Palladium-Catalyzed Room Temperature Acylative Cross-Coupling of Activated Amides with Trialkylboranes

Abstract
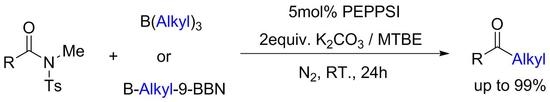
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedure for the PEPPSI-Catalyzed Cross-Coupling of N-Methyl-N-Tosylamides with Trialkylboranes
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Hie, L.; Nathel, N.F.F.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.-F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of amides to esters by the nickel-catalysed activation of amide C-N bonds. Nature 2015, 524, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Sterically controlled Pd-catalyzed chemoselective ketone synthesis via N-C cleavage in twisted amides. Org. Lett. 2015, 17, 4364–4367. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zou, G. Acylative Suzuki coupling of amides: Acyl-nitrogen activation via synergy of independently modifiable activating groups. Chem. Commun. 2015, 51, 5089–5092. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Holzer, W.; Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Structures of highly twisted amides relevant to amide N-C cross-coupling: Evidence for ground-state amide destabilization. Chem. Eur. J. 2016, 22, 14494–14498. [Google Scholar] [CrossRef] [PubMed]
- Osumi, Y.; Liu, C.; Szostak, M. N-Acylsuccinimides: Twist-controlled, acyl-transfer reagents in Suzuki–Miyaura cross-coupling by N–C amide bond activation. Org. Biomol. Chem. 2017, 15, 8867–8871. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Chen, Z.; Liu, T.; Wang, H.; Zeng, Z. N-Acylsuccinimides: Efficient acylative coupling reagents in palladium-catalyzed Suzuki coupling via C-N cleavage. Tetrahedron Lett. 2017, 58, 3819–3822. [Google Scholar] [CrossRef]
- Boit, T.B.; Weires, N.A.; Kim, J.; Garg, N.K. Nickel-catalyzed Suzuki-Miyaura coupling of aliphatic amides. ACS Catal. 2018, 8, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Hie, L.; Baker, E.L.; Anthony, S.M.; Desrosiers, J.-N.; Senanayake, C.; Garg, N.K. Nickel-catalyzed esterification of aliphatic amides. Angew. Chem. Int. Ed. 2016, 55, 15129–15132. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, R.M.; Sheppard, T.D. Recent developments in amide synthesis: Direct amidation of carboxylic acids and transamidation reactions. Eur. J. Org. Chem. 2013, 7453–7465. [Google Scholar] [CrossRef]
- Baker, E.L.; Yamano, M.M.; Zhou, Y.; Anthony, S.M.; Garg, N.K. A two-step approach to achieve secondary amide transamidation enabled by nickel catalysis. Nat. Commun. 2016, 7, 11554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, P.; Meng, G.; Ling, Y.; An, J.; Nolan, S.P.; Szostak, M. General method for the Suzuki–Miyaura cross-coupling of primary amide-derived electrophiles enabled by [Pd(NHC)(cin)Cl] at room temperature. Org. Lett. 2017, 19, 6510–6513. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, M. Nickel-catalyzed diaryl ketone synthesis by N-C cleavage: Direct Negishi cross-coupling of primary amides by site-selective N, N-di-Boc activation. Org. Lett. 2016, 18, 5872–5875. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Site-selective C-H/C-N activation by cooperative catalysis: Primary amides as arylating reagents in directed C–H arylation. ACS Catal. 2017, 7, 7251–7256. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Liu, R.; Lalancette, R.; Szostak, R.; Szostak, M. Palladium-catalyzed Suzuki−Miyaura cross-coupling of N-mesylamides by N-C cleavage: Electronic effect of the mesyl group. Org. Lett. 2017, 19, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, L.; Wang, F.; Zou, G. Highly efficient synthesis of aryl ketones by PEPPSI-palladium catalyzed acylative Suzuki coupling of amides with diarylborinic acids. Tetrahedron Lett. 2018, 59, 2299–2301. [Google Scholar] [CrossRef]
- Liu, C.; Meng, G.; Liu, Y.; Liu, R.; Lalancette, R.; Szostak, R.; Szostak, M. N-Acylsaccharins: Stable electrophilic amide-based acyl transfer reagents in Pd-catalyzed Suzuki-Miyaura coupling via N-C cleavage. Org. Lett. 2016, 18, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, Y.; Cui, M.; Jian, J.; Zeng, Z. Suzuki coupling of amides via palladium-catalyzed C-N cleavage of N-acylsaccharins. Adv. Synth. Catal. 2016, 358, 3876–3880. [Google Scholar] [CrossRef]
