
, ,  ,
,
Abstract
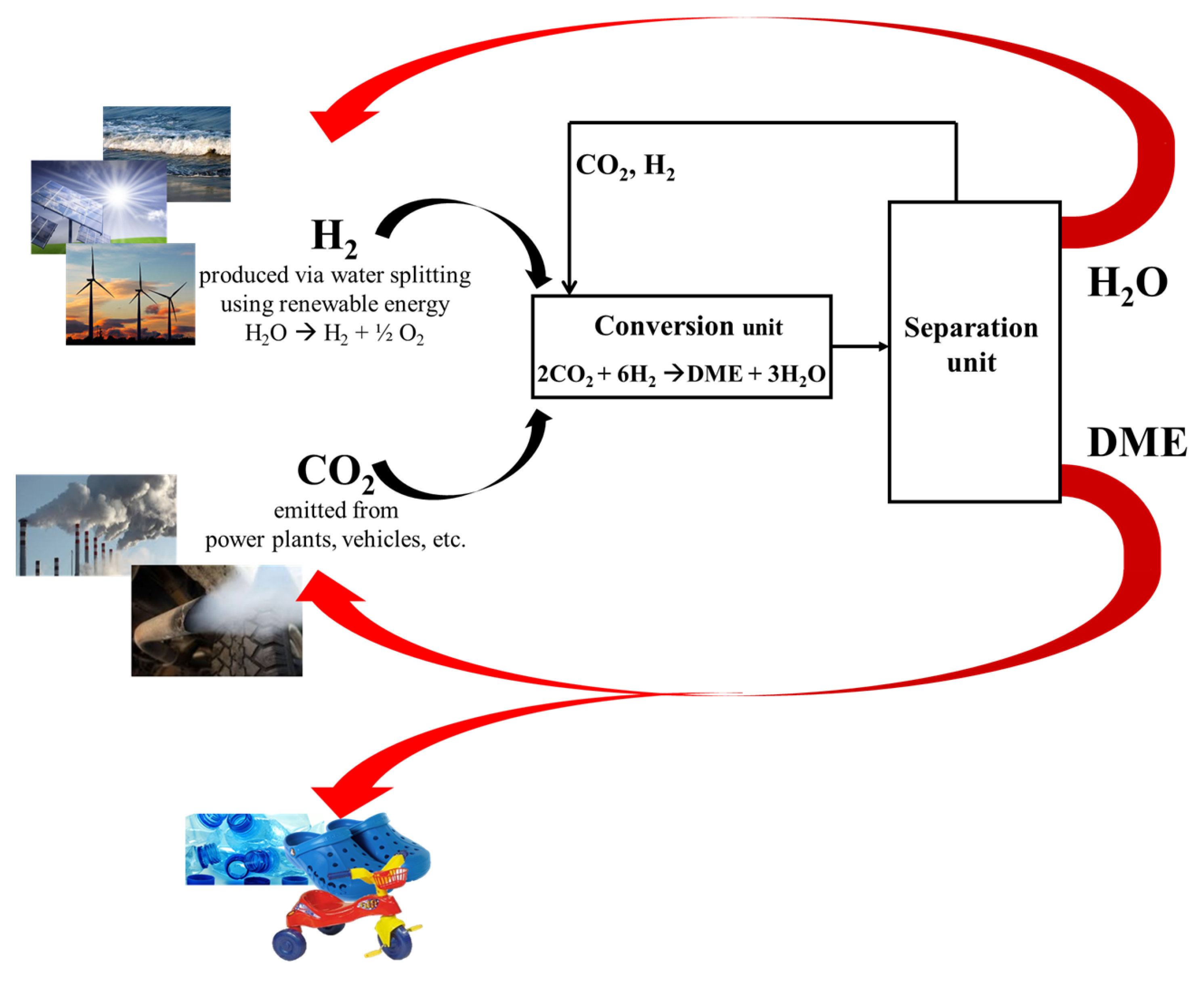
:1. Introduction: How Can CO2 Become the Future Carbon Source?
- (a)
- Production of hydrogen via water splitting by using renewable energy (e.g., solar energy);
- (b)
- Capture and safe storage of CO2 from power plants emission or even from atmosphere;
- (c)
- Hydrogenation of captured CO2 to produce methanol and/or DME (DME should be preferred because of its no-toxicity);
- (d)
- Utilization of DME for energy production or as intermediate in the chemical-chain industry;
- (e)
- Reuse the carbon dioxide from eco-friendly combustion of DME to re-produce itself.
2. DME as Valuable Fuel of the Future
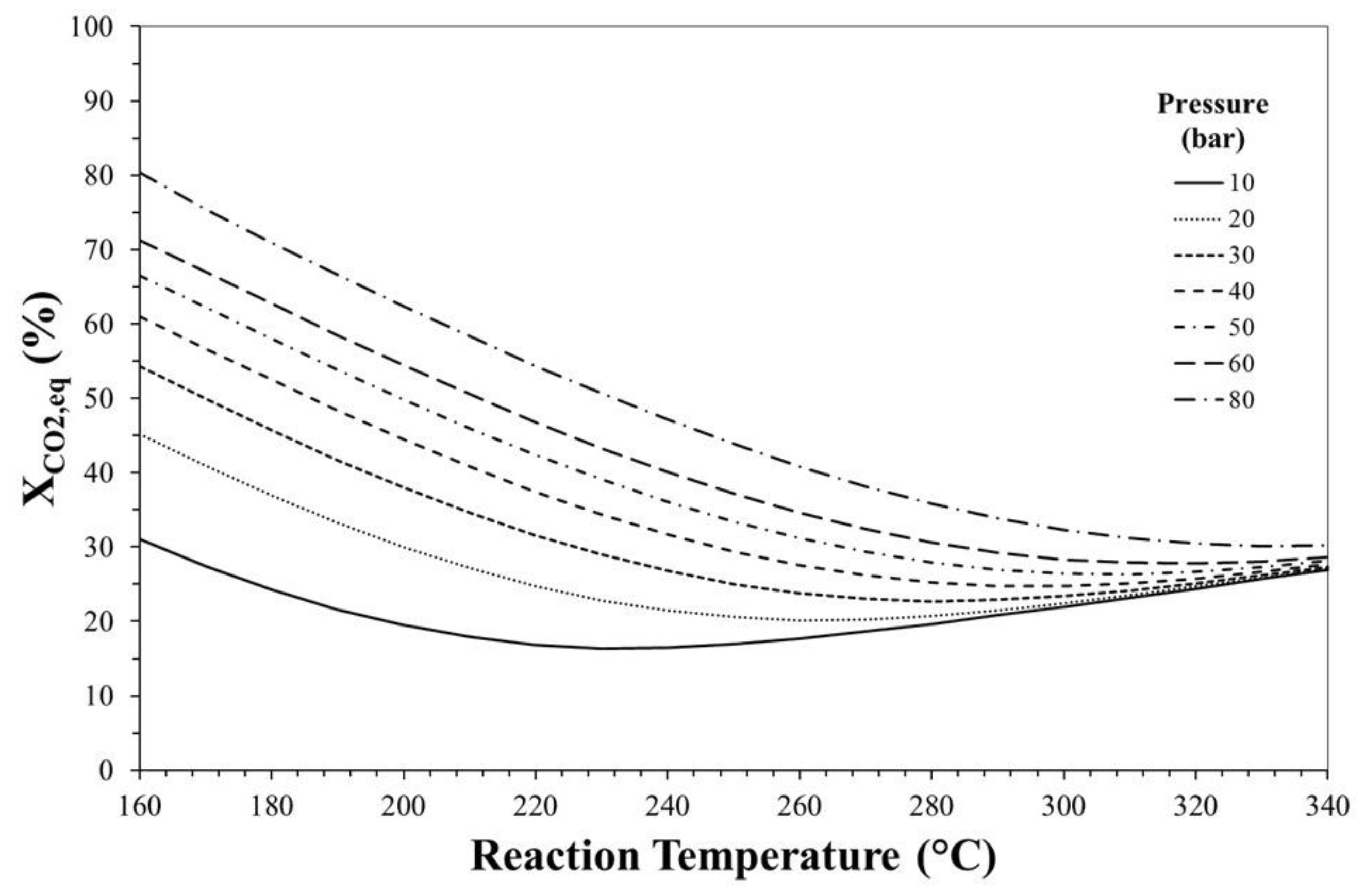
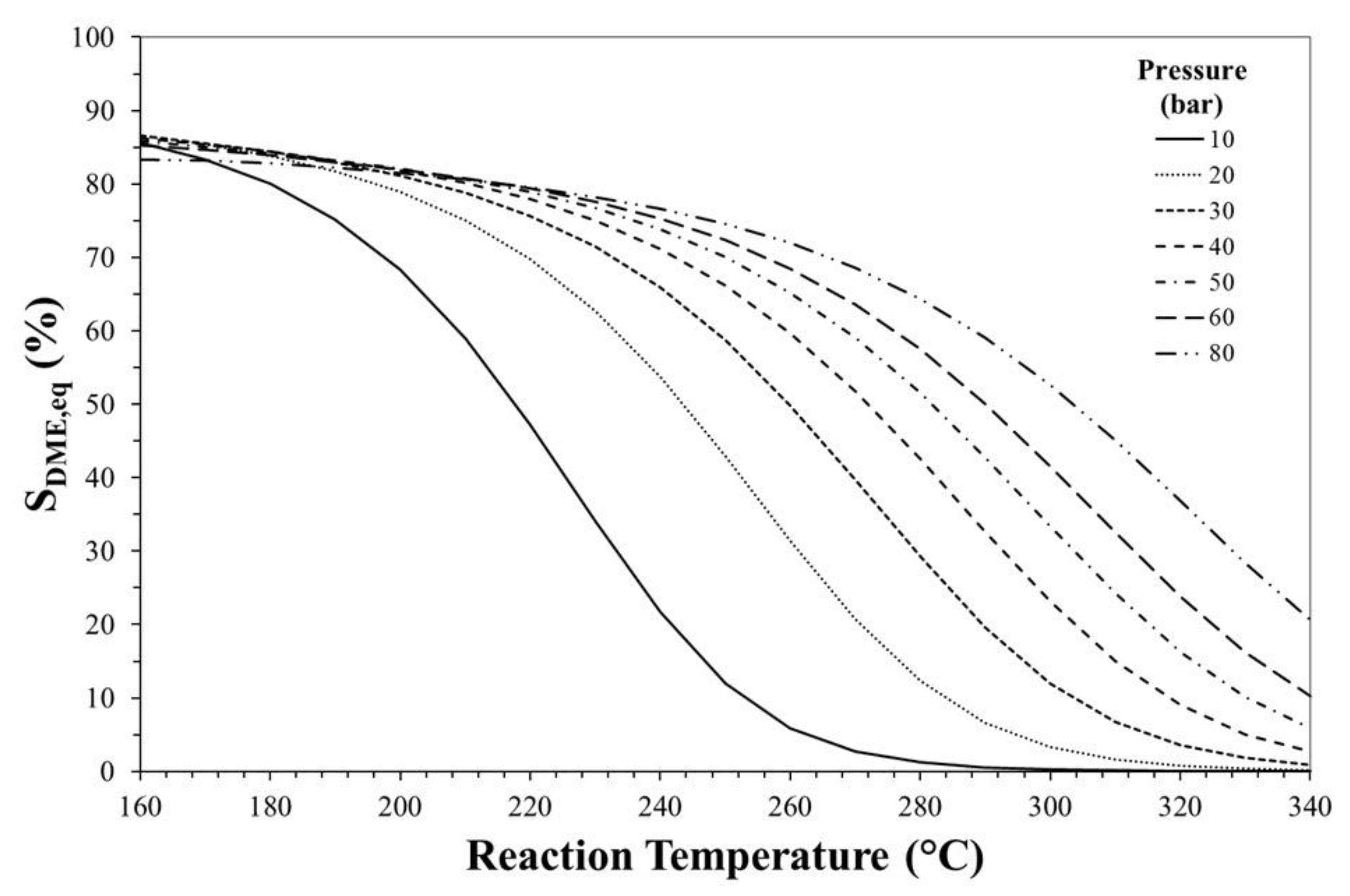
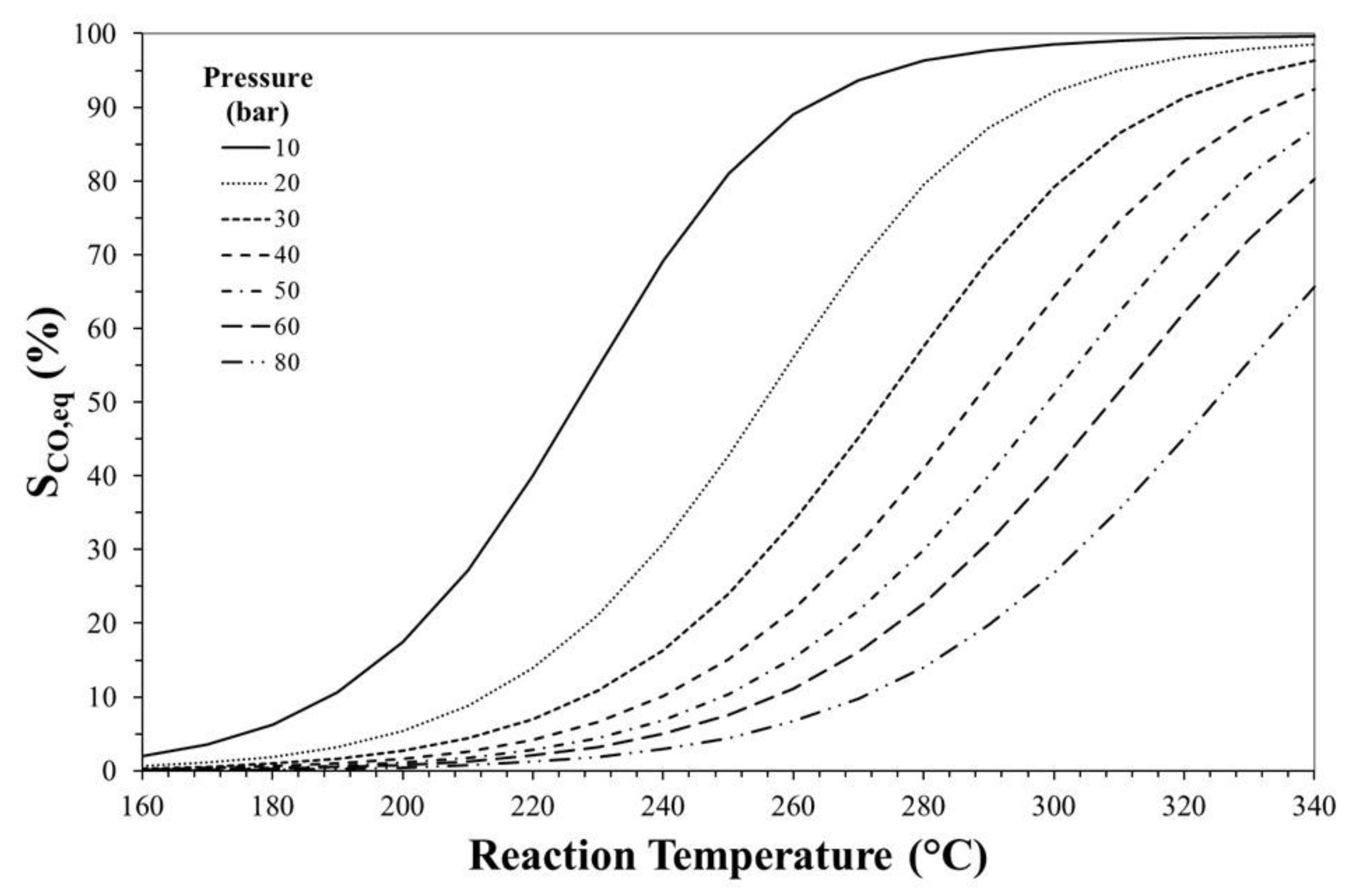
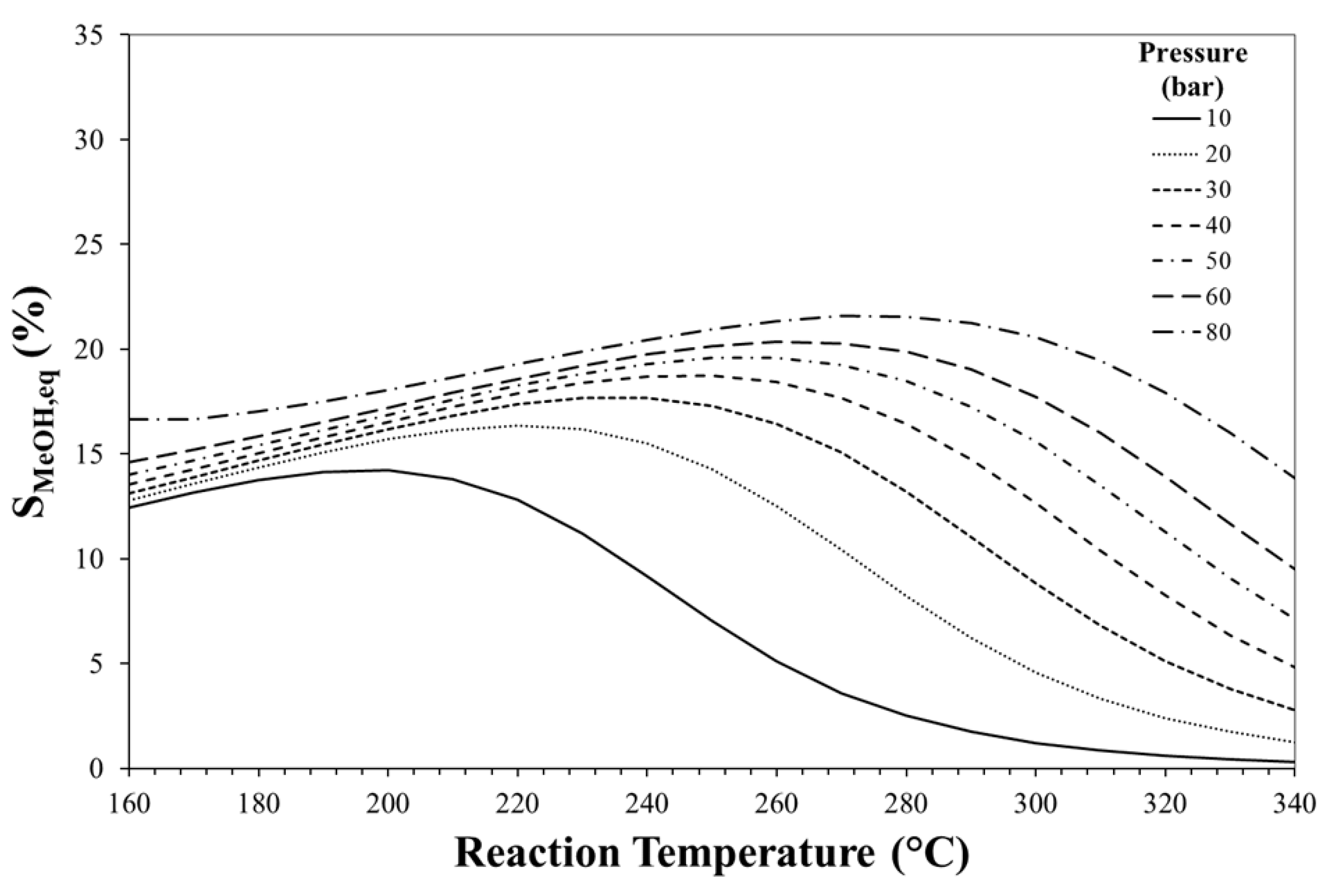
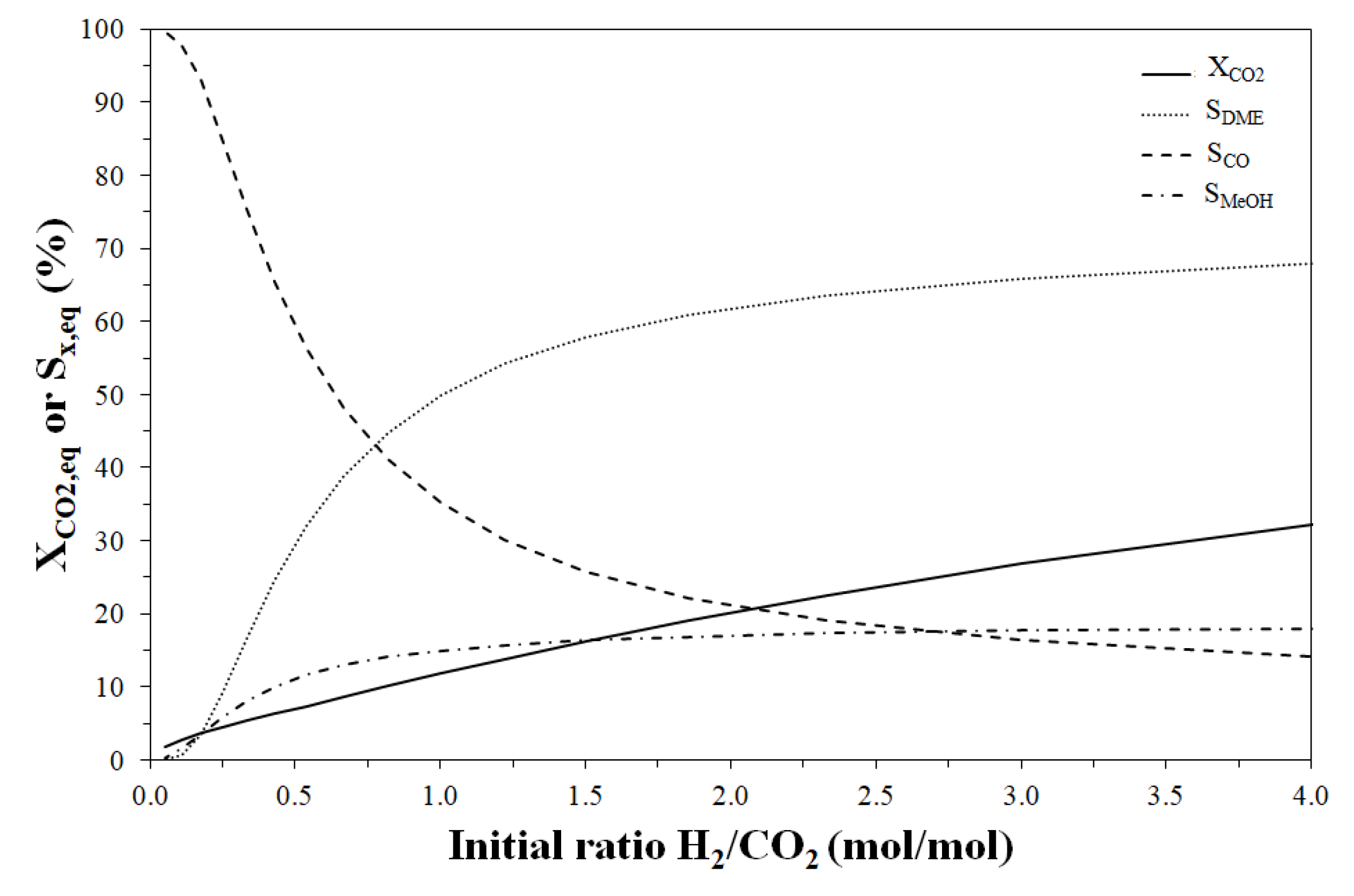
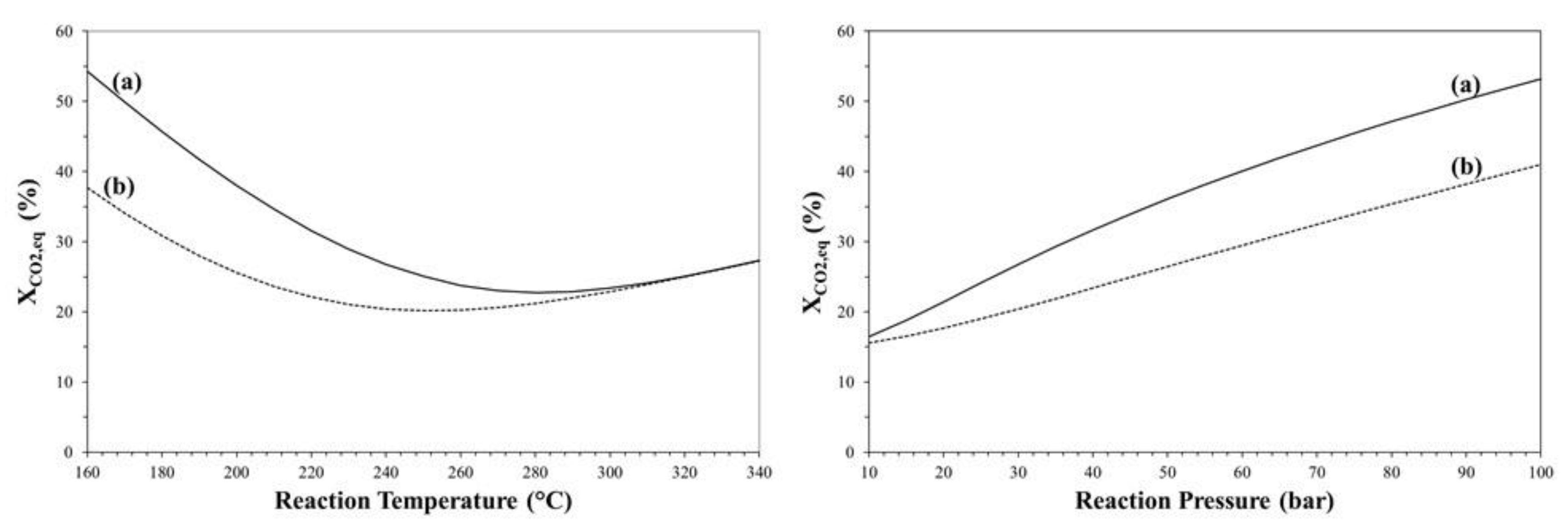
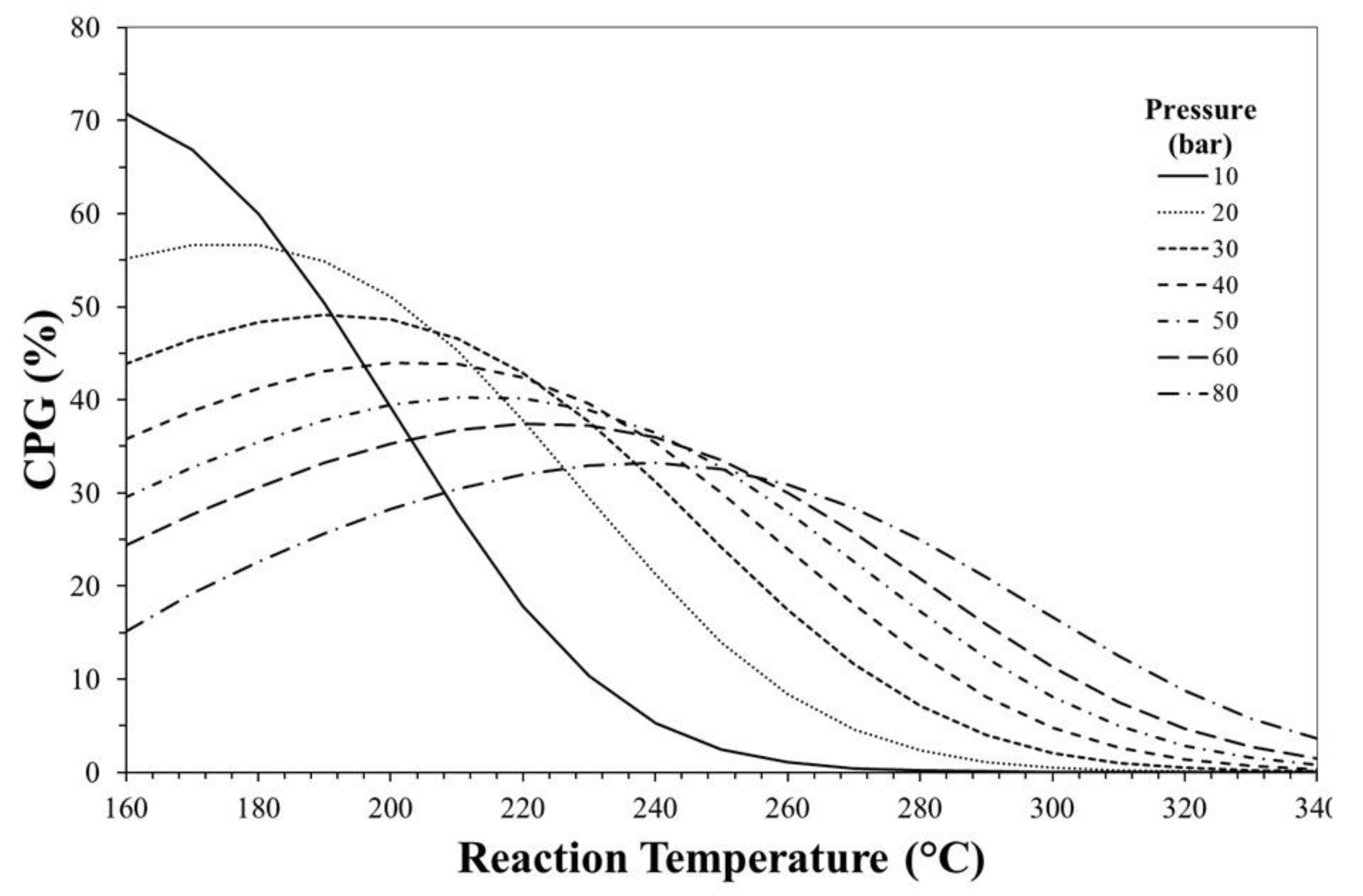
3. Thermodynamic Considerations on CO2-to-DME Process
The Effect of Methanol Dehydration Reaction Step on Thermodynamics
4. Catalytic Systems for DME Production
4.1. Catalysts for CO2-to-MeOH Step
4.2. Catalysts for MeOH-to-DME Step
Methanol Dehydration over Zeolites
4.3. Catalysts for One-Pot CO2 Hydrogenation to DME
5. Conclusions and Perspectives on the Catalyst Development
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, P.; Wang, M. Carbon dioxide emissions allocation: A review. Ecol. Econ 2016, 125, 47–59. [Google Scholar] [CrossRef]
- De Conik, H.; Benson, S.M. Carbon dioxide capture and storage: Issues and prospects. Annu. Rev. Environ. Resour. 2014, 39, 243–270. [Google Scholar] [CrossRef]
- Chianese, D.S.; Rotz, C.A.; Richard, T.L. Simulation of carbon dioxide emission from dairy farms to assess greenhouse gas reduction strategies. Trans. ASABE 2009, 52, 1301–1312. [Google Scholar] [CrossRef]
- Tseng, S.-C.; Hung, S.-W. A strategic decision-making model considering the social costs of carbon dioxide emissions for sustainable supply chain management. J. Environ. Manag. 2014, 133, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A. Beyond oil and gas: The methanol economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Arcoumanis, C.; Bae, C.; Crookes, R.; Kinoshita, E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 2008, 87, 1014–1030. [Google Scholar] [CrossRef]
- Ogawa, T.; Inoue, N.; Shikada, T.; Ohno, Y. Direct dimethyl ether synthesis. J. Nat. Gas Chem. 2003, 12, 219–227. [Google Scholar]
- Cheng, C.; Zhang, H.; Ying, W.; Fang, D. Intrinsic kinetics of one-step dimethyl ether synthesis from hydrogen-rich synthesis gas over bi-functional catalyst. Korean J. Chem. Eng. 2011, 28, 1511–1517. [Google Scholar] [CrossRef]
- Trippe, F.; Frohling, M.; Schultmann, F.; Stahl, R.; Henrich, E.; Dalai, A. Comprehensive techno-economic assessment of dimethyl ether (DME) synthesis and Fischer–Tropsch synthesis as alternative process steps within biomass-to-liquid production. Fuel Process. Technol. 2013, 106, 577–586. [Google Scholar] [CrossRef]
- Basu, A.; Fleisch, T.H.; McCarthy, C.I.; Udovich, C.A. Process and Fuel for Spark Ignition Engines. U.S. Patent 5,632,786, 27 May 1997. [Google Scholar]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Tamagnini, P.; Axelsson, R.; Lindberg, P.; Oxelfelt, F.; Wunschiers, R.; Lindblad, P. Renewable hydrogen production. Microbiol. Mol. Biol. Rev. 2006, 66, 1–20. [Google Scholar] [CrossRef]
- Chaubey, R.; Sahu, S.; James, O.O.; Maity, S. A review on development of industrial processes and emerging techniques for production of hydrogen from renewable and sustainable sources. Renew. Sustain. Energy Rev. 2013, 23, 443–462. [Google Scholar] [CrossRef]
- Parthasarathy, P.; Narayanan, K.S. Hydrogen production from steam gasification of biomass: Influence of process parameters on hydrogen yield—A review. Renew. Energy 2014, 66, 570–579. [Google Scholar] [CrossRef]
- Kalinci, Y.; Hepbasli, A.; Dincer, I. Biomass-based hydrogen production: A review and analysis. Int. J. Hydrog. Energy 2009, 34, 8799–8817. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.H.; Leung, D.Y.C.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Energy Mater. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef] [PubMed]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.S. Combustion performance and emission reduction characteristics of automotive DME engine system. Prog. Energy Combust. Sci. 2013, 39, 147–168. [Google Scholar] [CrossRef]
- Sidhu, S.; Graham, J.; Striebich, R. Semi-volatile and particulate emissions from the combustion of alternative diesel fuels. Chemosphere 2001, 42, 681–690. [Google Scholar] [CrossRef]
- Kim, M.Y.; Yoon, S.H.; Ryu, B.W.; Lee, C.S. Combustion and emission characteristics of DME as an alternative fuel for compression ignition engines with a high pressure injection system. Fuel 2008, 87, 2779–2786. [Google Scholar] [CrossRef]
- Egnell, R. Comparison of Heat Release and NOx Formation in a DI Diesel Engine Running on DME and Diesel Fuel; SAE Technical Paper 2001-01-0651; SAE 2001 World Congress: Detroit, MI, USA, 2001. [Google Scholar] [CrossRef]
- Teng, H.; McCandless, J.C.; Schneyer, J.B. Thermochemical Characteristics of Dimethyl Ether—An Alternative Fuel for Compression-Ignition Engines; SAE Technical Paper 2001-01-0154; SAE 2001 World Congress: Detroit, MI, USA, 2001. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Basu, A.; Gradassi, M.J.; Massin, J.G. Dimethyl ether: A fuel for the 21st century. Stud. Surf. Sci. Catal. 1997, 107, 117–125. [Google Scholar] [CrossRef]