- Liu, C.; Meng, G.; Szostak, M. N-Acylsaccharins as amide-based arylating reagents via chemoselective N-C cleavage: Pd-catalyzed decarbonylative Heck reaction. J. Org. Chem. 2016, 81, 12023–12030. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Wu, H.; Jian, J.; Wang, H.; Liu, C.; Daniel, S.; Zeng, Z. Palladium-catalyzed Sonogashira coupling of amides: Access to ynones via C-N bond cleavage. Chem. Commun. 2016, 52, 12076–12079. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, T.; Cui, M.; Li, Y.; Jian, J.; Wang, H.; Zeng, Z. Rhodium-catalyzed C-H functionalization with N-acylsaccharins. Org. Biomol. Chem. 2017, 15, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, R.; Szostak, M. Suzuki-Miyaura cross-coupling of N-acylpyrroles and pyrazoles: Planar, electronically activated amides in catalytic N-C cleavage. Org. Lett. 2017, 19, 3596–3599. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Lalancette, R.; Szostak, R.; Szostak, M. N-methylamino pyrimidyl amides (MAPA): Highly reactive, electronically-activated amides in catalytic N-C(O) cleavage. Org. Lett. 2017, 19, 4656–4659. [Google Scholar] [CrossRef] [PubMed]
- Masson-Makdissi, J.; Vandavasi, J.K.; Newman, S.G. Switchable selectivity in the Pd-catalyzed alkylative cross-coupling of esters. Org. Lett. 2018, 20, 4094–4098. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, J.; Rueping, M. Nickel-catalyzed C−O bond-cleaving alkylation of esters: Direct replacement of the ester moiety by functionalized alkyl chains. ACS Catal. 2017, 7, 4491–4496. [Google Scholar] [CrossRef]
- Chatupheeraphat, A.; Liao, H.-H.; Srimontree, W.; Guo, L.; Minenkov, Y.; Poater, A.; Cavallo, L.; Rueping, M. Ligand-controlled chemoselective C(acyl)−O Bond vs C(aryl)−C bond activation of aromatic esters in nickel catalyzed C(sp2)−C(sp3) cross-couplings. J. Am. Chem. Soc. 2018, 140, 3724–3735. [Google Scholar] [CrossRef] [PubMed]
- Simmons, B.J.; Weires, N.A.; Dander, J.E.; Garg, N.K. Nickel-catalyzed alkylation of amide derivatives. ACS Catal. 2016, 6, 3176–3179. [Google Scholar] [CrossRef]
- Liu, X.; Hsiao, C.-C.; Guo, L.; Rueping, M. Cross-coupling of amides with alkylboranes via nickel-catalyzed C-N bond cleavage. Org. Lett. 2018, 20, 2976–2979. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zou, G. Palladium-catalyzed acylative cross-coupling of amides with diarylborinic acids and sodium tetraarylborates. J. Organomet. Chem. 2015, 794, 136–145. [Google Scholar] [CrossRef]
- Si, S.; Wang, C.; Zhang, N.; Zou, G. Palladium-catalyzed room-temperature acylative Suzuki coupling of high-order aryl borons with carboxylic acids. J. Org. Chem. 2016, 81, 4364–4370. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Easily prepared air- and moisture-stable Pd–NHC (NHC=N-heterocyclic carbene) complexes: A reliable, user-friendly, highly active palladium precatalyst for the Suzuki-Miyaura reaction. Chem. Eur. J. 2006, 12, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Fortman, G.C.; Nolan, S.P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: A perfect union. Chem. Soc. Rev. 2011, 40, 5151–5169. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Nolan, S.P. Efficient cross-coupling of aryl chlorides with aryl Grignard reagents (Kumada reaction) mediated by a palladium/imidazolium chloride system. J. Am. Chem. Soc. 1999, 121, 9889–9890. [Google Scholar] [CrossRef]
- Kantchev, E.A.B.; Ying, J.Y. Practical one-pot, three-component synthesis of N-heterocyclic carbene (NHC) ligated palladacycles derived from N,N-dimethylbenzylamine. Organometallics 2009, 28, 289–299. [Google Scholar] [CrossRef]
- Brahmachari, G. Room Temperature Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780128011386. [Google Scholar]
- Brown, H.C.; Sharp, R.L. Hydroboration. XXIV. Directive effects in the hydroboration of some substituted styrenes. J. Am. Chem. Soc. 1966, 88, 5851–5854. [Google Scholar] [CrossRef]
- Brown, H.C.; Racherla, U.S. Organoboranes. 44. A convenient, highly efficient synthesis of triorganylboranes via a modified organometallic route. J. Org. Chem. 1986, 51, 427–432. [Google Scholar] [CrossRef]
- Samanta, S.; Mishra, B.K.; Pace, T.C.S.; Sathyamurthy, N.; Bohne, C.; Moorthy, J.N. β-Phenyl quenching of triplet excited ketones: How critical is the geometry for deactivation? J. Org. Chem. 2006, 71, 4453–4459. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.L.N.; Venkatesh, V.; Banerjee, D. Atom-efficient cross-coupling reactions of triarylbismuths with acyl chlorides under Pd(0) catalysis. Tetrahedron 2007, 63, 12917–12926. [Google Scholar] [CrossRef]
- Vu, M.D.; Das, M.; Liu, X.-W. Direct aldehyde Csp2-H functionalization through visible light mediated photoredox catalysis. Chem. Eur. J. 2017, 23, 15899–15902. [Google Scholar] [CrossRef] [PubMed]
- Zimbron, J.M.; Seeger-Weibel, M.; Hirt, H.; Gallou, F. Development of a robust and practical process for the Darzens condensation and α, β-epoxide rearrangement: Scope and limitations of the methodology. Synthesis 2008, 8, 1221–1226. [Google Scholar] [CrossRef]
- Liu, M.; Hyder, Z.; Sun, Y.; Tang, W.; Xu, L.; Xiao, J. Efficient synthesis of alkyl aryl ketones & ketals via palladium-catalyzed regioselective arylation of vinyl ethers. Org. Biomol. Chem. 2010, 8, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Suchand, B.; Satyanarayana, G. KOtBu-mediated domino isomerization and functionalization of aromatic allylic alcohols. Eur. J. Org. Chem. 2017, 3886–3895. [Google Scholar] [CrossRef]
- Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. In situ generated (hypo)iodite catalysts for the direct α-oxyacylation of carbonyl compounds with carboxylic acids. Angew. Chem. Int. Ed. 2011, 50, 5331–5334. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Tang, Y.; Ling, Z.; Cun, L.; Zhu, J.; Liao, J.; Deng, J. Chemoselective conjugate reduction of α, β-unsaturated ketones catalyzed by rhodium amido complexes in aqueous media. J. Org. Chem. 2010, 75, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Petrillo, D.E. Suzuki-Miyaura cross-coupling of potassium trifluoroboratohomoenolates. Org. Lett. 2008, 10, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Colbon, P.; Ruan, J.; Purdie, M.; Xiao, J. Direct acylation of aryl chlorides with aldehydes by palladium−pyrrolidine co-catalysis. Org. Lett. 2010, 12, 3670–3673. [Google Scholar] [CrossRef] [PubMed]
- Vautravers, N.R.; Regent, D.D.; Breit, B. Inter- and intramolecular hydroacylation of alkenes employing a bifunctional catalyst system. Chem. Commun. 2011, 47, 6635–6637. [Google Scholar] [CrossRef] [PubMed]
- Malanga, C.; Aronica, L.A.; Lardicci, L. Direct Nio mediated synthesis of ketones from acyl bromides and Grignard reagents. Tetrahedron Lett. 1995, 36, 9185–9188. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Equiv of 2a | Solvent | Base b | T (°C) | Time (h) | Yield (%) c |
|---|---|---|---|---|---|---|
| 1 | 1.1 | THF | K2CO3 | 60 | 5 | 61 |
| 2 | 1.1 | THF | / | 60 | 5 | NR d |
| 3 | 1.1 | THF | Cs2CO3 | 60 | 5 | 25 |
| 4 | 1.1 | THF | K3PO4 | 60 | 5 | 33 |
| 5 | 1.1 | THF | Na2CO3 | 60 | 8 | 15 |
| 6 | 1.1 | THF | NaHCO3 | 60 | 8 | Trace d |
| 7 | 1.1 | THF | NaOAc | 60 | 8 | Trace d |
| 8 | 1.1 | THF | Et3N | 60 | 8 | NR d |
| 9 | 1.1 | THF | Pyridine | 60 | 8 | NR d |
| 10 | 1.5 | THF | K2CO3 | 60 | 5 | 80 |
| 11 | 1.5 | Dioxane | K2CO3 | 60 | 8 | 35 |
| 12 | 1.5 | MTBE | K2CO3 | 55 | 8 | 90 |
| 13 | 1.5 | MeCN | K2CO3 | 60 | 8 | 17 |
| 14 | 1.5 | MTBE | K2CO3 | rt | 24 | 98 |
| 15 | 1.5 | MTBE | K3PO4 | rt | 24 | 84 |
| 16 | 1.5 | THF | K2CO3 | rt | 24 | 82 |
| 17 | 1.5 | THF | K3PO4 | rt | 12 | 40 |

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, W.; Zou, G. Palladium-Catalyzed Room Temperature Acylative Cross-Coupling of Activated Amides with Trialkylboranes. Molecules 2018, 23, 2412. https://doi.org/10.3390/molecules23102412
Shi W, Zou G. Palladium-Catalyzed Room Temperature Acylative Cross-Coupling of Activated Amides with Trialkylboranes. Molecules. 2018; 23(10):2412. https://doi.org/10.3390/molecules23102412
Chicago/Turabian StyleShi, Weijia, and Gang Zou. 2018. "Palladium-Catalyzed Room Temperature Acylative Cross-Coupling of Activated Amides with Trialkylboranes" Molecules 23, no. 10: 2412. https://doi.org/10.3390/molecules23102412