- Gupta, K.K.; Rehman, A.; Sarviya, R.M. Bio-fuels for the gas turbine: A review. Renew. Sustain. Energy Rev. 2010, 14, 2946–2955. [Google Scholar] [CrossRef]
- Lee, M.C.; Seo, S.B.; Chung, J.H.; Joo, Y.J.; Ahn, D.H. Combustion performance test of a new fuel DME to adapt to a gas turbine for power generation. Fuel 2008, 87, 2162–2167. [Google Scholar] [CrossRef]
- Lee, M.C.; Seo, S.B.; Chung, J.H.; Joo, Y.J.; Ahn, D.H. Industrial gas turbine combustion performance test of DME to use as an alternative fuel for power generation. Fuel 2009, 88, 657–662. [Google Scholar] [CrossRef]
- Lee, M.C.; Yoon, Y. Development of a gas turbine fuel nozzle for DME and a design method thereof. Fuel 2012, 102, 823–830. [Google Scholar] [CrossRef]
- Haryanto, A.; Fernando, S.; Murali, N.; Adhikari, S. Current status of hydrogen production techniques by steam reforming of ethanol: A review. Energy Fuels 2005, 19, 2098–2106. [Google Scholar] [CrossRef]
- Ghenciu, A.F. Review of fuel processing catalysts for hydrogen production in PEM fuel cell systems. Curr. Opin. Solid State Mater. Sci. 2002, 6, 389–399. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol steam reforming for hydrogen production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, K.; Suzuki, H. Steam reforming of dimethyl ether. Appl. Catal. A 2004, 260, 111–117. [Google Scholar] [CrossRef]
- Vicente, J.; Gayubo, A.G.; Ereña, J.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Improving the DME steam reforming catalyst by alkaline treatment of the HZSM-5 zeolite. Appl. Catal. B 2013, 130–131, 73–83. [Google Scholar] [CrossRef]
- Gazsi, A.; Ugrai, I.; Solymosy, F. Production of hydrogen from dimethyl ether on supported Au catalysts. Appl. Catal. A 2011, 391, 360–366. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Ott, K.C.; Borup, R.L.; Greene, H.L. Generating hydrogen-rich fuel-cell feeds from dimethyl ether (DME) using physical mixtures of a commercial Cu/Zn/Al2O3 catalyst and several solid–acid catalysts. Appl. Catal. B 2006, 65, 291–300. [Google Scholar] [CrossRef]
- Jeong, J.W.; Ahn, C.-I.; Lee, D.H.; Um, S.H.; Bae, J.W. Effects of Cu–ZnO Content on Reaction Rate for Direct Synthesis of DME from Syngas with Bifunctional Cu–ZnO/γ-Al2O3 Catalyst. Catal. Lett. 2013, 143, 666–672. [Google Scholar] [CrossRef]
- Asthana, S.; Samanta, C.; Bhaumik, A.; Banerjee, B.; Voolapalli, R.K.; Saha, B. Direct synthesis of dimethyl ether from syngas over Cu-based catalysts: Enhanced selectivity in the presence of MgO. J. Catal. 2016, 334, 89–101. [Google Scholar] [CrossRef]
- Wender, I. Reactions of synthesis gas. Fuel Process. Technol. 1996, 48, 189–297. [Google Scholar] [CrossRef]
- Huang, M.H.; Lee, H.M.; Liang, K.C.; Tzeng, C.C.; Chen, W.H. An experimental study on single-step dimethyl ether (DME) synthesis from hydrogen and carbon monoxide under various catalysts. Int. J. Hydrog. Energy 2015, 40, 13583–13593. [Google Scholar] [CrossRef]
- Sousa-Aguiar, E.F.; Appel, L.G.; Mota, C. Natural gas chemical transformations: The path to refining in the future. Catal. Today 2005, 101, 3–7. [Google Scholar] [CrossRef]
- Bae, J.W.; Potdar, H.S.; Kang, S.-H.; Jun, K.-W. Coproduction of methanol and dimethyl ether from biomass-derived syngas on a Cu–ZnO–Al2O3/γ-Al2O3 hybrid catalyst. Energy Fuels 2008, 22, 223–230. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Ereña, J.; Mier, D.; Arandes, J.M.; Olazar, M.; Bilbao, J. Kinetic Modeling of Dimethyl Ether Synthesis in a Single Step on a CuO–ZnO–Al2O3/γ-Al2O3 Catalyst. Ind. Eng. Chem. Res. 2007, 46, 5522–5530. [Google Scholar] [CrossRef]
- Ge, Q.; Huang, Y.; Qiu, F.; Li, S. Bifunctional catalysts for conversion of synthesis gas to dimethyl ether. Appl. Catal. A 1998, 167, 23–30. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Diao, J.; Jin, Y. The synergy effect of process coupling for dimethyl ether synthesis in slurry reactors. Chem. Eng. Technol. 2001, 24, 507–511. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Basu, A.; Sills, R.A. Introduction and advancement of a new clean global fuel: The status of DME developments in China and beyond. J. Nat. Gas Sci. Eng. 2012, 9, 94–107. [Google Scholar] [CrossRef]
- Wambach, J.; Baiker, A.; Wokaum, A. CO2 hydrogenation over metal/zirconia catalysts. Phys. Chem. Chem. Phys. 1999, 1, 5071–5080. [Google Scholar] [CrossRef]
- Sugawa, S.; Sayama, K.; Arakawa, H. Methanol synthesis from CO2 and H2 over silver catalyst. Energy Convers. Manag. 1995, 36, 665–668. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Advances in Catalysts and Processes for Methanol Synthesis from CO2. In CO2: A Valuable Source of Carbon; Springer: London, UK, 2013; pp. 147–169. [Google Scholar]
- Koppel, R.A.; Stocker, C.; Baiker, A. Copper-and silver–zirconia aerogels: Preparation, structural properties and catalytic behavior in methanol synthesis from carbon dioxide. J. Catal. 1998, 179, 515–527. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Mansfield, K.; Spencer, M.S. The methanol synthesis-how does it work. Chemtech 1990, 11, 692–699. [Google Scholar]
- Nakamura, J.; Nakamura, I.; Uchijima, T.; Kanai, Y.; Watanabe, T. Methanol synthesis over a Zn-deposited copper model catalyst. Catal. Lett. 1995, 31, 325–331. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Li, W. In situ IR studies on the mechanism of methanol synthesis from CO/H2 and CO2/H2 over Cu-ZnO-Al2O3 catalyst. Korean J. Chem. Eng. 2010, 27, 1093–1098. [Google Scholar] [CrossRef]
- Samei, E.; Taghizadeh, M.; Bahmani, M. Enhancement of stability and activity of Cu/ZnO/Al2O3 catalysts by colloidal silica and metal oxides additives for methanol synthesis from a CO2-rich feed. Fuel Process. Technol. 2012, 96, 128–133. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Influence of modifier (Mn, La, Ce, Zr and Y) on the performance of Cu/Zn/Al catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. Appl. Catal. A 2013, 468, 442–452. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Kozłowska, A.; Olszewski, P.; Lachowska, M.; Skrzypek, J.; Stoch, J. Effect of Mg and Mn oxide additions on structural and adsorptive properties of Cu/ZnO/ZrO2 catalysts for the methanol synthesis from CO2. Appl. Catal. A 2003, 249, 129–138. [Google Scholar] [CrossRef]
- Fujiwara, M.; Ando, H.; Tanaka, M.; Souma, Y. Hydrogenation of carbon dioxide over Cu–Zn–Cr oxide catalysts. Bull. Chem. Soc. Jpn. 1994, 67, 546–550. [Google Scholar] [CrossRef]
- Pasupulety, N.; Driss, H.; Alhamed, Y.A.; Alzahrani, A.A.; Daous, M.A.; Petrova, L. Studies on Au/Cu–Zn–Al catalyst for methanol synthesis from CO2. Appl. Catal. A 2015, 504, 308–318. [Google Scholar] [CrossRef]
- Arena, F.; Barbera, K.; Italiano, G.; Bonura, G.; Spadaro, L.; Frusteri, F. Synthesis, characterization and activity pattern of Cu–ZnO/ZrO2 catalysts in the hydrogenation of carbon dioxide to methanol. J. Catal. 2007, 249, 185–194. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-state interactions, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH. Appl. Catal. A 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The changing nature of the active site of Cu–Zn–Zr catalysts for the CO2 hydrogenation reaction to methanol. Appl. Catal. B 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. Glycine–nitrate combustion synthesis of CuO–ZnO–ZrO2 catalysts for methanol synthesis from CO2 hydrogenation. J. Catal. 2010, 271, 178–185. [Google Scholar] [CrossRef]
- Donphai, W.; Piriyawate, N.; Witoon, T.; Jantaratana, P.; Varabuntoonvit, V.; Chareonpanich, M. Effect of magnetic field on CO2 conversion over Cu-ZnO/ZrO2 catalyst in hydrogenation reaction. J. CO2 Util. 2016, 16, 204–211. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bonura, G.; Spadaro, L.; Frusteri, F. Basic evidences for methanol-synthesis catalyst design. Catal. Today 2009, 143, 80–85. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhan, H.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Wang, H.; Sun, Y. Influence of Zr on the performance of Cu/Zn/Al/Zr catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. J. Catal. 2013, 298, 51–60. [Google Scholar] [CrossRef]
- Yang, C.; Ma, Z.Y.; Zhao, N.; Wei, W.; Hu, T.D.; Sun, Y.H. Methanol synthesis from CO2-rich syngas over a ZrO2 doped CuZnO catalyst. Catal. Today 2006, 115, 222–227. [Google Scholar] [CrossRef]
- Melián-Cabrera, I.; Granados, L.; Fierro, J.L.G. Pd-modified Cu–Zn catalysts for methanol synthesis from CO2/H2 mixtures: Catalytic structures and performance. J. Catal. 2002, 210, 285–294. [Google Scholar] [CrossRef]
- Sahibzada, M. Pd-promoted Cu/ZnO catalyst systems for methanol synthesis from CO2/H2. Chem. Eng. Res. Des. 2000, 78, 943–946. [Google Scholar] [CrossRef]
- Melian-Cabrera, O.; Granados, M.L.; Terreros, P.; Fierro, J.L.G. CO2 hydrogenation over Pd-modified methanol synthesis catalysts. Catal. Today 1998, 45, 251–256. [Google Scholar] [CrossRef]
- Ban, H.; Li, C.; Asami, K.; Fujimoto, K. Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2. Catal. Commun. 2014, 54, 50–54. [Google Scholar] [CrossRef]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. The influence of La doping on the catalytic behavior of Cu/ZrO2 for methanol synthesis from CO2 hydrogenation. J. Mol. Catal. A 2011, 345, 60–68. [Google Scholar] [CrossRef]
- Batyrev, E.D.; van den Heuvel, J.C.; Beckers, J.; Jansen, W.P.A.; Castricum, H.L. The effect of the reduction temperature on the structure of Cu/ZnO/SiO2 catalysts for methanol synthesis. J. Catal. 2005, 229, 136–143. [Google Scholar] [CrossRef]
- Owen, G.; Hawkes, C.M.; Lloyd, D. Methanol synthesis from intermetallic precursors: Binary lanthanide-copper catalysts. Appl. Catal. 1987, 33, 405–430. [Google Scholar] [CrossRef]
- Bonura, G.; Arena, F.; Mezzatesta, G.; Cannilla, C.; Spadaro, L.; Frusteri, F. Role of the ceria promoter and carrier on the functionality of Cu-based catalysts in the CO2-to-methanol hydrogenation reaction. Catal. Today 2011, 171, 251–256. [Google Scholar] [CrossRef]
- Li, M.; Zeng, Z.; Liao, F.; Hong, X.; Tsang, S.C.E. Enhanced CO2 hydrogenation to methanol over CuZn nanoalloy in Ga modified Cu/ZnO catalysts. J. Catal. 2016, 343, 157–167. [Google Scholar] [CrossRef]
- Toyir, J.; Ramìrez de la Piscina, P.; Homs, N. Ga-promoted copper-based catalysts highly selective for methanol steam reforming to hydrogen; relation with the hydrogenation of CO2 to methanol. Int. J. Hydrogen Energy 2015, 40, 11261–11266. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhang, Y.; Cao, Y.; Zhang, S.; Wang, K.; Ding, F.; Wu, J. V-modified CuO–ZnO–ZrO2/HZSM-5 catalyst for efficient direct synthesis of DME from CO2 hydrogenation. Catal. Commun. 2014, 55, 49–52. [Google Scholar] [CrossRef]
- Zhang, Q.; Zuo, Y.-Z.; Han, M.-H.; Wang, J.-F.; Jin, Y.; Wei, F. Long carbon nanotubes intercrossed Cu/Zn/Al/Zr catalyst for CO/CO2 hydrogenation to methanol/dimethyl ether. Catal. Today 2010, 150, 55–60. [Google Scholar] [CrossRef]
- Deerattrakul, V.; Dittanet, P.; Sawangphruk, M.; Kongkachuichay, P. CO2 hydrogenation to methanol using Cu-Zn catalyst supported on reduced graphene oxide nanosheets. J. CO2 Util. 2016, 16, 104–113. [Google Scholar] [CrossRef]
- Fan, Y.J.; Wu, S.F. A graphene-supported copper-based catalyst for the hydrogenation of carbon dioxide to form methanol. J. CO2 Util. 2016, 16, 150–156. [Google Scholar] [CrossRef]
- Ahmed, N.; Shibata, Y.; Taniguchi, T.; Izumi, Y. Photocatalytic conversion of carbon dioxide into methanol using zinc–copper–M (III)(M = aluminum, gallium) layered double hydroxides. J. Catal. 2011, 279, 123–135. [Google Scholar] [CrossRef]
- Wang, G.; Zuo, Y.; Han, M.; Wang, J. Copper crystallite size and methanol synthesis catalytic property of Cu-based catalysts promoted by Al, Zr and Mn. React. Kinet. Mech. Catal. 2010, 101, 443–454. [Google Scholar] [CrossRef]
- Ladera, R.; Pérez-Alonso, F.J.; González-Carballo, J.M.; Ojeda, M.; Rojas, S.; Fierro, J.L.G. Catalytic valorization of CO2 via methanol synthesis with Ga-promoted Cu–ZnO–ZrO2 catalysts. Appl. Catal. B 2013, 142–143, 241–248. [Google Scholar] [CrossRef]
- Zhan, H.; Li, F.; Xin, C.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. Performance of the La–Mn–Zn–Cu–O Based Perovskite Precursors for Methanol Synthesis from CO2 Hydrogenation. Catal. Lett. 2015, 145, 1177–1185. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Tavan, Y.; Hosseini, S.H.; Ghavipour, M.; Nikou, M.R.K.; Shariati, A. From laboratory experiments to simulation studies of methanol dehydration to produce dimethyl ether—Part I: Reaction kinetic study. Chem. Eng. Process. 2013, 73, 144–150. [Google Scholar] [CrossRef]
- Khaleel, A. Titanium-doped alumina for catalytic dehydration of methanol to dimethyl ether at relatively low temperatures. Fuel 2011, 90, 2422–2427. [Google Scholar] [CrossRef]
- Liu, D.; Yao, C.; Zhang, J.; Fan, D.; Chen, D. Catalytic dehydration of methanol to dimethyl ether over modified γ-Al2O3 catalyst. Fuel 2011, 90, 1738–1742. [Google Scholar] [CrossRef]
- Mollavalli, M.; Yaripour, F.; Mohammadi-Jam, S.; Atashi, H. Relationship between surface acidity and activity of solid-acid catalysts in vapour phase dehydration of methanol. Fuel Process. Technol. 2009, 90, 1093–1098. [Google Scholar] [CrossRef]
- Xia, J.; Mao, D.; Zhang, B.; Chen, Q.; Zhang, Y.; Tang, Y. Catalytic properties of fluorinated alumina for the production of dimethyl ether. Catal. Commun. 2006, 7, 362–366. [Google Scholar] [CrossRef]
- Yaripour, F.; Shariatinia, Z.; Sahebdelfar, S.; Irandoukht, A. The effects of synthesis operation conditions on the properties of modified γ-alumina nanocatalysts in methanol dehydration to dimethyl ether using factorial experimental design. Fuel 2015, 139, 40–50. [Google Scholar] [CrossRef]
- Xu, M.; Lunsford, J.H.; Goodman, D.W.; Bhattacharyya, A. Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts. Appl. Catal. A 1997, 149, 289–301. [Google Scholar] [CrossRef]
- Yaripour, F.; Baghaei, F.; Schmidt, I.; Perregaard, J. Catalytic dehydration of methanol to dimethyl ether (DME) over solid-acid catalysts. Catal. Commun. 2005, 6, 147–152. [Google Scholar] [CrossRef]
- Jun, K.W.; Lee, H.S.; Roh, H.S.; Park, S.E. Highly water-enhanced H-ZSM-5 catalysts for dehydration of methanol to dimethyl ether. Bull. Korean Chem. Soc. 2003, 24, 106–108. [Google Scholar]
- Tao, J.; Jun, K.; Lee, K. Co-production of dimethyl ether and methanol from CO2 hydrogenation: Development of a stable hybrid catalyst. Appl. Organomet. Chem. 2001, 15, 105–108. [Google Scholar] [CrossRef]
- Alharbi, W.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Dehydration of methanol to dimethyl ether over heteropoly acid catalysts: The relationship between reaction rate and catalyst acid strength. ACS Catal. 2015, 5, 7186–7193. [Google Scholar] [CrossRef]
- Ladera, R.M.; Fierro, J.L.G.; Ojeda, M.; Rojas, S. TiO2-supported heteropoly acids for low-temperature synthesis of dimethyl ether from methanol. J. Catal. 2014, 312, 195–203. [Google Scholar] [CrossRef]
- Spivey, J. Review: Dehydration catalysts for the methanol/dimethyl ether reaction. Chem. Eng. Commun. 1991, 110, 123–142. [Google Scholar] [CrossRef]
- Ciftci, A.; Sezgi, N.A.; Dogu, T. Nafion-incorporated silicate structured nanocomposite mesoporous catalysts for dimethyl ether synthesis. Ind. Eng. Chem. Res. 2010, 49, 6753–6762. [Google Scholar] [CrossRef]
- Varisli, D.; Dogu, T. Production of clean transportation duel dimethylether by dehydration of methanol over nafion catalyst. Gazi Univ. J. Sci. 2008, 21, 37–41. [Google Scholar]
- Hosseininejad, S.; Afacan, A.; Hayes, R.E. Catalytic and kinetic study of methanol dehydration to dimethyl ether. Chem. Eng. Res. Des. 2012, 90, 825–833. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q. Solid acid catalysis using ion-exchange resins. Appl. Catal. A 2001, 221, 45–62. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Aloise, A.; Crea, F.; Antonucci, P.L.; Nagy, J.B.; Frusteri, F.; Giordano, G. Catalytic dry-reforming on Ni–zeolite supported catalyst. Catal. Today 2012, 179, 52–60. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Aloise, A.; Antonucci, P.L.; Giordano, G.; Nagy, J.B. Effect of support surface on methane dry-reforming catalyst preparation. Catal. Today 2013, 218, 18–29. [Google Scholar] [CrossRef]
- Frontera, P.; Aloise, A.; Macario, A.; Antonucci, P.L.; Crea, F.; Giordano, G.; Nagy, J.B. Bimetallic zeolite catalyst for CO2 reforming of methane. Top. Catal. 2010, 53, 265–272. [Google Scholar] [CrossRef]
- Macario, A.; Giordano, G. Catalytic conversion of renewable sources for biodiesel production: A comparison between biocatalysts and inorganic catalysts. Catal. Lett. 2013, 143, 159–168. [Google Scholar] [CrossRef]
- Macario, A.; Verri, F.; Diaz, U.; Corma, A.; Giordano, G. Pure silica nanoparticles for liposome/lipase system encapsulation: Application in biodiesel production. Catal. Today 2013, 204, 148–155. [Google Scholar] [CrossRef]
- Corma, A. State of the art and future challenges of zeolites as catalysts. J. Catal. 2003, 216, 298–312. [Google Scholar] [CrossRef]
- Vishwanathan, V.; Jun, K.; Kim, J.; Roh, H. Vapour phase dehydration of crude methanol to dimethyl ether over Na-modified H-ZSM-5 catalysts. Appl. Catal. A 2005, 276, 251–255. [Google Scholar] [CrossRef]
- Migliori, M.; Aloise, A.; Catizzone, E.; Giordano, G. Kinetic analysis of methanol to dimethyl ether reaction over H-MFI catalyst. Ind. Eng. Chem. Res. 2014, 53, 14885–14891. [Google Scholar] [CrossRef]
- Andzelm, J.; Goving, N.; Fitzgerald, G.; Maiti, A. DFT study of methanol conversion to hydrocarbons in a zeolite catalyst. Int. J. Quantum Chem. 2003, 91, 467–473. [Google Scholar] [CrossRef]
- Wang, W.; Seiler, M.; Hunger, M. Role of surface methoxy species in the conversion of methanol to dimethyl ether on acidic zeolites investigated by in situ stopped-flow MAS NMR spectroscopy. J. Phys. Chem. B 2001, 105, 12553–12558. [Google Scholar] [CrossRef]
- Migliori, M.; Aloise, A.; Giordano, G. Methanol to dimethylether on H-MFI catalyst: The influence of the Si/Al ratio on kinetic parameters. Catal. Today 2014, 227, 138–143. [Google Scholar] [CrossRef]
- Tajima, N.; Tsuneda, T.; Toyama, F.; Hirao, K. A new mechanism for the first carbon–carbon bond formation in the MTG process: A theoretical study. J. Am. Chem. Soc. 1998, 120, 8222–8229. [Google Scholar] [CrossRef]
- Teketel, S.; Olsbye, U.; Lillerud, K.P.; Beato, P.; Svelle, S. Co-conversion of methanol and light alkenes over acidic zeolite catalyst H-ZSM-22: Simulated recycle of non-gasoline range products. Appl. Catal. A 2015, 494, 68–76. [Google Scholar] [CrossRef]
- Li, J.; Wei, Z.; Chen, Y.; Jing, B.; He, Y.; Dong, M.; Jiao, H.; Li, X.; Qin, Z.; Wang, J.; Fan, W. A route to form initial hydrocarbon pool species in methanol conversion to olefins over zeolites. J. Catal. 2014, 317, 277–283. [Google Scholar] [CrossRef]
- Qi, L.; Wei, Y.; Xu, L.; Liu, Z. Reaction behaviors and kinetics during induction period of methanol conversion on HZSM-5 zeolite. ACS Catal. 2015, 5, 3973–3982. [Google Scholar] [CrossRef]
- Svelle, S.; Joensen, F.; Nerlov, J.; Olsbye, U.; Lillerud, K.P.; Kolboe, S.; Bjørgen, M. Conversion of methanol into hydrocarbons over zeolite H-ZSM-5: Ethene formation is mechanistically separated from the formation of higher alkenes. J. Am. Chem. Soc. 2006, 128, 14770–14771. [Google Scholar] [CrossRef] [PubMed]
- Campelo, J.M.; Lafont, F.; Marinas, J.M.; Ojeda, M. Studies of catalyst deactivation in methanol conversion with high, medium and small pore silicoaluminophosphates. Appl. Catal. A 2000, 192, 85–96. [Google Scholar] [CrossRef]
- Palumbo, L.; Bonino, F.; Beato, P.; Bjørgen, M.; Zecchina, A.; Bordiga, S. Conversion of methanol to hydrocarbons: Spectroscopic characterization of carbonaceous species formed over H-ZSM-5. J. Phys. Chem. C 2008, 112, 9710–9716. [Google Scholar] [CrossRef]
- Chua, Y.T.; Stair, P.C. An ultraviolet Raman spectroscopic study of coke formation in methanol to hydrocarbons conversion over zeolite H-MFI. J. Catal. 2003, 213, 39–46. [Google Scholar] [CrossRef]
- Mentzel, U.V.; Hojholt, K.; Holm, M.S.; Fehrmann, R.; Beato, P. Conversion of methanol to hydrocarbons over conventional and mesoporous H-ZSM-5 and H-Ga-MFI: Major differences in deactivation behavior. Appl. Catal. A 2012, 417–418, 290–297. [Google Scholar] [CrossRef]
- Nushyama, N.; Kawaguchi, M.; Hirota, Y.; Van Vu, D.; Egashira, Y.; Ueyama, K. Size control of SAPO-34 crystals and their catalyst lifetime in the methanol-to-olefin reaction. Appl. Catal. A Gen. 2009, 362, 193–199. [Google Scholar] [CrossRef]
- Deimund, M.A.; Schmidt, J.E.; Davis, M.E. Effect of pore and cage size on the formation of aromatic intermediates during the methanol-to-olefins reaction. Top. Catal. 2015, 58, 416–423. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. From 1-D to 3-D zeolite structures: Performance assessment in catalysis of vapour-phase methanol dehydration to DME. Microporous Mesoporous Mater. 2017, 243, 102–111. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. Dimethyl ether synthesis via methanol dehydration: Effect of zeolite structure. Appl. Catal. A 2015, 502, 215–220. [Google Scholar] [CrossRef]
- Khandan, N.; Kazemeini, M.; Aghaziarati, M. Determining an optimum catalyst for liquid-phase dehydration of methanol to dimethyl ether. Appl. Catal. A 2008, 349, 6–12. [Google Scholar] [CrossRef]
- Tang, Q.; Xu, H.; Zheng, Y.; Wang, J.; Li, H.; Zhang, J. Catalytic dehydration of methanol to dimethyl ether over micro–mesoporous ZSM-5/MCM-41 composite molecular sieves. Appl. Catal. A 2012, 413–414, 36–42. [Google Scholar] [CrossRef]
- Rutkowska, M.; Macina, D.; Piwowarska, Z.; Gajewska, M.; Diaz, U.; Chmielarz, L. Hierarchically structured ZSM-5 obtained by optimized mesotemplate-free method as active catalyst for methanol to DME conversion. Catal. Sci. Technol. 2016, 6, 4849–4862. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.-C.; Lee, Y.-J.; Jun, K.-W.; Hong, S.B. Influence of catalytic functionalities of zeolites on product selectivities in methanol conversion. Energy Fuels 2009, 23, 593–598. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. The effect of FER zeolite acid sites in methanol-to-dimethyl-ether catalytic dehydration. J. Energy Chem. 2017, 26, 406–415. [Google Scholar] [CrossRef]
- Kim, S.D.; Baek, S.C.; Lee, Y.; Jun, K.; Kim, M.J.; Yoo, I.S. Effect of γ-alumina content on catalytic performance of modified ZSM-5 for dehydration of crude methanol to dimethyl ether. Appl. Catal. A 2006, 309, 139–143. [Google Scholar] [CrossRef]
- Hassanpour, S.; Yaripour, F.; Taghizadeh, M. Performance of modified H-ZSM-5 zeolite for dehydration of methanol to dimethyl ether. Fuel Process. Technol. 2010, 91, 1212–1221. [Google Scholar] [CrossRef]
- Khandan, N.; Kazemeini, M.; Aghaziarati, M. Dehydration of methanol to dimethyl ether employing modified H-ZSM-5 catalysts. Iran. J. Chem. Eng. 2009, 6, 3–11. [Google Scholar]
- Hassanpour, S.; Taghizadeh, M.; Yaripour, F. Preparation, characterization, and activity evaluation of H-ZSM-5 catalysts in vapor-phase methanol dehydration to dimethyl ether. Ind. Eng. Chem. Res. 2010, 49, 4063–4069. [Google Scholar] [CrossRef]
- Amaroli, T.; Simon, L.J.; Digne, M.; Montanari, T.; Bevilacqua, M.; Valtchev, V.; Patarin, J.; Busca, G. Effect of crystal size and Si/Al ratio on the surface properties of H-ZSM-5 zeolites. Appl. Catal. A 2006, 306, 78–84. [Google Scholar] [CrossRef]
- García-Trenco, A.; Martínez, A. Direct synthesis of DME from syngas on hybrid CuZnAl/ZSM-5 catalysts: New insights into the role of zeolite acidity. Appl. Catal. A 2012, 411–412, 170–179. [Google Scholar] [CrossRef]
- Frusteri, F.; Cordaro, M.; Cannilla, C.; Bonura, G. Multifunctionality of Cu–ZnO–ZrO2/H-ZSM5 catalysts for the one-step CO2-to-DME hydrogenation reaction. Appl. Catal. B 2015, 162, 57–65. [Google Scholar] [CrossRef]
- Wang, D.; Han, Y.; Tan, Y.; Tsubaki, N. Effect of H2O on Cu-based catalyst in one-step slurry phase dimethyl ether synthesis. Fuel Process. Technol. 2009, 90, 446–451. [Google Scholar] [CrossRef]
- Diban, N.; Urtiaga, A.M.; Ortiz, I.; Ereña, J.; Bilbao, J.; Aguayo, A.T. Influence of the membrane properties on the catalytic production of dimethyl ether with in situ water removal for the successful capture of CO2. Chem. Eng. J. 2013, 234, 140–148. [Google Scholar] [CrossRef]
- Iliuta, I.; Larachi, F.; Fongarland, P. Dimethyl ether synthesis with in situ H2O removal in fixed-bed membrane reactor: Model and simulations. Ind. Eng. Chem. Res. 2010, 49, 6870–6877. [Google Scholar] [CrossRef]
- Haw, J.F.; Song, W.; Marcus, D.M.; Nicholas, J.B. The mechanism of methanol to hydrocarbon catalysis. Acc. Chem. Res. 2003, 36, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Stich, I.; Gale, J.D.; Terakura, K.; Payne, M.C. Role of the zeolitic environment in catalytic activation of methanol. J. Am. Chem. Soc. 1999, 121, 3292–3302. [Google Scholar] [CrossRef]
- Ivanova, S.; Lebrun, C.; Vanhaecke, E.; Pham-Huu, C.; Louis, B. Influence of the zeolite synthesis route on its catalytic properties in the methanol to olefin reaction. J. Catal. 2009, 265, 1–7. [Google Scholar] [CrossRef]
- Naik, S.P.; Ryu, T.; Bui, V.; Miller, J.D.; Drinnan, N.B.; Zmierczak, W. Synthesis of DME from CO2/H2 gas mixture. Chem. Eng. J. 2011, 167, 362–368. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, J.; Zhang, J. Effects of ZrO2 on the performance of CuO-ZnO-Al2O3/HZSM-5 catalyst for dimethyl ether synthesis from CO2 hydrogenation. J. Nat. Gas Chem. 2007, 16, 389–392. [Google Scholar] [CrossRef]
- Park, Y.; Baek, S.; Ihm, S. CO2 hydrogenation over copper-based hybrid catalysts for the synthesis of oxygenates. Fuel Chem. Div. Prepr. 2002, 47, 293–294. [Google Scholar]
- Ereña, J.; Garona, R.; Arandes, J.M.; Aguayo, A.T.; Bilbao, J. Effect of operating conditions on the synthesis of dimethyl ether over a CuO-ZnO-Al2O3/NaHZSM-5 bifunctional catalyst. Catal. Today 2005, 107–108, 467–473. [Google Scholar] [CrossRef]
- Wang, S.; Mao, D.; Guo, X.; Wu, X.; Lu, G. Dimethyl ether synthesis via CO2 hydrogenation over CuO–TiO2–ZrO2/HZSM-5 bifunctional catalysts. Catal. Commun. 2009, 10, 1367–1370. [Google Scholar] [CrossRef]
- Gao, W.; Wang, H.; Wang, Y.; Guo, W.; Jia, M. Dimethyl ether synthesis from CO2 hydrogenation on La-modified CuO-ZnO-Al2O3/HZSM-5 bifunctional catalysts. J. Rare Earths 2013, 31, 470–476. [Google Scholar] [CrossRef]
- Qi, G.X.; Fei, J.H.; Zheng, X.M.; Hou, Z.Y. DME synthesis from carbon dioxide and hydrogen over Cu–Mo/HZSM-5. Catal. Lett. 2001, 72, 121–124. [Google Scholar] [CrossRef]
- Sun, K.; Lu, W.; Wang, M.; Xu, X. Low-temperature synthesis of DME from CO2/H2 over Pd-modified CuO–ZnO–Al2O3–ZrO2/HZSM-5 catalysts. Catal. Commun. 2004, 5, 367–370. [Google Scholar] [CrossRef]
- Zha, F.; Tian, H.; Yan, J.; Chang, Y. Multi-walled carbon nanotubes as catalyst promoter for dimethyl ether synthesis from CO2 hydrogenation. Appl. Surf. Sci. 2013, 285, 945–951. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Mezzapica, A.; Frusteri, F. DME production by CO2 hydrogenation: Key factors affecting the behaviour of CuZnZr/ferrierite catalysts. Catal. Today 2017, 281, 337–344. [Google Scholar] [CrossRef]
- Ereña, J.; Garoña, R.; Arandes, J.M.; Aguayo, A.T.; Bilbao, J. Direct synthesis of dimethyl ether from (H2+ CO) and (H2+ CO2) feeds. Effect of feed composition. Int. J. Chem. React. Eng. 2005, 3. [Google Scholar] [CrossRef]
- Zha, F.; Ding, J.; Chang, Y.; Ding, J.; Wang, J.; Ma, J. Cu–Zn–Al Oxide Cores Packed by Metal-Doped Amorphous Silica–Alumina Membrane for Catalyzing the Hydrogenation of Carbon Dioxide to Dimethyl Ether. Ind. Eng. Chem. Res. 2012, 51, 345–352. [Google Scholar] [CrossRef]
- Liu, R.; Qin, Z.; Ji, H.; Su, T. Synthesis of dimethyl ether from CO2 and H2 using a Cu–Fe–Zr/HZSM-5 catalyst system. Ind. Eng. Chem. Res. 2013, 52, 16648–16655. [Google Scholar] [CrossRef]
- Frusteri, F.; Migliori, M.; Cannilla, C.; Frusteri, L.; Catizzone, E.; Aloise, A.; Giordano, G.; Bonura, G. Direct CO2-to-DME hydrogenation reaction: New evidences of a superior behaviour of FER-based hybrid systems to obtain high DME yield. J. CO2 Util. 2017, 18, 353–361. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S. Effect of irradiation power and time on ultrasound assisted co-precipitation of nanostructured CuO–ZnO–Al2O3 over HZSM-5 used for direct conversion of syngas to DME as a green fuel. Energy Convers. Manag. 2014, 83, 212–222. [Google Scholar] [CrossRef]
- Bonura, G.; Frusteri, F.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Catalytic features of CuZnZr–zeolite hybrid systems for the direct CO2-to-DME hydrogenation reaction. Catal. Today 2016, 277, 48–54. [Google Scholar] [CrossRef]
- Frusteri, F.; Bonura, G.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation. Appl. Catal. B 2015, 176, 522–531. [Google Scholar] [CrossRef]
- Bansode, A.; Urakawa, A. Towards full one-pass conversion of carbon dioxide to methanol and methanol-derived products. J. Catal. 2014, 309, 66–70. [Google Scholar] [CrossRef]
- Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Effect of precipitants during the preparation of Cu-ZnO-Al2O3/Zr-ferrierite catalyst on the DME synthesis from syngas. J. Ind. Eng. Chem. 2009, 15, 566–572. [Google Scholar] [CrossRef]
- Yang, G.; Tsubaki, N.; Shamoto, J.; Yoneyama, Y.; Zhang, Y. Confinement effect and synergistic function of H-ZSM-5/Cu-ZnO-Al2O3 capsule catalyst for one-step controlled synthesis. J. Am. Chem. Soc. 2010, 132, 8129–8136. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Thongkam, M.; Vitidsant, T.; Yoneyama, Y.; Tan, Y.; Tsubaki, N. A double-shell capsule catalyst with core–shell-like structure for one-step exactly controlled synthesis of dimethyl ether from CO2 containing syngas. Catal. Today 2011, 171, 229–235. [Google Scholar] [CrossRef]
- Nie, R.; Lei, H.; Pan, S.; Wang, L.; Fei, J.; Hou, Z. Core–shell structured CuO–ZnO@H-ZSM-5 catalysts for CO hydrogenation to dimethyl ether. Fuel 2012, 96, 419–425. [Google Scholar] [CrossRef]
- Sun, K.; Lu, W.; Qiu, F.; Liu, S.; Xu, X. Direct synthesis of DME over bifunctional catalyst: Surface properties and catalytic performance. Appl. Catal. A 2003, 252, 243–249. [Google Scholar] [CrossRef]
- García-Trenco, A.; Vidal-Moya, A.; Martínez, A. Study of the interaction between components in hybrid CuZnAl/HZSM-5 catalysts and its impact in the syngas-to-DME reaction. Catal. Today 2012, 179, 43–51. [Google Scholar] [CrossRef]
- Jin, D.; Zhu, B.; Hou, Z.; Fei, J.; Lou, H.; Zheng, X. Dimethyl ether synthesis via methanol and syngas over rare earth metals modified zeolite Y and dual Cu–Mn–Zn catalysts. Fuel 2007, 86, 2707–2713. [Google Scholar] [CrossRef]
- Stiefel, M.; Ahmad, R.; Amold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466–1474. [Google Scholar] [CrossRef]
- Chen, H.-J.; Fan, C.-W.; Yu, C.-S. Analysis, synthesis, and design of a one-step dimethyl ether production via a thermodynamic approach. Appl. Energy 2013, 101, 449–456. [Google Scholar] [CrossRef]
- Moradi, G.R.; Yaripour, F.; Vale-Sheyda, P. Catalytic dehydration of methanol to dimethyl ether over mordenite catalysts. Fuel Process. Technol. 2010, 91, 461–468. [Google Scholar] [CrossRef]
- Chen, W.-H.; Lin, B.-J.; Lee, H.-M.; Huang, M.-H. One-step synthesis of dimethyl ether from the gas mixture containing CO2 with high space velocity. Appl. Energy 2012, 98, 92–101. [Google Scholar] [CrossRef]
- Rownaghi, A.; Rezaei, F.; Stante, M.; Hedlund, J. Selective dehydration of methanol to dimethyl ether on ZSM-5 nanocrystals. Appl. Catal. B 2012, 119, 56–61. [Google Scholar] [CrossRef]
- Raoof, F.; Taghizadeh, M.; Eliassi, A.; Yaripour, F. Effects of temperature and feed composition on catalytic dehydration of methanol to dimethyl ether over γ-alumina. Fuel 2008, 87, 2967–2971. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Unit | DME | Diesel |
|---|---|---|---|
| Carbon content | mass% | 52.2 | 86 |
| Hydrogen content | mass% | 1–3 | 14 |
| Oxygen content | mass% | 34.8 | 0 |
| Carbon-to-hydrogen ratio | - | 0.337 | 0.516 |
| Liquid density | kg/m3 | 667 | 831 |
| Cetane number | - | >55 | 40–50 |
| Autoignition temperature | K | 508 | 523 |
| Stoichiometric air/fuel mass ratio | - | 9.6 | 14.6 |
| Normal boiling point | K | 248.1 | 450–643 |
| Enthalpy of vaporization | kJ/kg | 467.1 | 300 |
| Lower heating value | MJ/kg | 27.6 | 42.5 |
| Ignition limits | vol% in air | 3.4/18.6 | 0.6/6.5 |
| Elastic Modulus | N/m2 | 6.37 × 108 | 14.86 × 108 |
| Liquid kinematic viscosity | cSt | <0.1 | 3 |
| Surface tension (at 298 K) | N/m | 0.012 | 0.027 |
| Vapour pressure (at 298 K) | kPa | 530 | <10 |
| Reaction No. | Reaction Stoichiometry | |
|---|---|---|
| 1 | CO2 + 3H2 = CH3OH + H2O | −49.5 kJ/molCO2 |
| 2 | 2CH3OH = CH3OCH3 + H2O | −23.4 kJ/molDME |
| 3 | CO2 + H2 = CO + H2O | +41.2 kJ/molCO2 |
| 4 | CO + 2H2 = CH3OH | −90.6 kJ/molCO |
| Catalyst M/ZrO2 | Prep. a | Precursors | BET Surface (m2∙g−1) | Metal Surface (m2∙g−1) | Product Selectivity b | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| CH3OH | CO | CH4 | ||||||
| Cu | impreg. | Copper formate | - | - | - | - | - | 11-13 |
| Cu | impreg. | Copper tetrammine | 107 | 1.8 | 35 | 65 | 0 | 14 |
| Cu | co-prec. | Nitrates | 64 | - | 68 | 32 | 0 | 15,16 |
| Cu | co-prec. | Nitrates | 174 | 7.1 | 66 | 34 | 0 | 14,17 |
| Cu | co-prec. | Chloride/Sulfate | 48.4 | - | 53 | 47 | 0 | 18 |
| Cu | Ho-prec. | Nitrates | 161 | 3.0 | 69 | 31 | 0 | 17 |
| Cu | Prec. | Nitrates | 86 | - | 15 | 57 | 28 | 19 |
| Cu | Sol-gel | Acetate | 215 | - | 40 | 60 | 0 | 20 |
| Cu | alloy | Cu70Zr30 | 47 | 5.0 | 64 | 36 | 0 | 21 |
| HT-Cu | Sol-gel, 2 | Acetate | 128 | 0.8 | 100-55 | 22 | ||
| HT-Cu | Sol-gel, 1 | Acetate | 100 | - | 100-55 | 22 | ||
| HT-Cu | Sol-gel, 1 | HNO3 | 143 | 1.3 | 100-55 | 22 | ||
| LT-Cu | Sol-gel, 1 | HNO3 | 139 | 5.0 | 100-55 | 22 | ||
| Cu/CZ1 | Sol-gel | 253 | - | 52 | 47 | 23 | ||
| Cu/CZ2 | Sol-gel | 268 | 17.8 | 96 | 4 | 23 | ||
| Cu/CZ3 | Sol-gel | 241 | 28.5 | 97 | 3 | 23 | ||
| Cu/CZ4 | Sol-gel | 234 | 31.3 | 97 | 3 | 23 | ||
| Cu/CZ5 | Sol-gel | 225 | 41.2 | 96 | 4 | 23 | ||
| Cu/ZnO | Sol-gel | Acetates | 150 | - | 64 | 36 | 0 | 20 |
| Cu/Zn 0.1 | Ox-co-prec. | Nitrates | 39 | 3.4 | 36/40 | 24 | ||
| Cu/Zn 0.2 | Ox-co-prec. | Nitrates | 36 | 14.9 | 37/46 | 24 | ||
| Cu/Zn 0.3 | Ox-co-prec. | Nitrates | 70 | 12.6 | 38/42 | 24 | ||
| Cu/Zn 0.4 | Ox-co-prec. | Nitrates | 45 | 9.6 | 37/43 | 24 | ||
| Cu/Zn 0.5 | Carb-co-prec. | 33/38 | 24 | |||||
| Cu/V | Prec. | Nitrates | 185 | - | 13 | 67 | 20 | 19 |
| Cu/Ag | Co-prec. | Nitrates | 281 | 4.1 | 81 | 19 | 0 | 25 |
| Ag | Co-prec. | Nitrates | 112 | - | 100 | 0 | 0 | 25 |
| Ag | Impreg. | Nitrates | 125 | - | 70 | 30 | 0 | 26 |
| HT-Ag | Sol-gel, 2 | Acetate | 99 | - | 100-55 | 22 | ||
| HT-Ag | Sol-gel, 1 | Acetate | 77 | - | 100-55 | 22 | ||
| HT-Ag | Sol-gel, 1 | HNO3 | 125 | - | 100-55 | 22 | ||
| LT-Ag | Sol-gel, 1 | HNO3 | 112 | - | 90-48 | 22 | ||
| Au | Co-prec. | HAuCl4/ZrO(NO3)2 | 185 | - | 24 | 76 | 0 | 26,27 |
| Au | alloy | Au25Zr75 | 47 | - | 20 | 74 | 6 | 27 |
| Pt | Impreg. | HPtCl6/chloride | - | - | 5 | 2 | 93 | 28 |
| Pd | alloy | Pd33Zr67 | 17 | 5.6 | 30 | 27 | 43 | 29 |
| Ni | alloy | Ni64Zr36 | 8 | - | 0 | 0 | 100 | 30 |
| Rh | Impreg. | Nitrate/chloride | - | - | 5 | 32 | 63 | 31 |
| Rh | Impreg. | Nitrate/chloride | - | - | 0 | 0 | 100 | 32,33 |
| Ru | Impreg. | Ru(III)AcAc | - | - | 0 | 0 | 100 | 34 |
| Re | Impreg. | - | - | 4.1 | 11 | 58 | 29 | 35 |
| Rh-Mo | Impreg. | Molybdate/chloride | 54 | 0 | 100 | 36 | ||
| Rh-Mo-Li | Impreg. | Molybdate/chloride/nitrate | 47 | 0 | 100 | 36 | ||
| Rh-Mo-K | Impreg. | Molybdate/chloride/nitrate | 51 | 0 | 100 | 36 | ||
| Rh-Mo-Re | Impreg. | Molybdate/chloride/perrhenate | 52 | 0 | 100 | 36 | ||
| Rh-Mo-Co | Impreg. | Molybdate/chloride/nitrate | 53 | 0 | 100 | 36 | ||
| Rh-Mo-Ce | Impreg. | Molybdate/chloride/nitrate | 57 | 0 | 100 | 36 | ||
| Catalyst | Preparation Method | H2/CO2 | GHSV (NmL∙g−1∙h−1) | P;T (MPa; °C) | XCO2 (%) | YCO (%) | YMeOH (%) | YDME (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Cu/Zn/Al | PM | 3 | 3000 | 5;260 | 31 | 2 | 9.3 | 19 | [145] |
| HZSM5 | |||||||||
| Cu/Zn/Al | |||||||||
| γ-Al2O3 | 20 | 11.6 | 8 | 0.4 | |||||
| Cu/Zn/Al/Zr ZSM5 | WM | 3 | 3100 | 3;260 | 24.1 | 7 | 10.6 | 6.4 | [146] |
| Cu/Zn/Zr Ga-Sil1 | CP | 3 | 1200 | 3;250 | 19.0 | 6.4 | 4 | 8.6 | [147] |
| Cu/Ti/Zr ZSM5 | WM | 3 | 1500 | 3;250 | 15.6 | 6.1 | 2.0 | 7.4 | [148] |
| Cu/Zn/Zr/V ZSM5 | CP | 3 | 1500 | 3;270 | 32.5 | 9.1 | 4.3 | 19.1 | [77] |
| Cu/Zn/Al/Zr ZSM5 | PM | 3 | 6000 | 5;270 | 27.5 | - | 5.0 | 16 | [149] |
| Cu/Zn/Al/La ZSM5 | PM | 3 | 3000 | 3;250 | 43.8 | 0.11 | 1.9 | 31.2 | [150] |
| Cu/Mo ZSM5 | IM | 2 | 1500 | 3;240 | 12.4 | 2 | 0.7 | 9.5 | [151] |
| Cu/Zn/Zr/Pd ZSM5 | CP | 3 | 1800 | 3;200 | 18.7 | 2.4 | 2.5 | 13.8 | [152] |
| Cu/Zn/Al ZSM5+CNTs | PM | 3 | 1800 | 3;260 | 46.2 | 8.9 | 16.4 | 21 | [153] |
| Cu/Zn/Zr FER | CP | 3 | 8800 | 5;260 | 23.6 | 9.2 | 3.5 | 10.6 | [154] |
| Cu/Zn/Al | CP | 3 | 750 | 4;275 | 35 | 23 | [155] | ||
| ZSM5 | |||||||||
| Cu/Zn/Al | |||||||||
| γ-Al2O3 | 40 | - | - | 10 | |||||
| Cu/Zn/Al Amorphous silica-alumina | CP | 3 | 1800 | 3;270 | 47.1 | 12.3 | 14.7 | 20.1 | [156] |
| Cu/Fe/Zr ZSM5 | PM | 5 | 1500 | 3;260 | 28.4 | 2.2 | 4.2 | 18.3 | [157] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives. Molecules 2018, 23, 31. https://doi.org/10.3390/molecules23010031
Catizzone E, Bonura G, Migliori M, Frusteri F, Giordano G. CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives. Molecules. 2018; 23(1):31. https://doi.org/10.3390/molecules23010031
Chicago/Turabian StyleCatizzone, Enrico, Giuseppe Bonura, Massimo Migliori, Francesco Frusteri, and Girolamo Giordano. 2018. "CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives" Molecules 23, no. 1: 31. https://doi.org/10.3390/molecules23010031